

Abstract
Genetic diversity and population structure of Plasmodium vivax parasites can predict the origin and spread of novel variants within a population enabling population specific malaria control measures. We analyzed the genetic diversity and population structure of 425 P. vivax isolates from Sri Lanka, Myanmar, and Ethiopia using 12 trinucleotide and tetranucleotide microsatellite markers. All three parasite populations were highly polymorphic with 3–44 alleles per locus. Approximately 65% were multiple-clone infections. Mean genetic diversity (HE) was 0.7517 in Ethiopia, 0.8450 in Myanmar, and 0.8610 in Sri Lanka. Significant linkage disequilibrium was maintained. Population structure showed two clusters (Asian and African) according to geography and ancestry. Strong clustering of outbreak isolates from Sri Lanka and Ethiopia was observed. Predictive power of ancestry using two-thirds of the isolates as a model identified 78.2% of isolates accurately as being African or Asian. Microsatellite analysis is a useful tool for mapping short-term outbreaks of malaria and for predicting ancestry.
Introduction
Malaria caused by Plasmodium vivax is often regarded as a benign and self-limiting infection. However, there is increasing evidence that the overall burden, economic impact, and the severity of disease caused by P. vivax have been underestimated.1 The current knowledge on genetic diversity and population structure of P. vivax lags behind that of P. falciparum, mostly because of lack of appropriate genetic markers for the P. vivax genome2 and its inability to be propagated continuously in the laboratory except in non-human primates.3 Sequencing of the P. vivax genome was completed recently3 and provides a valuable resource adding stimulus to the much needed study of this neglected species.
Analysis of allelic variation at multiple independent loci provides an effective means to determine population structure.4–6 Microsatellites7 have been estimated to mutate at high rates,8,9 and the mechanism of mutation appears to be strand slippage at replication10 leading to either a lengthening or shortening of the DNA. This variation in length is assumed to give rise to new electrophoretically distinguishable alleles. Thus, genetic diversity and evolutionary history inferred from microsatellite data are based on similarity of electrophoretic mobility, rather than by descent.
The distinct pathologic and epidemiologic nature of P. vivax malaria is caused by several biological characteristics of P. vivax, which makes it differ from P. falciparum. P. vivax forms hypnozoite stages in the liver, which may persist for months or years before initiating relapses (secondary infections), which provide a mechanism for the parasite to hibernate during less optimal transmission periods.11 Differences in Anopheles mosquito dynamics, which provides P. vivax with a broader temperature tolerance, enables transmission even in temperate climates not tolerated by P. falciparum.3 Relapses and broader temperature tolerance could be expected to increase gene flow between different geographic regions. However, the distinctive phenotypic features found in parasite populations from different countries suggests barriers to gene flow, or alternatively, strong selection maintaining differentiation at loci underlying these traits.5
The ability of P. vivax malaria to re-emerge in regions where malaria eradication or control efforts in the past have been successful is already evident in countries such as Uzbekistan,12 Azerbaijan,13 the Republic of Korea14 and northern Afghanistan.15 Because of increased international travel and migration, the risk of re-introduction of P. vivax in areas where species of Anopheles with greater vectorial capacity are present is high. The analysis of the parasites genetic make-up would be useful in identifying the geographic origins of these parasites,16 and facilitate meaningful control and preventive measures to be implemented.
In this study, we analyzed the genetic diversity and population structure of 425 P. vivax patient isolates collected in Sri Lanka (2003–2008), Myanmar (2007), and Ethiopia (2006–2008) using 12 highly polymorphic trinucleotide and tetranucleotide microsatellite markers, which have been validated and tested.17 We considered the geographic and temporal influences on the genetic diversity and population structure of these P. vivax patient isolates. We further evaluated the use of these microsatellite haplotypes as a model for predicting the ancestry of P. vivax isolates using the Bayesian algorithm software STRUCTURE.
Materials and Methods
Field isolates.
Plasmodium vivax microsatellite typing was conducted on 425 field isolates collected in Sri Lanka (140), Myanmar (167), and Ethiopia (118). A field isolate is defined in this study as a sample of parasites derived from a single-infected patient collected on a single occasion. Most patients had fever and were diagnosed as having P. vivax malaria by microscopic examination of Giemsa-stained thick and thin blood smears. Transmission levels of malaria differed in the three countries, with the incidence rates in 2006 ranging between 50 and 200, 5 and 49, and 0 and 4 per thousand population in Ethiopia, Myanmar, and Sri Lanka respectively.18 Malaria transmission has decreased steadily in Sri Lanka since 2001 from 55,922 cases to 189 cases of P. vivax malaria in 2007 (Annual Report Anti-Malaria Campaign, Ministry of Health, Sri Lanka). Malaria is endemic or hypoendemic in Myanmar. Although morbidity and mortality rates for malaria have decreased gradually in Myanmar, malaria still contributes to a large proportion of deaths in the southeast Asia region.19 Malaria transmission in Ethiopia is unstable and seasonal with occasional devastating epidemics.20
The 140 field isolates of P. vivax from Sri Lanka were collected from patients attending medical health clinics in Trincomalee, Batticaloa, Ampara (Eastern Province), Anuradhapura, Polonnaruwa (Northcentral Province), Kurunegala (Northwestern Province), Vavuniya (Northern Province), and Colombo. Venous blood samples were collected from these patients in 2003 (SL47, n = 01), 2004 (SLA1–SLH4, n = 38), 2005 (SL45, SLA7–SLP2E1, n = 61), 2006 (SLP2A2–SLP2H3, n = 18), 2007 (SL24–SL44, n = 21), and 2008 (SL46, n = 01). The 21 isolates collected during 2007 were from an outbreak of malaria that occurred in Trincomalee.
