

Abstract
Background
Helicosporidia are achlorophyllous, non-photosynthetic protists that are obligate parasites of invertebrates. Highly specialized, these pathogens feature an unusual cyst stage that dehisces inside the infected organism and releases a filamentous cell displaying surface projections, which will penetrate the host gut wall and eventually reproduce in the hemolymph. Long classified as incertae sedis or as relatives of other parasites such as Apicomplexa or Microsporidia, the Helicosporidia were surprisingly identified through molecular phylogeny as belonging to the Chlorophyta, a phylum of green algae. Most phylogenetic analyses involving Helicosporidia have placed them within the subgroup Trebouxiophyceae and further suggested a close affiliation between the Helicosporidia and the genus Prototheca. Prototheca species are also achlorophyllous and pathogenic, but they infect vertebrate hosts, inducing protothecosis in humans. The complete plastid genome of an Helicosporidium species was recently described and is a model of compaction and reduction. Here we describe the complete mitochondrial genome sequence of the same strain, Helicosporidium sp. ATCC 50920 isolated from the black fly Simulium jonesi.
Methodology/Principal Findings
The circular mapping 49343 bp mitochondrial genome of Helicosporidium closely resembles that of the vertebrate parasite Prototheca wickerhamii. The two genomes share an almost identical gene complement and display a level of synteny that is higher than any other sequenced chlorophyte mitochondrial DNAs. Interestingly, the Helicosporidium mtDNA feature a trans-spliced group I intron, and a second group I intron that contains two open reading frames that appear to be degenerate maturase/endonuclease genes, both rare characteristics for this type of intron.
Conclusions/Significance
The architecture, genome content, and phylogeny of the Helicosporidium mitochondrial genome are all congruent with its close relationship to Prototheca within the Trebouxiophyceae. The Helicosporidium mitochondrial genome does, however, contain a number of novel features, particularly relating to its introns.
Introduction
Helicosporidia are single cell parasitic eukaryotes infecting a wide range of insects ([1] and references therein). These entomopathogens feature three different life stages: cysts, filamentous cells and vegetative cells. When the infectous cysts burst open within the gut of their host, they release a filamentous cell with surface barbs along with three egg-shaped accessory cells [2]. The barbed filaments proceed to invade the gut cells, passing though them and emerging into the hemolymph [3]. In their vegetative state within the hemolymph, Helicosporidia reproduce by several rounds of autosporogenic division within the pellicle of the mother cell, with each autosporulation producing up to eight daugther cells [4]. Generally, the infection leads to the death of the host, but the exact mode of transmission remains poorly known.
First described in 1921 by Keilin [5], the Helicosporidia were long ignored in classification systems due to their mysterious origins ([3], [6] and references therein). Initially, they were ascribed to the Protozoa, then transferred to the Fungi, before being reclassified as Protozoa, specifically within the Cnidosporidia. Cnidosporidia were a longstanding group consisting of Helicosporidia, Microsporidia, and Myxosporidia. The latter two groups are now known to be fungi and animals, respectively, so it is fitting that the Helicosporidia should eventually be determined to be closely related to plants, or more specifically to green algae. This was first suggested based on the astute observation that the morphology and in vitro development of Helicosporidia resemble that of the achlorophyllous non-photosynthetic green algae of the genus Prototheca [3]. This taxonomic affiliation was quickly supported by molecular phylogeny of several nucleus-encoded genes [6], [7], [8], [9], and further supported by the subsequent finding that Helicosporidia harbour a functional yet heavily reduced chloroplast genome [10], [11].
Phylogenetic analyses have, where the sampling diversity was sufficient, consistently suggested an affiliation to Prototheca [6], [7], [8], [9], [12], in agreement with their morphological characters [3]. Prototheca is a member of the green algal class Trebouxiophyceae, and is also achlorophyllous and pathogenic. This association is intriguing since protothecans infect only vertebrates inducing protothecosis in humans [13], whereas the Helicosporidia are known so far to invade only invertebrates.
To learn more about these intriguing but poorly studied parasites, and further compare them with their likely closest relatives in the genus Prototheca, here we report the complete sequence of the Helicosporidium sp. ATCC 50920 mitochondrial genome. The mitochondrial genome is a useful tool for such comparisons because complete mitochondrial genomes are available from representatives of most major groups of green algae, including Prototheca. The architecture of the 49343 pb-long Helicosporidium mitochondrial genome and the 60 genes it encodes are highly similar to those of Prototheca wickerhamii and display a level of synteny that have not been previously observed between any two chlorophyte mitochondrial DNAs (mtDNAs). The Helicosporidium mtDNA also has several interesting characteristics that are not only absent from Prototheca, but are rare in mitochondria as a whole, including a rare case of group I intron spliced in trans and introns that encode multiple ORFs.
