

Abstract
This paper introduces the use of mass spectrometry to analyze peptide arrays for applications in profiling enzyme specificity. The strategy is illustrated with arrays containing 361 acetylated peptides to profile the activities of several histone deacetylases (HDACs). The arrays reveal distinct substrates that are preferred by members of the HDAC family. This example is particularly relevant because the label-dependent assays now used for these enzymes constrain the range of substrates that can be assayed and can perturb the intrinsic activities of the enzymes.
Keywords: Enzymes, Post-translational modifications, SAMDI, Screening
The application of peptide arrays for profiling biochemical activities has expanded since Frank and colleagues first reported the SPOT synthesis methodology, wherein peptides are synthesized directly on a cellulose membrane in a cost-efficient manner.[1] These arrays can be treated with enzymes and then analyzed using absorbance, fluorescence, or radio-isotopic assays to rank the activities of the peptides.[2] The requirement for labels can make it challenging to develop assays for certain enzymes and can also lead to false-positive and false-negative results. These limitations have motivated the development of label free formats based on optical methods[3] and mass spectrometry,[4] including our strategy to combine matrix-assisted laser desorption-ionization mass spectrometry with self-assembled monolayer substrates (i.e. the SAMDI method).[5] In this paper we demonstrate the combination of SAMDI-MS with peptide arrays, with an emphasis on the profiling of substrate specificities of several members of the HDAC family, which play a primary role in the regulation of gene expression.
The assays now used to measure HDAC activity have been important for mechanistic studies and for identifying hit compounds in high throughput screens, but they have limitations when applied to studies of the substrate specificities. One group of assays identifies active substrates by labeling the deacetylated lysine with haptens that allow the isolation and subsequent sequencing of the peptide substrate. Hence, the original peptide substrates must exclude residues susceptible to false positive labeling such as natural lysine, arginine, methionine, and cysteine.[6] A second group of assays conjugates a fluorophore to the carboxy-side of the acetylated lysine. Following HDAC treatment, the chromophore can be proteolytically released only in the deacetylated peptides, providing a convenient assay for HDAC activity compatible with microtiter plates.[7] With this format, the peptide sequence cannot be varied at the carboxy-side of the acetylated lysine and interactions between the fluorophore and enzyme can contribute to activity, making it difficult to understand the intrinsic enzyme specificities. In a clear example of this limitation, the assay revealed that resveratrol activates the SIRT1 deacetylase,[8] yet later work showed this effect to be dependent on the fluorescent label.[9, 10] Still, the fluorophore-based assays are now commercially available—with the Fluor de Lys (fluorescent deacetylation of lysine) kit provided by BIOMOL (herein FdL)—and widely used in the research community.
In this paper, we use an array comprising 361 six-mer peptides having the sequence Ac-GXKAcZGC-NH2 (where X, Z = all amino acids except cysteine) to profile the local substrate specificities of several HDACs. An inspection of crystal structures of enzyme-substrate complexes suggests that six-mer peptides will suffice to determine interactions between the substrate and the enzyme active site.[11] The peptides are selected to reveal the dependence of activity on the amino acids present on both sides of the acetylated residue. We immobilized the peptides in an array format to a self-assembled monolayer of alkanethiolates on gold and used SAMDI-MS to monitor the acetylation states of the peptides.[5, 12]
We used FMOC chemistry to synthesize peptides on SynPhase D-series lanterns in 96-well filter plates to give 361 lantern resins each having a single peptide. The peptides were deprotected, cleaved, and stored as aqueous suspensions in a 384 well plate at −20°C. To prepare the array, we applied 1 μL of each peptide solution onto a glass slide having an array of gold circles modified with a maleimide-terminated self-assembled monolayer.[13] The immobilization reactions were allowed to proceed for 60 minutes at 37°C and then rinsed and used in assays (Figure 1A).
Figure 1.