All 167 field isolates of P. vivax from Myanmar were collected between July and November 2007 from patients attending medical health clinics at Kayin State, which is situated along the Myanmar–Thailand border (MYM3–MYM229, n = 96), Kachin State (MYKC19–MYKC1478, n = 56), and Rakhine State, along the Myanmar-Bangladesh border (MYY148–MYY1264, n = 15). Capillary blood from a fingerprick was collected from each patient on a filter paper.
The 118 P. vivax field isolates from Assendabo, Ethiopia, were collected from fever patients attending health clinics during 2006 (E1–E61, n = 59), 2007 (E84V–E2400V, n = 52), and 2008 (E434–E488V, n = 07). Capillary blood from a fingerprick was collected from each patient on a filter paper.
Either a venous or capillary blood sample was collected from each infected patient after informed consent was obtained. Filter paper samples from Ethiopia were de-linked from patient identifiers and provided as discarded samples from the clinic. DNA templates for polymerase chain reaction (PCR) amplification were isolated from venous blood by using the Nucleon genomic DNA extraction kit (Tepnel Life Sciences, Manchester, United Kingdom) and from filter papers by using either the fast methanol-based protocol21 or the QIAamp DNA purification Mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Extracted DNA was dissolved in sterile distilled water and stored at −20°C. Because of limited availability of genomic DNA in these samples, the extracted DNA was further subjected to whole genome amplification (WGA) using a REPLI-g Mini-kit (Qiagen). The study protocol was reviewed and approved by the Human Subjects Committee of the Harvard School of Public Health (#P10299-111/0209GENE) and the Ethical Review Committee of the Faculty of Medicine, University of Colombo (EC/08/092).
Microsatellite typing of P. vivax.
Twelve validated highly polymorphic microsatellite markers17 (MS1, MS2, MS3, MS4, MS5, MS7, MS8, MS10, MS12, MS15, MS16, and MS20) were used for typing all isolates. Apart from MS2, which has a tetranucleotide repeat array, all other markers consist of trinucleotide tandem repeat motifs. These 12 markers are situated on different chromosomes (Table 1).22 The 12 markers were PCR-amplified using oligonucleotide primers with the forward primer labeled with fluorescent dyes (6-FAM, VIC, NED, and PET) as shown in Table 1. The PCRs with all primer pairs (6.4 pmol working solution of each) were performed on a PTC-200 thermo cycler (MJ Research, Waltham, MA) with 1.5 µL of genomic DNA template, 1 unit of Platinum Taq polymerase, 0.7 µL of 50 mM MgCl2, 2.0 µL of 10× reaction buffer, and 1 mM of each dNTP (all supplied by Invitrogen, Carlsbad, CA) in a final reaction volume of 20 µL. Cycling parameters were the same for all primer pairs: 1 cycle at 94°C for 2 minutes; 40 cycles at 94°C for 30 seconds, 58°C for 40 seconds, and 72°C for 30 seconds; and a final cycle at 72°C for 5 minutes. Length variation of labeled PCR products was measured on an ABI PRISM 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA) by using ABI GS500LIZ internal size standards and the GENESCAN, GENOTYPER, and GENEMAPPER software (Applied Biosystems).
Table 1.
Characterization of the 12 polymorphic Plasmodium vivax microsatellite loci (n = 425)
| Locus | Chromosome | Core repeat sequence in the Salvador-1 strain | Primers (5'→3') | Size range, basepairs | No. alleles | HE | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| SL | M | E | SL | M | E | |||||
| MS1 | 3 (CM000444) | (GAA)11 | F:6-FAMTCAACTGTTGGAAGGGCAAT R:ctgtcttTTGCTGCGTTTTTGTTTCTG | 201–249 | 16 | 17 | 13 | 0.8997 | 0.9022 | 0.7692 |
| MS2 | 6 (CM000447) | (TAAA)2TATA(TAAA)6TATA (TAAA)19 | F:VICGAGCTAGCCAAAGGTTCAACA R:ctgtcttTGGGGAGAGACTCCCTTTTC | 182–286 | 16 | 21 | 13 | 0.8514 | 0.8929 | 0.8014 |
| MS3 | 4 (CM000445) | (GAA)11 | F:NEDGAAGATCCTGTGGAGGAGCA R:ctgtcttCTCCTTCGCTCCTTTCCTTT | 152–200 | 15 | 16 | 14 | 0.8835 | 0.8445 | 0.8617 |
| MS4 | 6 (CM000447) | (AGT)18 | F:PETCGATTTACTGTTGACGCTGAA R:ctgtcttCAAAGGAACATGCTCGATGA | 171–228 | 11 | 18 | 14 | 0.7848 | 0.8116 | 0.7850 |
| MS5 | 6 (CM000447) | CCTCTT(CCT)11 | F:NEDCGTCCTCTATCGCGTACACA R:ctgtcttAAAGGGAGAGGAGCGAAAAC | 139–232 | 17 | 16 | 6 | 0.8068 | 0.7933 | 0.6577 |
| MS7 | 12 (CM000453) | (GAA)9 | F:6-FAMTTGCAGAAAATGCAGAGAGC R:ctgtcttAGGGTCTTCAGCGTGTTGTT | 124–178 | 17 | 17 | 9 | 0.8526 | 0.9004 | 0.6523 |
| MS8 | 12 (CM000453) | (CAG)2(CAA)11 | F:NEDAGAGGAGGCAGAAATGCAGA R:ctgtcttAGCCCCTTTGCGTTCTTTAT | 165–345 | 42 | 44 | 37 | 0.9701 | 0.9666 | 0.9534 |
| MS10 | 13 (CM000454) | GAA(GGA)2AGA(GGA)9AGA(GGA)4AGAGGAAGA(GGA)3AGAGGAAGA(GGAAAA)4(GGA)2(AGA)11(GGA)3(AGA)2GGAGA(GGA)2 | F:PETTTATCCCTGCTGGATGTGAA R:ctgtcttTCCTTCAGGTGGGACTTGTT | 174–249 | 19 | 23 | 18 | 0.8973 | 0.9095 | 0.9035 |
| MS12 | 5 (CM000446) | (TTC)10(TGC)4 | F:6-FAMAATGCGCATCCTATGTCTCC R:ctgtcttCTGCTGTTGTTGTTGCTGCT | 191–263 | 20 | 24 | 19 | 0.9305 | 0.9397 | 0.8983 |
| MS15 | 5 (CM000446) | (TCT)10 | F:6-FAMTGTTTGCAAAGGAATCCACA R:ctgtcttCGGCCAGATGAAAAGGATAA | 236–263 | 10 | 9 | 8 | 0.8502 | 0.8570 | 0.7536 |