Results
Main Features of the Mitochondrial Genome
The Helicosporidium mitochodondrial genome (GenBank: GQ339576) was sequenced as part of an ongoing genome project on Helicosporidium sp. strain ATCC 50920 in which 402658 reads totalizing 146.7 Mbp were generated by 454 Titanium pyrosequencing. Over 87.5% of these reads (364 bp average) were assembled into 4360 contigs representing about 10.6 Mbp. The mitochondrial genome was represented by a single contig comprising 53785 reads, amounting to 1.96 Mbp, or 396-fold coverage of the genome
The mitochondrial genome maps as a circular molecule of 49343 bp (Figure 1) featuring an overall A+T content of 74.4% (Table 1). The 60 genes it encodes are distributed with a marked strand polarity, but are not as symmetrical as those of Prototheca. The Helicosporidium mtDNA contains a total of four introns, all group I, which split the rnl and cox1 genes in three exons each. The Helicosporidium mtDNA also features three intronic open reading frames (ORFs) and two freestanding ORFs that are longer than 150 codons. Intergenic regions in the Helicosporidium mitochondrial genome range from 0 to 2355 bp, with an average of 183 bp, and no overlapping genes. The Helicosporidium mtDNA is more densely packed than that of Prototheca and is leaner by about 6 kbp despite maintaining a near-identical gene complement, differing only by a single tRNA, trnG(gcc) (Tables S1 and S2). Both genomes features trnT(ugu), a tRNA-encoding gene also found within the mtDNA of the ulvophycean alga Pseudendoclonium (Table S2). Like Prototheca and Pseudendoclonium mtDNAs, the Helicosporidium mitochondrial genome harbors a self-sufficient tRNA gene complement able to decode all codons assuming super Wobble codon/anticodon interactions. Codon usage in Helicosporidium mtDNA (Table S3) is also similar to that of Prototheca mtDNA, which parallels their very similar A+T content.
Figure 1. Gene map of Helicosporidium mtDNA.

Genes (filled boxes) located outside/inside the map are transcribed clockwise/counterclockwise. Introns are denoted by open boxes whereas intronic ORFs are illustrated as half-height boxes within the open boxes. tRNA genes are indicated by the one-letter amino acid code followed by the anticodon in parentheses (Me, elongator methionine; Mf, initiator methionine). ORFs smaller than 150 amino acids are not shown. Shared clusters between the Helicosporidium and Prototheca mitochondrial genomes are denoted by alterning black and gray brackets.
Table 1. Main features of Helicosporidium and other chlorophyte mtDNAs.
| Chlorophyte mtDNA | Size (bp) | A+T content (%) | Gene contenta | Gene densityb | Coding seq. (%)c | Introns (I/II) | Intron ORFs (I/II) |
| Prasinophyceae | |||||||
| Nephroselmis | 45223 | 67.2 | 65 | 1/696 | 80.6 | 4/0 | 4/0 |
| Ostreococcus | 44237 | 61.8 | 65 | 1/590 | 92.1 | 0/0 | 0/0 |
| Trebouxiophyceae | |||||||
| Helicosporidium | 49343 | 74.4 | 60 | 1/822 | 75.9 | 4/0 | 3/0 |
| Prototheca | 55328 | 74.2 | 61 | 1/907 | 70.6 | 5/0 | 2/0 |
| Ulvophyceae | |||||||
| Oltmannsiellopsis | 56761 | 66.6 | 54 | 1/1051 | 68.7 | 2/1 | 2/1 |
| Pseudendoclonium | 95880 | 60.7 | 57 | 1/1682 | 58.7 | 7/0 | 6/0 |
| Chlorophyceae | |||||||
| C. eugametos | 22897 | 65.4 | 12 | 1/1908 | 84.6 | 9/0 | 7/0 |
| C. reinhardtii | 15758 | 54.8 | 12 | 1/1313 | 83.1 | 0/0 | 0/0 |
| Chlorogonium | 22704 | 62.2 | 12 | 1/1892 | 89.1 | 6/0 | 6/0 |
| Scenedesmus | 42919 | 63.7 | 42 | 1/1022 | 60.6 | 2/2 | 1/0 |
| Uncertain affiliation | |||||||
| Pedinomonas | 25137 | 77.8 | 22 | 1/1143 | 60.5 | 0/1 | 0/0 |
Duplicated genes, unique ORFs and intron ORFs were not taken into account.
Duplicated genes were taken into account (size/number of genes).
Conserved genes (unique and duplicated), ORFs, introns and introns ORFs were considered as coding sequences.
The two freestanding ORFs in Helicosporidium mtDNA (orf160b and orf185) showed no significant homology in BLAST searches (E-values ≤1E-05). Although orf160b shares no identifiable similarity to any known ORF, it is located at the same genetic locus as ymf45 (orf174) in Prototheca mtDNA, i.e. between the nad2-rps10 and rps3 genes. Given their small size, the two Helicosporidium mtDNA freestanding ORFs might not encode any relevant biological product and rather represent random open reading frames, but the conserved position of Helicosporidium orf160b and Prototheca orf174 does suggest they are rapidly diverging homologues.
Synteny
The Helicosporidium mitochondrial genome features a high level of synteny with that of Prototheca. The two genomes share 12 gene clusters encompassing a total of 45 genes (Figure 1). This level of synteny has not been previously observed between mitochondrial genomes of any other chlorophytes. The mitochondrial genomes of the prasinophytes Nephroselmis and Ostreococcus share 10 clusters comprising a total of 36 genes, those of the ulvophytes Pseudendoclonium and Oltmannsiellopsis share only two gene pairs (4 genes) despite displaying a similar gene complement [14], whereas in the Chlorophyceae the two more similar mtDNAs (Chlorogonium and C. eugametos) share 3 clusters (8 genes). The Helicosporidium mtDNA shares six clusters (13 genes) and five clusters (11 genes) with the prasinophycean mtDNAs of Ostreococcus and Nephroselmis, respectively, and none with the ulvophycean or chlorophycean mitochondrial genomes.