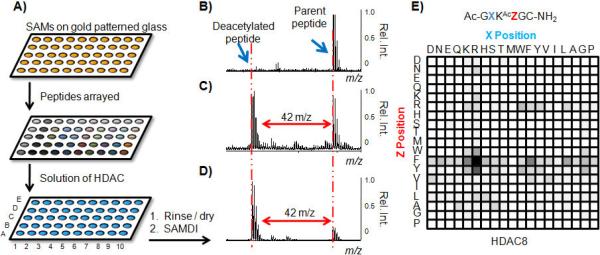
Peptide arrays are treated with an HDAC and analyzed by SAMDI-MS to produce substrate specificity profiles. a) Arrays are prepared by applying peptides to maleimide-presenting SAMs on glass slides patterned with gold circles. SAMDI spectra reveal whether a peptide is b) not active, c) partially active, d) or highly active. HDAC8 substrate specificity profile reveals the dependence of activity on the identity of the residues neighboring the acetylated lysine residue e). Rel. Int. = Relative Intensity.
In a first example, we applied HDAC8 (500 nM in HDAC Buffer) to the array for 60 minutes at 37°C and then rinsed the array with water and ethanol. The arrays were treated with matrix (2,4,6-trihydroxyacetophenone, 20 mg/mL in acetone) and analyzed by SAMDI-MS to acquire a mass spectrum of each peptide. The majority of spectra revealed mass peaks corresponding only to the substrate, evidence that the peptide sequence is a poor substrate (Figure 1B). Less than 5% of the peptides showed partial deacetylation (Figure 1C) and a few showed nearly complete deacetylation (Figure 1D). This experiment reveals that HDAC8 is specific for short peptides having the tripeptide motif RKAcF (Figure 1E). We repeated some reactions in solution on a small subgroup of peptides (including hits and non-hits) in a pull down format[14] and found that the extent of the reactions was highly comparable to the array format, suggesting that the monolayer does not interfere with the reaction (Figure S1).
We repeated the array experiment with several additional HDACs, including the other class I HDACs (1, 2, and 3). We found that HDAC1 and HDAC3, when assayed alone, showed no activity towards any of the peptides in the array (Figure 2A, data not shown for HDAC1), in agreement with certain prior reports,[15] but not with others.[16] When we treated the array with HDAC3 in the presence of an activating cofactor (HDAC3/SMRT), the complex was active and the profile displayed significantly less discrimination than did HDAC8 (Figure 2B). HDAC2, despite high sequence similarity to HDAC1 and HDAC3,[17] did possess catalytic activity and had a clear preference for substrates having an Arg in the X position and either Arg or Tyr in the Z position (Figure 2C). This result suggests that HDAC2 may not rely on protein interactions for activity as do HDAC1 and HDAC3, although a structural comparison is needed to address this point. Finally, SIRT1, a member of the sirtuin class that requires NAD+ for activity, displayed high specificity with a strong preference for Arg in both the X and Z positions (Figure 2D). We note that this substrate could not be assayed using the hapten technique since the Arg residue would react with the labeling step and result in a false positive signal.[6]
Figure 2.
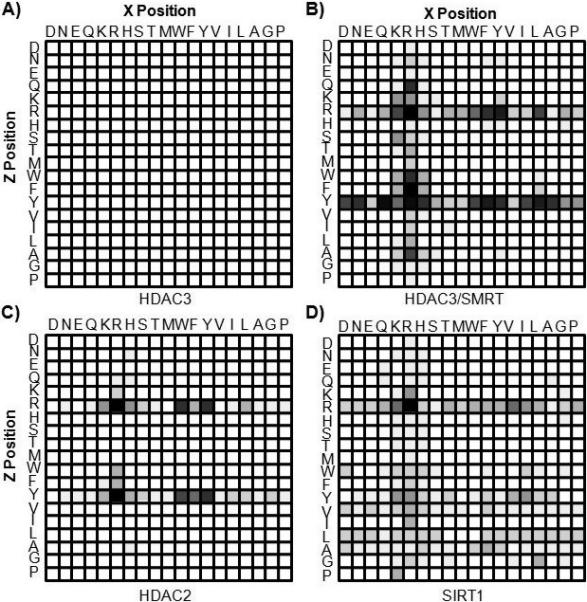
Arrays were treated with several HDACs to provide substrate specificity profiles: a) HDAC3 b) HDAC3/SMRT c) HDAC2 and d) SIRT1.