| MS16 | 9 (CM000450) | (ACA)9GCA(ACA)3GCA(ACA)7GCA(ACA)3GCAATC(ACA)2ACC(ACA)4ACC(ACA)3GCAATC(ACA)13 | F:PETTGTTGTGGTTGTTGATGGTGA R:ctgtcttGTCGGGGAGAACAACAACAT | 132–330 | 21 | 10 | 3 | 0.6893 | 0.3682 | 0.1995 |
| MS20 | 10 (CM000451) | (GAA)11GAG(GAA)13(CAA)4GAA(CAA)5 | F:VICGCACAACAAATGCAAGATCC R:ctgtcttGTGGCAGTGGCTCATCTTCT | 150–246 | 31 | 30 | 20 | 0.9164 | 0.9540 | 0.7843 |
Data analysis.
The single or predominant allele at each locus was considered for computing allele frequencies.23 All 12 markers are single-copy loci. Because blood-stage malaria parasites are haploid, the presence of one or more additional alleles at a particular locus was interpreted as a co-infection with two or more genetically distinct clones (multiple-clone infections) in the same isolate.5,24 An additional allele was scored if the peak was at least one-third the height of the predominant allele (highest peak) as shown by the electropherogram traces, a method that has been validated.5,6 Only the predominant allele was used to define haplotypes in multiple-clone infections.5,6,24
The genetic diversity of the parasite populations from Sri Lanka, Myanmar, and Ethiopia was determined by calculating the virtual heterozygosity (HE) at each locus in each separate population. Virtual heterozygosity is the average probability that a pair of alleles randomly obtained from the population is different and was defined as HE = [n/(n − 1)][1 − Σp2i], with n being the number of isolates analyzed and pi the frequency of the ith allele in the population. Virtual heterozygosity ranges between 0 and 1, with values close to 1 reflecting high genetic diversity levels in a population. The eBurst version 3 software25,26 was used to search for nearly identical multilocus haplotypes (those differing by a single locus).27
A standardized index of association (ISA) was used to test for evidence of overall multilocus linkage disequilibrium in each parasite population from Sri Lanka, Myanmar, and Ethiopia. This test compares the variance (VD) of the number of alleles shared between all pairs of haplotypes observed in the population (D) with the variance expected under random association of alleles (VE) and is defined as ISA = (VD/VE − 1)(r − 1), with r being the number of loci analyzed.28 VE is derived from 10,000 simulated data sets in which alleles were randomly reshuffled among haplotypes. Significant linkage disequilibrium is detected if VD is > 95% of the values derived from the reshuffled data sets. Data was analyzed using LIAN software version 3.5.29,30
The Bayesian clustering model to assign isolates to K populations according to allele frequencies at each locus was applied using STRUCTURE 2.2 software31 to test whether microsatellite haplotypes clustered according to the geographic origins of the isolates. The program was run 10 times at each of 8 different K values (K1–6) with a burn-in period of 50,000 iterations followed by 105 iterations. The admixture model was used in all analyses. The number of populations was inferred by plotting the log probability of the data [Ln P(D)] for each K value. The strongest statistical support was associated with K = 2 (P > 0.999). For predicting the power of ancestry, two-thirds of isolates (n = 283) were selected randomly as a training model. These isolates with known geographic origin were used by STRUCTURE to distinguish between Asian and African samples. The remaining isolates (N = 142) were analyzed in a blinded manner. The program was run using different migration rates (MIGRPRIOR = 0.001, 0.01, 0.05, and 0.1); best results were obtained with MIGRPRIOR = 0.1. Isolates with predominant ancestry (> 70%) were considered as members of that particular population.
Results
Microsatellite diversity in natural P. vivax isolates from Sri Lanka.
All parasite isolates from Sri Lanka were highly polymorphic and showed unique haplotypes, with the number of alleles per locus ranging from 10 to 42 (mean = 19.6; Table 1 and Supplemental Tables 1 and 2, available at www.ajtmh.org). No single haplotype was shared between isolates. Adjacent haplotypes (differing at a single locus) were seen between two pairs of isolates from Sri Lanka (SL32-SL40 and SL28-34), which were from one location in Sri Lanka (Trincomalee) collected during an outbreak in 2007. The average genetic distance (1 − proportion of microsatellite alleles shared by pairs of haplotypes) of isolates from Sri Lanka collected during the 2007 outbreak in Tricomalee (outbreak isolates = 0.7845) was significantly lower than that calculated for pairwise comparisons involving outbreak isolates and the rest of the isolates collected from Sri Lanka (non-outbreak isolates = 0.854; P < 0.0001, by two-sample randomization test performed with Poptools 2.5 software).32 No identical haplotypes occurred in either outbreak or non-outbreak isolates. 3.4% (N = 57) alleles were private (present only in the Sri Lankan population) and not shared between populations. 55.0% (N = 77) of isolates were mixed-clone infections.