Given the level of synteny between the Helicosporidium and Prototheca mitochondrial genomes, a minimum of 24 permutations by inversion between the 60 genes they share would be sufficient to convert the structure of one genome into that of the other. In contrast, at least 30 permutations (63 genes shared) and 46 permutations (50 genes shared) would be required to interconvert the structure of the Nephroselmis and Ostreococcus mtDNAs and of the Pseudendoclonium and Oltmannsiellopsis mtDNAs, respectively. However this number does not account for the creation of the inverted repeats in Ostreococcus due to the limitations of the GRIMM algorithm and is therefore an underestimate. In the Chlorophyceae, the gene-poor mtDNAs of Chlorogonium and C. eugametos are more closely related to each other (12 genes shared, 7 permutations) than to that of C. reinhardtii (12 genes shared, 19 and 18 permutations, respectively). The 42-gene mtDNA of Scenedesmus was not compared to the other gene-poor mtDNAs from the Chlorophyceae.
Introns
The Helicosporidium mitochondrial genome contains a total of four group I introns inserted into the cox1 and rnl genes (Figures 2 and S1). Although Prototheca mtDNA also has five introns within these two genes, none are located at cognate sites within Helicosporidium mtDNA. Interestingly, the Hsp.cox1.1 intron (Figure 2A) contains two distinct open reading frames, orf166 and orf239, located in different variable loops (L4 and L8, respectively). Both ORFs display a single dodecapeptide LAGLIDADG motif, indicative of a putative endonuclease function for these proteins. However, functional LAGLIDADG endonucleases contain two dodecapeptide motifs [15], raising the interesting possibility that the two ORFs generate an heterodimer constituted of one product in the N-terminal domain and of the other product in the C-terminal portion of the endonuclease. Alternatively, the two ORFs may also code for two independent homodimeric endonucleases. Homodimeric LAGLIDADG endonucleases are commonly found in group I introns, although the presence of two endonucleases in a single intron is extremely rare.
Figure 2. Predicted secondary structures of Helicosporidium cox1 group I introns.

The group I introns displayed according to Burke et al [36] were classified according to Michel and Westhof [37]. The Hsp.cox1.1 intron could not be assigned unambiguously to a subgroup of IB introns. Splice sites between exon and intron residues are denoted by arrows. Canonical Watson-Crick base pairings are denoted by dashes whereas guanine-uracyl pairings are marked by dots. Numbers inside variable loops indicate the sizes of these loops. The size of the L8 loop in the Hsp.cox1.2 intron is uncertain; the junction between the two parts of this trans-spliced intron occurs in the L8 loop. The putative LAGLIDADG endonucleases encoded within the intronic ORFs each contain a single copy of this motif.
Perhaps the most surprising finding is that the Helicosporidium mtDNA contains a trans-spliced group I intron. The Hsp.cox1.2 intron (Figure 2B) is fragmented into two pieces that are located on different strands and separated by three genes (trnR(ucu), nad5, rps19) spanning over 3 kb. This fragmentation is a genuine feature of the genome, supported by an assembly in which the mean coverage is about 400X. Because the cox1 gene is conserved among all chlorophyte mtDNAs and because its product, the first subunit of the mitochondrial cytochrome oxidase, is likely essential to the mitochondrion, we expected the trans-splicing of this intron to occur at the mRNA level so that a functional protein could be produced. To test this, we performed RT-PCRs on Helicosporidium RNA treated with DNAse using primers specific for the second and third cox1 exons. As expected, we obtained a 343 bp fragment consisting of the spliced exons (Figure 3). Sequencing of the 343 bp amplicon confirmed the 5′ and 3′ exon/intron splice junctions of the Hsp.cox1.2 intron illustrated in Figure 2B. When DNA was used as template for PCRs with the same primers, no amplicon was observed.
Figure 3. Electrophoretic analysis of RT-PCRs performed on Helicosporidium total RNA.
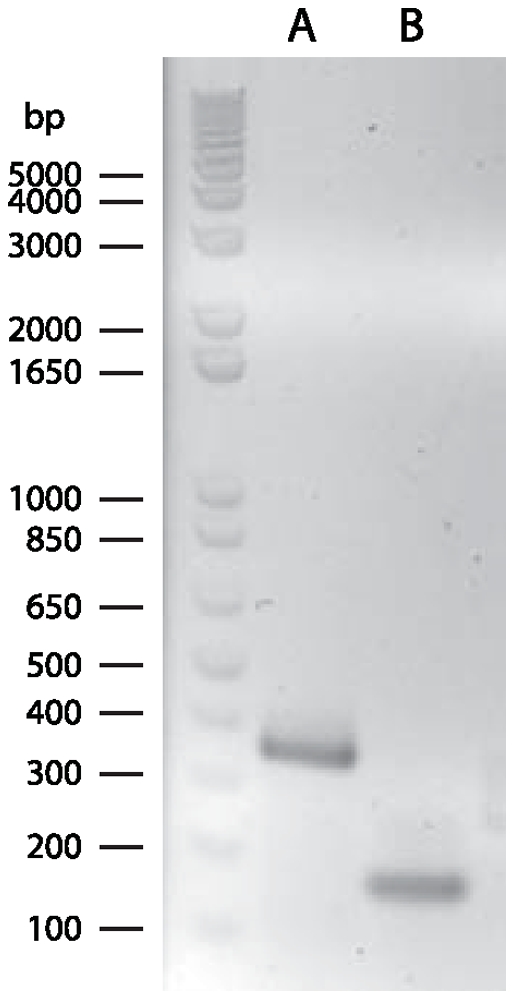
The amplicon in lane A corresponds to the expected size (343 bp) between the two internal cox1 primers in exons 2 and 3 (Hecox1F & Hecox1R) after trans-splicing of the Hsp.cox1.2 group I intron. The amplicon in lane B corresponds to the in cis positive control (166 bp) performed with internal atp1 primers (07487R & 00085R).