The activity profiles for HDACs 2, 3/SMRT, and 8 point to the importance of an aromatic residue immediately C-terminal to the acetylated lysine, and point to the suggestion that the FdL label might increase the activity of the substrate by mimicking an aromatic residue. To address this possibility, we compared commercially available fluorescent substrates with their unlabeled counterparts. Because the structure of the fluorophore is not disclosed (and in some cases neither is the peptide sequence) we obtained a MALDI-MS of two FdL substrates—based on residues 379-382 of p53 (Ac-RHKAcKAc-FdL and Ac-RHKKAc-FdL)—to determine the mass of the fluorophore. These masses, together with the excitation and emission wavelengths provided by Biomol, suggest that the fluorophore is a simple amino-methylcoumarin derivative, which has been previously used in other fluorescent HDAC assays,[7] although we admit our claim is not confirmed. We then used mass spectrometry to compare the activities of both the FdL and non-fluorogenic substrates. The HDAC3/SMRT complex showed comparable activity for both labeled and unlabeled Ac-RHKAcKAc. HDAC8, in contrast, efficiently deacetylated the FdL substrates but was inactive towards the corresponding non-labeled peptides (Figure 3). Similarly, HDAC3 alone was active towards a FdL substrate—though the sequence of this substrate is not disclosed—whereas we were unable to identify any active non-labeled substrates for this enzyme.
Figure 3.
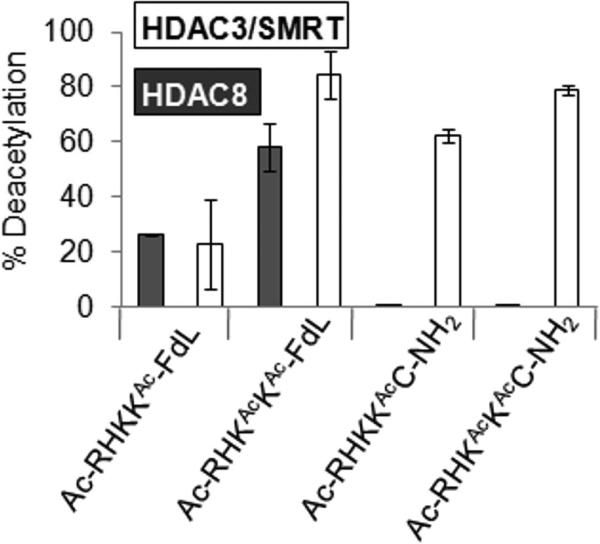
Comparison of the Fluor de Lys (FdL) assay with the SAMDI method. HDAC8 and HDAC3/SMRT were incubated with Ac-RHKKAc-FdL, AcRHKAcKAc-FdL, Ac-RHKKAcC-NH2 and Ac-RHKAcKAcC-NH2 in microtitre plates. The FdL requires the use of a developer solution that contains a protease to cleave the fluorophore to produce a fluorescent signal. For SAMDI, the reactions are stopped by heating to 80°C for 45 seconds and 1 μL of the reaction mixture is immobilized onto a maleimide-presenting SAM. The FdL assay is analyzed on a fluorescent plate scanner and the fluorescent signal is compared to positive and negative controls. The SAMDI method is analyzed by SAMDI-MS and peptides are directly characterized by observing shifts of m/z 42 corresponding to the loss of an acetyl group. HDAC8 is only active on the substrates when the fluorophore is present. HDAC3/SMRT is active on the substrates in the presence or absence of the fluorophore and the extent of reactions are comparable to the FdL assay.
To address the ability of the SAMDI assay to provide semi-quantitative data that can properly assess the relative activities of peptides, we synthesized forty peptides in the deacetylated state. We separately immobilized the acetylated and deacetylated peptides onto SAMs and compared the intensities of the peptide peaks to the background ethylene glycol peak (Figure S2). Importantly, we find that the intensities of the acetylated and deacetylated peptides differ by less than 6%, showing that yields for each peptide can be determined and compared.