The virtual heterozygosity (HE) values for the entire parasite population from Sri Lanka ranged from 0.6893 to 0.9701 (mean ± SE = 0.8610 ± 0.0216; Table 1). High diversity values were present even when considered according to the year of collection of the isolates (Table 2).
Table 2.
Genetic diversity of 12 microsatellite markers of Plasmodium isolates according to time and site of collection (n = 425)
| Country | Year | Location | No. | HE | ISA | P |
|---|---|---|---|---|---|---|
| Sri Lanka | 2003 | Colombo | 1 | |||
| 2004 | Anuradhapura | 20 | 0.8491 | 0.0113 | 0.145 | |
| Polonnaruwa | 16 | 0.8611 | 0.0143 | 0.121 | ||
| Mannar | 1 | |||||
| Ampara | 1 | |||||
| Total | 38 | 0.8602 | 0.0120 | 0.02 | ||
| 2005 | Trincomalee | 20 | 0.8215 | 0.0127 | 0.146 | |
| Batticaloa | 17 | 0.7953 | 0.0669 | < 0.001 | ||
| Ampara | 4 | 0.7778 | 0.2104 | 0.002 | ||
| Polonnaruwa | 4 | 0.8750 | 0.0350 | 0.386 | ||
| Vavuniya | 7 | 0.8492 | 0.0548 | 0.07 | ||
| Kurunegala | 3 | |||||
| Mannar | 3 | |||||
| Monaragala | 2 | |||||
| Colombo | 1 | |||||
| Total | 61 | 0.8542 | 0.0275 | < 0.001 | ||
| 2006 | Kurunegala | 17 | 0.6906 | 0.0116 | 0.156 | |
| Monaragala | 1 | |||||
| Total | 18 | 0.6912 | 0.0083 | 0.229 | ||
| 2007 | Trincomalee | 21 | 0.7845 | 0.2709 | < 0.001 | |
| 2008 | Colombo | 1 | ||||
| Total | 140 | 0.8610 | 0.0236 | < 0.001 | ||
| Myanmar | 2007 | Kayin State | 96 | 0.8536 | 0.0130 | < 0.001 |
| Kachin State | 56 | 0.8180 | 0.0313 | < 0.001 | ||
| Rakhine State | 15 | 0.8317 | 0.0164 | 0.139 | ||
| Total | 167 | 0.8450 | 0.0149 | < 0.001 | ||
| Ethiopia | 2006 | Assendabo | 59 | 0.7432 | 0.0199 | < 0.001 |
| 2007 | Assendabo | 52 | 0.7314 | 0.0180 | 0.006 | |
| 2008 | Assendabo | 7 | 0.6190 | 0.0697 | 0.044 | |
| Total | 118 | 0.7517 | 0.0137 | < 0.001 |
Significant multilocus linkage disequilibrium was found in the isolates (n = 140) from Sri Lanka (ISA = 0.0236, P < 0.001; Table 2). ISA was also calculated separately for the isolates considering their time of collection, and significant linkage disequilibrium was seen for subpopulations from 2004, 2005, and 2007: ISA = 0.0120, 0.0275, and 0.2709, respectively; P < 0.05). The association was extremely high in the 2007 subpopulation (all collected from a single site) in comparison to the other subpopulations. Further analysis of ISA conducted according to geographic location of isolates and times of collection showed significant associations at individual sites: Batticaloa (2005, n = 17; ISA = 0.0669), Trincomalee (2007, n = 21; ISA = 0.2709), and Ampara (2005, n = 4; ISA = 0.2104); the ISA in the 2005 collection from Trincomalee (n = 20; ISA = 0.0127) was not significant (Table 2).
Microsatellite diversity in natural P. vivax isolates from Myanmar.
The parasite isolates from Myanmar were also highly polymorphic, with the number of alleles per locus ranging from 9 to 44 (mean = 20.4; Table 1 and Supplemental Tables 1 and 2). All isolates showed unique haplotypes with 5.6% (n = 112) of the alleles being private to Myanmar. A total of 67.1% (n = 112) were mixed-clone infections. The virtual heterozygosity (HE) values for the parasite population from Myanmar (n = 167) ranged from 0.3682 to 0.9666 (mean ± SE = 0.8450 ± 0.0460; Table 1) and showed significant multilocus linkage disequilibrium with ISA = 0.0149, P < 0.001 (Table 2). Significant ISA was encountered in collections from Kayin State (2007, n = 96, ISA = 0.0130, P < 0.001) and Kachin State (2007, n = 56, ISA = 0.0313, P < 0.001), but not from Rakhine State (2007, n = 15, ISA = 0.0164, P = 0.139).
Microsatellite diversity in natural P. vivax isolates from Ethiopia.