To our knowledge, trans-spliced group I introns were never reported until this year, and presently only three other instances are known: all in the mitochondrial cox1 gene. The first of these is in the mtDNA of the lycophyte Isoetes engelmannii [16] while the other two were identified by reinvestigation of the Trichoplax adhaerens mitochondrial genome [17]. However, the Hsp.cox1.2 intron is not inserted at a cognate site with any of these trans-spliced introns (Figure S2). Also, aside from its canonical group I intron structure, the Hsp.cox1.2 intron does not display strong identity with the Isoetes and Trichoplax introns even though it does share the same L8 trans-splicing location as the two Trichoplax introns.
Repeated Elements
The Helicosporidium mtDNA is not rich in repeated elements. Most of the repeated nucleotide strings detectable in the Helicosporidium mitochondrial genome are confined to AT-rich intergenic regions, are short, and are composed of adenosine and/or thymine residues arranged either in stretches or as alternating bases. The distribution of repeated elements oberved in Helicosporidium mtDNA (Figure S3) parallels that of Prototheca mtDNA in which the presence of A+T-rich repeats arrayed in tandem has been previously reported [18]. As in Prototheca mtDNA, the repeated elements are dispersed throughout the whole genome sequence in intergenic regions and in introns.
Phylogeny
The availability of the Helicosporidium mitochondrial genome provides us with another opportunity to probe the phylogenetic position of the Helicosporidia, and a useful opportunity because all major subgroups of green algae are available for analysis (this is not true for many nuclear genes, and the plastid genome does not contribute substantially to the question since the Prototheca plastid genome has not been sequenced and that of Helicosporidium is so reduced as to be difficult to compare with its photosynthetic relatives). As expected from and congruent with previous phylogenetic analyses [3], [6], [7], [8], [9], [12], phylogenies inferred from amino acid sequences derived from the seven protein-encoding genes that are shared between all mitochondrial genomes of chlorophytes supported a close affiliation between Helicosporidium and Prototheca. The two pathogenic achlorophyllous algae were joined together in all analyses (Figure 4). This affiliation was not dependent on the method of phylogenetic reconstruction, and was recovered in ML, Bayesian and even MP analyses. Given the overall level of support for the Helicosporidium/Prototheca affiliation, the placement of the helicosporidian parasites within the Trebouxiophyceae is most likely genuine.
Figure 4. Phylogenetic position of Helicosporidium sp. ATCC 50920 as inferred from amino acid sequences derived from the seven protein-encoding genes that are shared between all sequenced chlorophyte mtDNAs.

The best ML-tree inferred with PHYML 3.0 under the LG+Γ4+F+I model of amino acid substitutions is shown here (13 taxa, 2362 positions, 1373 phylogenetically informative). The charophyte green algae Mesostigma viride and Chaetosphaeridium globosum were used as outgroups. Bootstrap values over 80% are shown above the corresponding nodes. Branch lenghts are drawn to scale. Pedinomonas minor has not yet been assigned unambiguously to one the four classes of chlorophyte green algae.
Discussion
The mitochondrial genome of the obligately parasitic green alga Helicosporidium stands out in two different ways. First, it strongly supports the relationship between Helicosporidium and Prototheca, but not just because it provides a large molecular data set from which phylogenies can be inferred, but also because of their shared genomic structure. Based on the low levels of gene order conservation in other green algal mitochondrial genomes, we might expect to see few blocks of conservation between Helicosporidium and Prototheca. Clearly this was not the case for Helicosporidium and Prototheca, because their mtDNAs display a surprisingly high level of similarity in form. This close resemblance is probably best explained by a recent split between these two species. The only chlorophyte mtDNAs that display a comparable level of similarity are those of the prasinophytes Nephroselmis and Ostreococcus, but even here the level of conservation is much lower, with one featuring an inverted repeat that is missing from the other. In the Chlorophyceae, the gene-poor mtDNAs of Chlorogonium and C. eugametos also display an appreciable level of synteny, with 8 of their 12 genes (66%) being located in shared clusters, although this percentage is still lower than that observed between Helicosporidium and Prototheca mtDNAs (75%), and there are far fewer combinations of 12 genes than of 60.
A second standout feature of the Helicosporidium mtDNA is in its introns, and in particular the presence of a group I intron that splices in trans. Although trans-splicing in various group II introns has been known to occur for some time (reviewed in [19], [20]), the first examples of trans-spliced group I introns have only been described recently [16], [17]. Like other known trans-spliced group I introns, the predicted secondary structure of the Hsp.cox1.2 intron (Figure 2) closely conforms to a canonical group I intron, suggesting it most likely arose from a cis-spliced group I intron that was broken into two pieces, but that could still fold at the mRNA level to produce a functional ribozyme. Because this fragmentation occurred within a variable loop, its effect on the intron self-splicing capability may have been minimal (although presumably if such an event had little impact it would occur more frequently than it does). It is unclear what effect such a fragmentation might have on the viability of the intron if it occurred in core regions like the P7 pairings. However, as ribozymes derived from group I introns can catalyse trans-excision-splicing reactions in other RNA molecules [21], [22], [23], their functional core may be somewhat malleable.