This paper describes an approach to efficiently characterize the local substrate specificities of the HDAC enzymes. The use of mass spectrometry expands the range of biochemical activities that can be assayed on peptide arrays and also serves to avoid false positive activities that stem from the either the presence of a fluorophore in the substrate or residues susceptible to interactions with haptens. This work reveals overlap in the specificity profiles of several HDACs, which supports claims that the high sequence similarity between these enzymes[18] leads to a lack of clear differential specificities. Of particular note is the similar specificity that SIRT1 shares with HDAC2 and HDAC3/SMRT, despite different catalytic mechanisms in the activity. We currently do not know whether the similar profiles indicate a high redundancy amongst the enzyme family, a dependency on co-localization to target specific substrates, or evolutionary pressure that selected this local specificity to maintain minimal background activity on other acetylated substrates. Finally, we emphasize that fluorescent substrates can alter the recognition of substrates by certain HDACs and therefore caution must be taken when assessing HDAC specificity using these assays.
Acknowledgments
This work was supported by the NIH (R01 GM084188)
Footnotes
Experimental Section:
Complete details for the source of the reagents, synthesis of the peptide library, peptide array fabrication, SAMDI-MS methods, and HDAC activity assay are provided in the supporting information.
References
- 1.Frank R. Tetrahedron. 1992;48:9217. [Google Scholar]
- 2.Reineke U, Volkmer-Engert R, Schneider-Mergener J. Current Opinion in Biotechnology. 2001;12:59. doi: 10.1016/s0958-1669(00)00178-6. [DOI] [PubMed] [Google Scholar]
- 3.Cooper MA. Analytical and Bioanalytical Chemistry. 2003;377:834. doi: 10.1007/s00216-003-2111-y. [DOI] [PubMed] [Google Scholar]
- 4.Roddy TP, Horvath CR, Stout SJ, Kenney KL, Ho PI, Zhang JH, Vickers C, Kaushik V, Hubbard B, Wang YK. Analytical Chemistry. 2007;79:8207. doi: 10.1021/ac062421q. [DOI] [PubMed] [Google Scholar]
- 5.Mrksich M. Acs Nano. 2008;2:7. doi: 10.1021/nn7004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garske AL, Denu JM. Biochemistry. 2006;45:94. doi: 10.1021/bi052015l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann K, Brosch G, Loidl P, Jung M. Nucleic Acids Research. 1999;27:2057. doi: 10.1093/nar/27.9.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Nature. 2003;425:191. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 9.Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, Kennedy BK. Journal of Biological Chemistry. 2005;280:17038. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 10.Borra MT, Smith BC, Denu JM. Journal of Biological Chemistry. 2005;280:17187. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 11.Zhao KH, Chai XM, Marmorstein R. Structure. 2003;11:1403. doi: 10.1016/j.str.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Gurard-Levin ZA, Mrksich M. Biochemistry. 2008;47:6242. doi: 10.1021/bi800053v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houseman BT, Gawalt ES, Mrksich M. Langmuir. 2003;19:1522. [Google Scholar]
- 14.Min DH, Yeo WS, Mrksich M. Analytical Chemistry. 2004;76:3923. doi: 10.1021/ac049816z. [DOI] [PubMed] [Google Scholar]
- 15.Guenther MG, Barak O, Lazar MA. Molecular and Cellular Biology. 2001;21:6091. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riester D, Hildmann C, Grunewald S, Beckers T, Schwienhorst A. Biochemical and Biophysical Research Communications. 2007;357:439. doi: 10.1016/j.bbrc.2007.03.158. [DOI] [PubMed] [Google Scholar]
- 17.Yang XJ, Seto E. Nature Reviews Molecular Cell Biology. 2008;9:206. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang DF, Helquist P, Wiech NL, Wiest O. Journal of Medicinal Chemistry. 2005;48:6936. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]