Parasite populations from Ethiopia were also highly polymorphic, having between 3 and 37 alleles per locus (mean = 14.5; Table 1 and Supplemental Tables 1 and 2). All isolates had unique haplotypes with only 0.07% (n = 1) of the alleles being private to Ethiopia. A total of 73.70% (n = 87) were mixed-clone infections. The virtual heterozygosity (HE) values for the entire parasite population from Ethiopia ranged from 0.1995 to 0.9534 (mean ± SE = 0.7517 ± 0.0568; Table 1). Comparable levels of genetic diversity were found for isolates collected in 2006 (n = 59; HE = 0.7432 ± 0.0551), 2007 (n = 52; HE = 0.7314 ± 0.0695), and 2008 (n = 7; HE = 0.6190 ± 0.0839). Significant multilocus linkage disequilibrium was also seen among the isolates from Ethiopia (n = 118), with ISA = 0.0137, P < 0.001 (Table 2) remaining significant even when considered according to the time of collection of the isolates: ISA = 0.0199, P < 0.001 for 2006, ISA = 0.0180, P = 0.006 for 2007, and ISA = 0.0697, P = 0.044 for 2008 (n = 7).
Geographic structure of P. vivax.
Analysis of 425 isolates from Sri Lanka, Myanmar (Asia) and Ethiopia (Africa) identified haplotypes that were all unique. Private alleles for Asia and Africa were 13.4% and 0.07% respectively. Multiple-clone infections were 64.9% (n = 276).
The clustering patterns obtained with K = 2 shown in Figure 1 showed clustering of P. vivax isolates into Asian and African origins with much admixture in both regions. Adding on a third population (K = 3, P < 0.001) did not help to further differentiate between populations from Sri Lanka and Myanmar (Figure 2). However, the first 21 (Figure 2) isolates from Sri Lanka collected from one site (Trincomalee) during a P. vivax malaria outbreak in 2007 and isolates from Assendabo, Ethiopia (308–425; Figure 2) collected during seasonal outbreaks in 2006, 2007, and 2008 are shown clearly in different clusters. The population structure of P. vivax according to ancestry at K = 2 is shown in Figure 3. The red cluster shows 62.7% (n = 74) of isolates with predominant (> 70%) ancestry from Africa, and the green cluster shows only a 48.8% (n = 150) membership fraction from Asia.
Figure 1.

A, Population structure of Plasmodium vivax plotted in a single line according to geographic origin (Asian or African) inferred from microsatellite typing of 425 isolates at K = 2. Isolates are numbered as 1–140 from Sri Lanka, 141–307 from Myanmar, and 308–425 from Ethiopia. B, Population structure of P. vivax plotted in multiple lines according to geographic origin (Asian or African) inferred from microsatellite typing of 425 isolates at K = 2. Isolates 1–140 were collected in Sri Lanka: 1–21 in Trincomalee (2007); 22–41 in Trincomalee (2005); 42–61 in Anuradhapura (2004); 62–77 in Polonnaruwa (2004); 78–81 in Polonnaruwa (2005); 82–88 in Vavuniya (2005); 89 in Mannar (2004); 90–92 in Mannar (2005); 93–109 in Batticaloa (2005); 100 in Ampara (2004); 111–114 in Ampara (2005); 115–116 in Monaragala (2005); 117 in Monaragala (2006); 118–120 in Kurunegala (2005); 121–137 in Kurunegala (2006); 138 in Colombo (2005); 139 in Colombo (2008); 140 in Colombo (2003). Isolates 141–307 were collected in Myanmar: 141–236 in Kayin State (2007); 237–292 in Kachin State (2007); 293–307 in Rakhine State (2007). Isolates 308–425 were collected in Ethiopia: 308–366 in Assendabo (2006); 367–418 in Assendabo (2007); 419–425 in Assendabo (2008). This figure appears in color at www.ajtmh.org.
Figure 2.
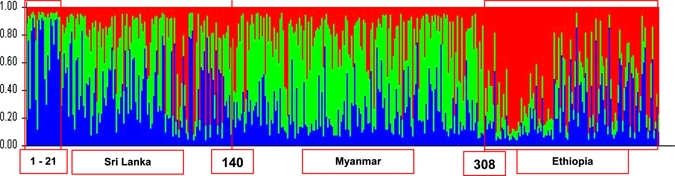
Population structure of Plasmodium vivax plotted in a single line according to geographic location inferred from microsatellite typing of 425 isolates at K = 3. Isolates 1–140 were collected in Sri Lanka; 141–307 in Myanmar; and 308–425 in Ethiopia. This figure appears in color at www.ajtmh.org.
Figure 3.
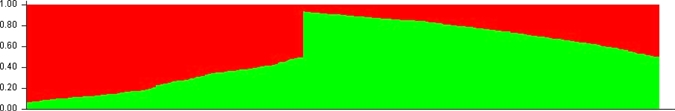
Population structure of Plasmodium vivax according to ancestry inferred from microsatellite typing of 425 isolates at K = 2 (single and multiple line plots). Predominant ancestry (> 70%) for Asian and African isolates in each cluster was as follows: red = 62.7% (n = 74) African and 12.7% (n = 39) Asian; green = 48.8% (n = 150) Asian and 5.9% (n = 7) African. This figure appears in color at www.ajtmh.org.
Predictive power of ancestry of P. vivax isolates.
We further assessed the predictive power of ancestry for P. vivax isolates using two-thirds of the isolates (n = 283) as a model. Of the 142 isolates that were tested in this model, 47 were from Sri Lanka, 56 from Myanmar, and 39 from Ethiopia. The percentages of test isolates that were correctly identified to have a predominant ancestry (> 70%) from either Asian or African origin were 72.3% (n = 34) for Sri Lanka (Asian), 78.6% (n = 44) for Myanmar (Asian), and 84.6% (n = 33) for Ethiopia (African), giving an overall predictive power of 78.2% (n = 111).