The four trans-spliced group I introns known so far most likely arose independently. Not only do they appear dissimilar at the nucleotide level outside of their canonical group I intron structure, but despite their shared location within the mitochondrial cox1 gene, none are inserted at cognate sites. Also to be transferred horizontally from one organism to another, at least two recombinational events would be required, one for each trans-spliced segment. This appears unlikely, especially considering that such recombinational events would likely involve the adjacent exons. As Helicosporidium, Trichoplax and Isoetes are evolutionary distant and belong to very different lineages, recombination between their genes, even as conserved as cox1, is not a very compelling hypothesis. It is perhaps not surprising that these rare introns were first discovered within the cox1 gene, given its conservation and its importance for the mitochondrion. Other existing instances of trans-splicing group I introns in less conserved genes may have been overlooked, and reinvestigation of sequenced mitochondrial genomes, as performed by Burger and coauthors on the Trichoplax mtDNA [17], may reveal more of these segmented yet functional ribozymes.
The homing endonucleases encoded within intronic ORFs confer mobility to the intron host by permitting double strand breaks of a target DNA. Very often, these endonucleases are lost and the introns lacking these ORFs are no longer considered mobile. It is very rare however to find an intron containing two distinct ORFs coding for putative endonucleases. Given that different homing endonucleases usually have different DNA targets, the presence of two such proteins tentatively confers a greater protential for mobility and self-propagation of the intron. It is unknown if the two ORFs present in the Helicosporidium Hsp.cox1.1 intron code for functional and expressed endonucleases. If so, it would be interesting to determine whether they act separately or as a heterologous unit.
Conclusions
The structure and content of the Helicosporidium mitochondrial genome, as well as the phylogenetic inferences derived from the sequences it encodes, support the specific relationship to the genus Prototheca. The introns of this genome also have a number of interesting characteristics rarely seen in other organelle genomes. Our results, combined with the previously published plastid genome sequence of Helicosporidium sp. ATCC 50920 [21], complete the deciphering of this peculiar species's organellar genetic imprint. The sequencing of the Helicosporidium sp. ATCC 50920 nuclear genome would provide us with a global picture that, hopefully, would yield clues into the adaption of this alga from a free-living entity to that of an entomoparasite. Also, a comparative approach with its protothecan relatives would give us interesting insights into the nature of their selective parasitism.
Materials and Methods
PCRs and RT-PCRs
PCRs were performed using 22- to 24-mers and the EconoTaq PLUS GREEN kit from Lucigen (Middleton, WI, USA) with 35 cycles of denaturation (1 min at 94°C), annealing (1 min at 55°C) and elongation (3 min at 72°C). RT-PCRs were performed using the SuperScript One-Step RT-PCR with Platinum Taq kit from Invitrogen (Carlsbad, CA, USA) with an initial cDNA synthesis cycle (30 min at 50°C) followed by a 2 min denaturation cycle at 94°C. A total of 35 amplification cycles of denaturation (15 sec at 94°C), annealing (30 sec at 55°C), and elongation (1 min at 70°C) were then performed. The Hecox1F (5′-CTCTTCCTGTATTAGCTGGTGG-3′) and Hecox1R (5′-GCAATAATCATTGTAGCTGCAG-3′) and the 07487R (5′-CAATTGTAGACGTACCAGTTGG-3′) and 00085R (5′-GTTTGCATAGGTTGGCTTACAG-3′) primers were used in RT-PCRs.
Genome Sequencing
The Helicosporidium mtDNA was sequenced using the massively parallel GS-FLX DNA pyrosquencing platform from Roche 454 Life Sciences (Branford, CT, USA).
The creation of the Helicosporidium mtDNA GS-FLX shotgun library and the GS-FLX 454 pyrosequencing (using the GS-FLX Titanium reagents) were carried out by the McGill University and Génome Québec Innovation Centre. The Newbler assemblies obtained from Génome Québec were converted to, edited, and assembled with CONSED 19 [24]. Ambiguous regions in the assemblies were either (1) edited according to their conceptual translations or (2) amplified by PCR with 22-mers primers flanking the ambiguous regions, sequenced using traditional Sanger chemistry by Macrogen (Seoul, Korea) and then edited according to Sanger base calling.
Genome Annotation and Analysis
Genes were identified by Blast homology searches [25] against a local copy of the National Center for Biotechnology Information (NCBI) nonredundant database using the NCBI BLASTALL suite (http://www.ncbi.nlm.nih.gov/Ftp/blast). Positions of open reading frames and protein-coding genes were determined using GETORF from EMBOSS 6.0.1 [26] and ORFFINDER at NCBI, whereas positions of tRNA-encoding genes were determined with tRNAscan-SE [27]. Insertions sites of group I introns and their predicted secondary structures were determined manually. Codon usage in protein-encoding genes was determined with CUSP from the EMBOSS package. Repeated elements were first visualized with PipMaker [28]. Then, repeated elements arrayed in tandem were identified with ETANDEM from the EMBOSS package whereas dispersed repeated elements were located with REPuter 2.74 [29]. Potential hairpin structures were screened for with PALINDROME from the EMBOSS package. Minimal number of permutations by inversions between mitochondrial genomes were inferred with GRIMM [30]. For this analysis, the trans-spliced exons of the cox1 gene in Helicosporidium mtDNA and the fragmented rRNA genes in chlorophycean mtDNAs were coded as distinct fragments. Also, as GRIMM cannot handle duplicate genes, one copy of the Ostreococcus inverted repeats was removed.