Discussion
Microsatellite polymorphisms are derived mainly from variability in length rather than in the primary sequence. Genetic variation at many microsatellite loci are characterized by high heterozygosity and the presence of multiple alleles,33 which is well demonstrated among the 425 isolates from Sri Lanka, Myanmar and Ethiopia. Most of this microsatellite diversity is probably caused by strand slippage during mitotic replication of the parasites, which causes transient dissociation of the replicating DNA strands followed by misaligned re-association, resulting in either the addition or deletion of repeat units.10
Genetic diversity measured by virtual heterozygosity was high in all three populations, which is expected with microsatellite data.5,19,34,35 This high diversity extended to all individual loci except for 1 of the 12 analyzed. Interestingly, the MS16 marker showed diversity levels varying from HE = 0.1995 in the parasite population from Ethiopia to HE = 0.6908 in the population from Sri Lanka. This microsatellite marker is located on chromosome 9 of the P. vivax genome in a gene coding for a hypothetical protein (PVX_092630, Seq. ID CM000450-PlasmoDB 5.5). Whether the hypothetical protein coded for by this gene is under selection requires further investigation because fluctuating selection pressures within populations may contribute to reduced variability in regions of reduced recombination.36
Little is known of the global genetic diversity and population structure of P. vivax. This lack of information is due mostly to the lack of appropriate genetic markers for the P. vivax genome, which has hampered in-depth analysis of the population structure and evolutionary history of the parasite and prevented efforts to map determinants contributing to important parasite phenotypes.2 In this study, with the use of 12 highly polymorphic microsatellite markers distributed genome wide, we were able to identify the population structure of P. vivax according to their geographic location (Asian versus African, K = 2). Although the addition of a third population (K = 3) did not improve differentiation between isolates from Sri Lanka and Myanmar (Figure 2), a strong clustering of the outbreak isolates from Sri Lanka (first portion of cluster 1 in Figure 2 shown predominantly in blue) and Ethiopia (cluster 3, predominantly in red) was observed, which is consistent with epidemic transmission of clones. The significantly lower average genetic distance between the outbreak and non-outbreak isolates from Sri Lanka (0.7845 versus 0.854; P < 0.0001) also substantiates this result. Unfortunately, comparisons between outbreak and non-outbreak isolates from Ethiopia could not be made because non-outbreak isolates were not available.
Figure 3 shows the P. vivax populations sorted according to Asian or African ancestry, with much admixture in both fractions, especially the Asian fraction. Almost 63% of isolates from Ethiopia had a predominant ancestry (> 70%) from Africa, and only 45% from Myanmar and 54% from Sri Lanka were shown to be from Asia. Many alleles were shown to be private for Asia (13.4%; Supplemental Table 2) than Africa (0.07%), which corroborates the historical origins of P. vivax in Asia.2,37,38
Microsatellites are known to be a valuable resource for the study of genetic diversity because they are generally hypervariable, codominant, locus-specific, and not directly subject to host immunity.39 However, they have high mutation rates and significant mutation-rate heterogeneity among loci, which means that it might be difficult to translate estimates of genetic distance into absolute timescales.33 More importantly, alleles generated by microsatellite data represent similarities in electrophoretic mobility rather than by descent. Therefore, use of microsatellites in population genetics and evolution has its limitations. However, the population structure generated using these microsatellites show promise for microsatellite data being a good tool to identify parasite differentiation occurring over a short period, such as for mapping outbreaks.
The predictive power of ancestry using two-thirds of isolates as a model gave good results for isolates from all three populations. Predominant ancestry (> 70%) for African or Asian origins were 85% African and 3% Asian for isolates from Ethiopia, 79% Asian and 5% African for isolates from Myanmar, and 72% Asian and 11% African for isolates from Sri Lanka. Thus, microsatellite data as a model for predicting the origin of P. vivax parasites holds promise and would provide useful information regarding genetic structure of parasites, in context to the resurgence of P. vivax malaria in many regions worldwide.16 This approach would be a useful complement to conventional epidemiologic methods with important implications for control and prevention of this parasite.
High levels of genetic diversity in P. vivax have been demonstrated in Papua New Guinea, Thailand, India, Colombia, and Laos by use of dinucleotide microsatellites.5,34,40 High diversity levels have also been observed in India35 and Myanmar19 with the use of genetic markers. Previous studies with P. vivax parasites from Sri Lanka,17 Brazil,4 and Myanmar19 demonstrated high genetic diversity and multiple clone infections despite low levels of transmission in these areas. The co-occurrence of significant linkage disequilibrium in the presence of high diversity and multiple clone infections was also demonstrated in parasites from both Sri Lanka17 and Brazil.4 Findings of the present study involving three geographic sites add further strength to the above observations. Linkage disequilibrium in parasite populations may arise as an artifact of the admixture of two or more subpopulations with different allele frequencies. Therefore, isolates from each population were analyzed individually according to their times and sites of collection. Apart from some sites in Sri Lanka (Trincomalee, 2005, n = 20), Anuradhapura (2004, n = 20), Polonnaruwa (2004 and 2005, n = 16 and 4, respectively), Vavuniya (2005, n = 7), and Kurunegala (2006, n = 16) and a single site in Myanmar (Rakhine State, 2007, n = 12), significant linkage disequilibrium was consistently present among the remaining sites (Table 2).
The predominance of inbreeding in P. vivax with high proportions of multiple-clone infections and genetic diversity is unusual. High rates of selfing may occur in the presence of multiple clone infections if only a small proportion of the co-infecting genetically distinct clones produce viable gametocytes that are taken up by mosquitoes or when strand slippage events occur so frequently during mitotic replication of parasites that new haplotypes would be generated without affecting the overall patterns of association between loci.4,6
In conclusion, microsatellite-based analysis of P. vivax parasites from Sri Lanka, Myanmar, and Ethiopia showed extensive genetic diversity co-occurring with significant multilocus linkage disequilibrium. These parasite populations were seen to cluster according to their geographic locations and ancestry. The predictive power of ancestry with this microsatellite data as a model is promising and could be useful in identifying the origin of P. vivax malaria cases and enabling meaningful control and preventive measures to be implemented. Although microsatellite data may have some limitations in its use in population genetics and evolution of malaria parasites, it appears to serve as an excellent tool for mapping short-term outbreaks of malaria.
Supplementary Material
Acknowledgments
We thank the director of the Anti-Malaria Campaign, Sri Lanka, for authorizing collection of samples; G.M.G. Kapilananda (Department of Parasitology, Faculty of Medicine, University of Colombo) and Nira Mahesh (Department of Immunology and Infectious Diseases, School of Public Health, Harvard University) for technical assistance; Yihenew Tesfaye (Boston University/Harvard School of Public Health project), Abayneh Dega, Enuku Keno Doti, Teklu Aklilu (Assendabo Health Center), and Myat Htut Nyunt (Myanmar) for assistance in providing samples.
Note: Supplemental tables appear at www.ajtmh.org.
Footnotes
Financial support: This study was supported by National Institutes of Health grant 5R03TW007966-02.
Authors’ addresses: Sharmini Gunawardena, Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA and Department of Parasitology, Faculty of Medicine, University of Colombo, Colombo, Sri Lanka. Nadira D. Karunaweera, Department of Parasitology, Faculty of Medicine, University of Colombo, Colombo, Sri Lanka. Marcelo U. Ferreira, Department of Parasitology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil. Myatt Phone-Kyaw, Parasitology Research Division, Department of Medical Research (Lower Myanmar), Myanmar. Richard J. Pollack and Dyann F. Wirth, Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA. Michael Alifrangis, Flemming Konradsen, and Mette L. Schousboe, Centre for Medical Parasitology, Department of International Health, Immunology and Microbiology, University of Copenhagen, and Department of Infectious Diseases and Department of Clinical Microbiology, Copenhagen University Hospital (Rigshospitalet), Copenhagen, Denmark. Rupika S. Rajakaruna, Department of Zoology, University of Peradeniya, Peradeniya, Sri Lanka. Priyanie H. Amerasinghe, International Water Management Institute, Delhi, India. Gawrie N. L. Galappaththy and Rabindra R. Abeyasinghe, Anti-Malaria Campaign, Ministry of Health, Colombo, Sri Lanka. Daniel L. Hartl, Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA.
References
- 1.Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007;77:79–87. [PMC free article] [PubMed] [Google Scholar]
- 2.Das A, Bajaj R, Mohanty S, Swain V. Genetic diversity and evolutionary history of Plasmodium falciparum and P. vivax. Curr Sci. 2007;92:1516–1524. [Google Scholar]
- 3.Carlton JM, Adams JH, Silva JC. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira MU, Karunaweera ND, Silva-Nunes M, Da Silva NS, Wirth DF, Hartl DL. Population structure and transmission dynamics of Plasmodium vivax in rural Amazonia. J Infect Dis. 2007;195:1218–26. doi: 10.1086/512685. [DOI] [PubMed] [Google Scholar]
- 5.Imwong M, Nair S, Pukrittayakamee S. Contrasting genetic structure in Plasmodium vivax populations from Asia and South America. Int J Parasitol. 2007;37:1013–1022. doi: 10.1016/j.ijpara.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Karunaweera ND, Ferreira MU, Munasinghe A, Barnwell JW, Collins WE, King CL, Kawamoto F, Hartl DL, Wirth DF. Extensive microsatellite diversity in the human malaria parasite Plasmodium vivax. Gene. 2008;410:105–112. doi: 10.1016/j.gene.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 7.Schlötterer C. Genome evolution: are microsatellites really simple sequences? Curr Biol. 1998;8:R132–R134. doi: 10.1016/s0960-9822(98)70989-3. [DOI] [PubMed] [Google Scholar]
- 8.Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. High resolution of human evolution with polymorphic microsatellites. Nature. 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 9.Forbes SH, Hogg JT, Buchanan FC, Crawford AM, Allendorf FW. Microsatellite evolution in congeneric mammals: domestic and bighorn sheep. Mol Biol Evol. 1995;12:1106–1113. doi: 10.1093/oxfordjournals.molbev.a040284. [DOI] [PubMed] [Google Scholar]
- 10.Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 11.Joshi H, Prajapati SK, Verma A, Kang’a S, Carlton JM. Plasmodium vivax in India. Trends Parasitol. 2008;24:228–235. doi: 10.1016/j.pt.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Severini C, Menegon M, Di Luca M, Abdullaev I, Majori G, Razakov SA, Gradoni L. Risk of Plasmodium vivax malaria reintroduction in Uzbekistan: genetic characterization of parasites and status of potential malaria vectors in the Surkhandarya region. Trans R Soc Trop Med Hyg. 2004;98:585–592. doi: 10.1016/j.trstmh.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Leclerc MC, Menegon M, Cligny A, Noyer JL, Mammadov S, Aliyev N, Gasimov E, Majori G, Severini C. Genetic diversity of Plasmodium vivax isolates from Azerbaijan. Malar J. 2004;3:40. doi: 10.1186/1475-2875-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim H, Pacha LA, Lee W, Lee J, Gaydos JC, Sames WJ, Lee HS, Bradley K, Jeung G, Tobler SK, Klein TA. Malaria in the Republic of Korea, 1993–2007. Variables related to re-emergence and persistence of Plasmodium vivax among Korean populations and U.S. Forces in Korea. Mil Med. 2009;174:762–769. doi: 10.7205/milmed-d-01-6208. [DOI] [PubMed] [Google Scholar]
- 15.Faulde MK, Hoffmann R, Fazilat KM, Hoerauf A. Malaria re-emergence in northern Afghanistan. Emerg Infect Dis. 2007;13:1402–1404. doi: 10.3201/eid1309.061325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Severini C, Menegon M, Gradoni L, Majori G. Use of the Plasmodium vivax merozoite surface protein 1 gene sequence analysis in the investigation of an introduced malaria case in Italy. Acta Trop. 2002;84:151–157. doi: 10.1016/s0001-706x(02)00186-9. [DOI] [PubMed] [Google Scholar]
- 17.Karunaweera ND, Ferreira MU, Hartl DL, Wirth DF. Fourteen polymorphic microsatellite DNA markers for the human malaria parasite Plasmodium vivax. Mol Ecol Notes. 2007;7:172–175. [Google Scholar]
- 18.World Malaria Report Estimated Burden of Malaria in 2006. 2008. http://www.who.int/healthinfo/bodestimates/en/index.html Available at. Accessed November 12, 2008.