Phylogenetic Analyses
In addition to the Helicosporidium mitochondrial genome sequenced by the authors [GenBank:GQ339576], the following mtDNAs used in this study were retrieved from GenBank: Chaetosphaeridium globosum [GenBank:NC_004118], Chlamydomonas eugametos [GenBank:NC_001872], Chlamydomonas reinhardtii [GenBank:NC_001638], Chlorogonium elongatum [Genbank:Y13643, Y13644, Y07814], Mesostigma viride [GenBank:NC_008240], Nephroselmis olivacea [GenBank:NC_008239], Oltmannsiellopsis viridis [GenBank:NC_008256], Ostreococcus tauri [GenBank:NC_008290], Pedinomonas minor [GenBank:NC_000892], Prototheca wickerhamii [GenBank:NC_001613], Pseudendoclonium akinetum [GenBank:NC_005926], Scenedesmus obliquus [GenBank:NC_002254]. Mitochondrial protein sequences were inferred from the conceptual translation of the seven protein-encoding genes that are share between all chlorophyte mtDNAs. The amino acid sequences were aligned using T-COFFEE 7.81 [31], the ambiguous regions within these alignments filtered with GBLOCKS 0.91 b [32], and the filtered individual sequences concatenated. Maximum Likelihood computations were performed using PHYML 3.0 [33] under the LG+Γ4+F+I model of amino acid substitution selected with ProtTest 2.0 [34]. Bayesian inferences were performed with PhyloBayes 3.2 [35] under the CAT+ Γ4 model of amino acid substitution running two concurrent chains terminated using PhyloBayes automatic stopping rule (maxdiff <0.3).
Supporting Information
Predicted secondary structures of Helicosporidium rnl group I introns. The rnl group I introns displayed according to Burke et al were classified according to Michel and Westhof. Splice sites between exon and intron residues are denoted by arrows. Canonical Watson-Crick base pairings are denoted by dashes whereas guanine-uracyl pairings are marked by dots. Numbers inside variable loops indicate the sizes of these loops. The putative LAGLIDADG endonuclease encoded within the Hsp.rnl.1 intronic ORF contains a single copy of this motif.
(0.91 MB EPS)
Trans-spliced GI introns insertion sites. The insertion sites of the Helicosporidium, Isoetes and Trichoplax trans-spliced group I introns are indicated by arrows on the Cox1 amino acid alignment (positions 225 to 464 shown). Numbers on the right of the alignment indicate the amino acid positions on the corresponding sequences. The different trans-spliced introns are indicated by roman numerals: I, Helicosporidium (Hsp.cox1.2); II, Trichoplax (cox1 intron 4); III, Trichoplax (cox1 intron 5); IV, Isoetes (coxi1305). The Trichoplax intron insertion sites were taken from Burger et al.
(4.97 MB EPS)
Densities of repeated elements in Helicosporidium and other chlorophyte mitochondrial genomes. Repeated elements identified with REPuter are connected by lines on the corresponding mtDNA circular representations (adapted from Kurtz). Repeats of at least 15, 30 and 45 nt are shown on the left, middle and right panels respectively. For this analysis, one copy of the Ostreococcus mtDNA inverted repeats has been removed. Also, the linear C. reinhardtii mitochondrial genome is represented here as a circle.
(2.22 MB PDF)
Gene repertoires of Helicosporidium and other chlorophyte mtDNAs. a Nol, Nephroselmis olivacea; Ota, Ostreococcus tauri; Hsp, Helicosporidium sp.; Pwi, Prototheca wickerhamii; Ovi, Oltmannsiellopsis viridis; Pak, Pseudendoclonium akinetum; Sob, Scenedesmus obliquus; Pmi, Pedinomonas minor; Cre, Chlamydomonas reinhardtii; Ceu, Chlamydomonas eugametos; Cel, Chlorogonium elongatum. Presence/absence of a gene is denoted by +/−. b Gene fragmented in corresponding mtDNA.
(0.10 MB DOC)
tRNA gene repertoires of Helicosporidium and other chlorophyte mtDNAs. a Nol, Nephroselmis olivacea; Ota, Ostreococcus tauri; Hsp, Helicosporidium sp.; Pwi, Prototheca wickerhamii; Ovi, Oltmannsiellopsis viridis; Pak, Pseudendoclonium akinetum; Sob, Scenedesmus obliquus; Pmi, Pedinomonas minor; Cre, Chlamydomonas reinhardtii; Ceu, Chlamydomonas eugametos; Cel, Chlorogonium elongatum. Presence/absence of a gene is denoted by +/−. b Me, elongator methionine; Mf, initiator methionine. c Genome specifies a single trnM(cau).