- 19.Moon SU, Lee HW, Kim JY, Na BK, Cho SH, Lin K, Sohn WM, Kim TS. High frequency of genetic diversity of Plasmodium vivax field isolates in Myanmar. Acta Trop. 2009;109:30–36. doi: 10.1016/j.actatropica.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Shargie EB, Gebre T, Ngondi J, Graves PM, Mosher AW, Emerson PM, Ejigsemahu Y, Endeshaw T, Olana D, WeldeMeskel A, Teferra A, Tadesse Z, Tilahun A, Yohannes G, Richards FO. Malaria prevalence and mosquito net coverage in Oromia and SNNPR regions of Ethiopia. BMC Public Health. 2008;8:321. doi: 10.1186/1471-2458-8-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moll K, Ljungstrom I, Perlmann H, Scherf A, Wahlgren M. Methods in Malaria Research. Manassas, VA: American Type Culture Collection; 2008. (Fast methanol-based DNA extraction from blood spots in filter paper). ISBN 0-930009-64-9. [Google Scholar]
- 22.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Jr, Treatman C, Wang H. PlasmoDB: a functional genomic database for malaria parasites. Version 5.5. 2008. http://plasmodb.org Available at. Accessed November 12, 2008. [DOI] [PMC free article] [PubMed]
- 23.Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP. Microsatellite markers reveal a spectrum of population structures in the malaria parasite P. falciparum. Mol Biol Evol. 2000;17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 24.Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–125. doi: 10.1017/s0031182099004552. [DOI] [PubMed] [Google Scholar]
- 25.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST version 3. 2004. http://eburst.mlst.net Available at. Accessed November 12, 2008.
- 27.Cooper G, Amos W, Hoffman D, Rubinsztein DC. Network analysis of human Y microsatellite haplotypes. Hum Mol Genet. 1996;5:1759–1766. doi: 10.1093/hmg/5.11.1759. [DOI] [PubMed] [Google Scholar]
- 28.Hudson RR. Analytical results concerning linkage disequilibrium in models with genetic transformation and recombination. J Evol Biol. 1994;7:535–548. [Google Scholar]
- 29.Haubold B, Hudson RR. LIAN 3.5. PubMLST. 2000. http://pubmlst.org/perl/mlstanalyse/mlstanalyse.pl? site=pubmlst &page=lian& referer=pubmlst.org Available at. Accessed October 12, 2008. [DOI] [PubMed]
- 30.Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage analysis. Bioinformatics. 2000;16:847–848. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- 31.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hood G. Poptools, Version 2.5. 2002. http://www.cse.csiro.au/poptools Available at. Accessed August 18, 2009.
- 33.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 34.Imwong M, Sudimack D, Pukrittayakamee S, Osorio L, Carlton JM, Day NP, White NJ, Anderson TJ. Microsatellite variation, repeat array length and population history of Plasmodium vivax. Mol Biol Evol. 2006;23:1016–1018. doi: 10.1093/molbev/msj116. [DOI] [PubMed] [Google Scholar]
- 35.Kim JR, Imwong M, Nandy A, Chotivanich K, Nontprasert A, Tonomsing N, Maji A, Addy M, Day NPJ, White NJ, Pukrittayakamee S. Genetic diversity of Plasmodium vivax in Kolkata, India. Malar J. 2006;5:71. doi: 10.1186/1475-2875-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charlesworth B. Measures of divergence between populations and the effect of forces that reduce variability. Mol Biol Evol. 1998;15:538–543. doi: 10.1093/oxfordjournals.molbev.a025953. [DOI] [PubMed] [Google Scholar]
- 37.Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, Collins WE, Lal AA. A monkey's tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci USA. 2005;102:1980–1985. doi: 10.1073/pnas.0409652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornejo OE, Escalante AA. The origin and age of Plasmodium vivax. Trends Parasitol. 2006;22:558–563. doi: 10.1016/j.pt.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell B, Suwanarusk R, Lek-Uthai U. Plasmodium vivax genetic diversity: microsatellite length matters. Trends Parasitol. 2006;22:399–401. doi: 10.1016/j.pt.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 40.Gomez JC, McNamara DT, Bockarie MJ, Baird JK, Carlton JM, Zimmerman PA. Identification of a polymorphic Plasmodium vivax microsatellite marker. Am J Trop Med Hyg. 2003;69:377–389. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.