(0.09 MB DOC)
Codon usage in the 32 protein-encoding genes of Helicosporidium mtDNA. a Percentage of each amino acid specified by the specified codon. b Anticodon of Helicosporidium mtDNA-encoded tRNA recognizing the corresponding codon. The following tRNAs with an uracyl in the first position of their anticodon are assumed to decode all four members of the four-codon families: alanine, GCN; glycine, GGN; leucine, CUN; proline, CCN; serine, UCN; threonine, ACN; valine, GUN. c Amino acids are labelled by their one-letter IUPAC code. Termination codons are indicated by asterisks. d In chlorophytes mtDNAs, the gene coding for tRNAThr (ugu) has been found only within the mitochondrial genomes of Helicosporidium, Prototheca and Pseudendoclonium. e The initiator and elongator tRNAMet (cau) are encoded by different genes.
(0.07 MB DOC)
Acknowledgments
We thank our collaborator on the Helicosporidium genome project, Dr Drion Boucias from the Department of Entomology and Nematology at the University of Florida, for providing Helicosporidium sp. ATCC 50920 total DNA and RNA.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by a Natural Sciences and Engineering Research Council (NSERC) operating grant to PJK. JFP is the recipient of the Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT)/Génome Québec Louis-Berlinguet Postdoctoral Fellowship. PJK is a Fellow of the Canadian Institute for Advanced Research (CIFAR), and a Senior Scholar of the Michael Smith Foundation for Health Research (MSFHR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Conklin T, Bläske-Lietze VU, Becnel JJ, Boucias DG. Infectivity of two isolates of Helicosporidium spp. (Chlorophyta: Trebouxiophyceae) in heterologous host insects. Fla Entomol. 2005;88:431–439. [Google Scholar]
- 2.Bläske-Lietze VU, Shapiro AM, Denton JS, Botts M, Becnel JJ, et al. Development of the insect pathogenic alga Helicosporidium. J Eukaryot Microbiol. 2006;53:165–176. doi: 10.1111/j.1550-7408.2006.00090.x. [DOI] [PubMed] [Google Scholar]
- 3.Boucias DG, Becnel JJ, White SE, Bott M. In vivo and in vitro development of the protist Helicosporidium sp. J Eukaryot Microbiol. 2001;48:460–470. doi: 10.1111/j.1550-7408.2001.tb00180.x. [DOI] [PubMed] [Google Scholar]
- 4.Bläske-Lietze VU, Boucias DG. Pathogenesis of Helicosporidium sp. (Chlorophyta: Trebouxiophyceae) in susceptible noctuid larvae. J Invertebr Pathol. 2005;90:161–168. doi: 10.1016/j.jip.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Keilin D. On the life history of Helicosporidium parasiticum, n. g., n. sp., a new type of protist parasitic in the larva of Dasyhelea obscura Winn. (Diptera, Ceratopogonidae) and in some other arthropods. Parasitology. 1921;13:97–113. [Google Scholar]
- 6.Tartar A, Boucias DG, Adams BJ, Becnel JJ. Phylogenetic analysis identifies the invertebrate pathogen Helicosporidium sp. as a green alga (Chlorophyta). Int J Syst Evol Microbiol. 2002;52:273–279. doi: 10.1099/00207713-52-1-273. [DOI] [PubMed] [Google Scholar]
- 7.Tartar A, Boucias DG, Becnel JJ, Adams BJ. Comparison of plastid 16S rRNA (rrn16) genes from Helicosporidium spp.: evidence supporting the reclassification of Helicosporidia as green algae (Chlorophyta). Int J Syst Evol Microbiol. 2003;53:1719–1723. doi: 10.1099/ijs.0.02559-0. [DOI] [PubMed] [Google Scholar]
- 8.de Koning AP, Keeling PJ. Nucleus-encoded genes for plastid-targeted proteins in Helicosporidium: functional diversity of a cryptic plastid in a parasitic alga. Eukaryot Cell. 2004;3:1198–1205. doi: 10.1128/EC.3.5.1198-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Koning AP, Tartar A, Boucias DG, Keeling PJ. Expressed sequence tag (EST) survey of the highly adapted green algal parasite, Helicosporidium. Protist. 2005;156:181–190. doi: 10.1016/j.protis.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Tartar A, Boucias DG. The non-photosynthetic, pathogenic green alga Helicosporidium sp. has retained a modified, functional plastid genome. FEMS Microbiol Lett. 2004;233:153–157. doi: 10.1016/j.femsle.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 11.de Koning AP, Keeling PJ. The complete plastid genome sequence of the parasitic green alga Helicosporidium sp. is highly reduced and structured. BMC Biol. 2006;4:12. doi: 10.1186/1741-7007-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keeling PJ, Inagaki Y. A class of eukaryotic GTPase with a punctate distribution suggesting multiple functional replacements of translation elongation factor 1alpha. Proc Natl Acad Sci U S A. 2004;101:15380–15385. doi: 10.1073/pnas.0404505101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lass-Florl C, Mayr A. Human protothecosis. Clin Microbiol Rev. 2007;20:230–242. doi: 10.1128/CMR.00032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pombert JF, Beauchamp P, Otis C, Lemieux C, Turmel M. The complete mitochondrial DNA sequence of the green alga Oltmannsiellopsis viridis: evolutionary trends of the mitochondrial genome in the Ulvophyceae. Curr Genet. 2006;50:137–147. doi: 10.1007/s00294-006-0076-z. [DOI] [PubMed] [Google Scholar]
- 15.Silva GH, Belfort M, Wende W, Pingoud A. From monomeric to homodimeric endonucleases and back: engineering novel specificity of LAGLIDADG enzymes. J Mol Biol. 2006;361:744–754. doi: 10.1016/j.jmb.2006.06.063. [DOI] [PubMed] [Google Scholar]
- 16.Grewe F, Viehoever P, Weisshaar B, Knoop V. A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 2009;37:5093–5104. doi: 10.1093/nar/gkp532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger G, Yan Y, Javadi P, Lang BF. Group I-intron trans-splicing and mRNA editing in the mitochondria of placozoan animals. Trends Genet. 2009;25:381–386. doi: 10.1016/j.tig.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Wolff G, Plante I, Lang BF, Kuck U, Burger G. Complete sequence of the mitochondrial DNA of the chlorophyte alga Prototheca wickerhamii. Gene content and genome organization. J Mol Biol. 1994;237:75–86. doi: 10.1006/jmbi.1994.1210. [DOI] [PubMed] [Google Scholar]
- 19.Bonen L. Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion. 2008;8:26–34. doi: 10.1016/j.mito.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Lambowitz AM, Zimmerly S. Mobile group II introns. Annu Rev Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- 21.Dotson PP, 2nd, Johnson AK, Testa SM. Tetrahymena thermophila and Candida albicans group I intron-derived ribozymes can catalyze the trans-excision-splicing reaction. Nucleic Acids Res. 2008;36:5281–5289. doi: 10.1093/nar/gkn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dotson PP, 2nd, Sinha J, Testa SM. A Pneumocystis carinii group I intron-derived ribozyme utilizes an endogenous guanosine as the first reaction step nucleophile in the trans excision-splicing reaction. Biochemistry. 2008;47:4780–4787. doi: 10.1021/bi7020525. [DOI] [PubMed] [Google Scholar]
- 23.Einvik C, Fiskaa T, Lundblad EW, Johansen S. Optimization and application of the group I ribozyme trans-splicing reaction. Methods Mol Biol. 2004;252:359–371. doi: 10.1385/1-59259-746-7:359. [DOI] [PubMed] [Google Scholar]
- 24.Gordon D. Viewing and Editing Assembled Sequences Using Consed. In: Baxevanis AD, Davison DB, editors. Current Protocols in Bioinformatics. New York: John Wiley & Co; 2004. pp. 11.12.11–11.12.43. [DOI] [PubMed] [Google Scholar]
- 25.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 27.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, et al. PipMaker↓a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesler G. GRIMM: genome rearrangements web server. Bioinformatics. 2002;18:492–493. doi: 10.1093/bioinformatics/18.3.492. [DOI] [PubMed] [Google Scholar]
- 31.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 32.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 33.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 34.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 35.Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- 36.Burke JM, Belfort M, Cech TR, Davies RW, Schweyen RJ, et al. Structural conventions for group I introns. Nucleic Acids Res. 1987;15:7217–7221. doi: 10.1093/nar/15.18.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michel F, Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Predicted secondary structures of Helicosporidium rnl group I introns. The rnl group I introns displayed according to Burke et al were classified according to Michel and Westhof. Splice sites between exon and intron residues are denoted by arrows. Canonical Watson-Crick base pairings are denoted by dashes whereas guanine-uracyl pairings are marked by dots. Numbers inside variable loops indicate the sizes of these loops. The putative LAGLIDADG endonuclease encoded within the Hsp.rnl.1 intronic ORF contains a single copy of this motif.
(0.91 MB EPS)
Trans-spliced GI introns insertion sites. The insertion sites of the Helicosporidium, Isoetes and Trichoplax trans-spliced group I introns are indicated by arrows on the Cox1 amino acid alignment (positions 225 to 464 shown). Numbers on the right of the alignment indicate the amino acid positions on the corresponding sequences. The different trans-spliced introns are indicated by roman numerals: I, Helicosporidium (Hsp.cox1.2); II, Trichoplax (cox1 intron 4); III, Trichoplax (cox1 intron 5); IV, Isoetes (coxi1305). The Trichoplax intron insertion sites were taken from Burger et al.
(4.97 MB EPS)
Densities of repeated elements in Helicosporidium and other chlorophyte mitochondrial genomes. Repeated elements identified with REPuter are connected by lines on the corresponding mtDNA circular representations (adapted from Kurtz). Repeats of at least 15, 30 and 45 nt are shown on the left, middle and right panels respectively. For this analysis, one copy of the Ostreococcus mtDNA inverted repeats has been removed. Also, the linear C. reinhardtii mitochondrial genome is represented here as a circle.
(2.22 MB PDF)
Gene repertoires of Helicosporidium and other chlorophyte mtDNAs. a Nol, Nephroselmis olivacea; Ota, Ostreococcus tauri; Hsp, Helicosporidium sp.; Pwi, Prototheca wickerhamii; Ovi, Oltmannsiellopsis viridis; Pak, Pseudendoclonium akinetum; Sob, Scenedesmus obliquus; Pmi, Pedinomonas minor; Cre, Chlamydomonas reinhardtii; Ceu, Chlamydomonas eugametos; Cel, Chlorogonium elongatum. Presence/absence of a gene is denoted by +/−. b Gene fragmented in corresponding mtDNA.
(0.10 MB DOC)
tRNA gene repertoires of Helicosporidium and other chlorophyte mtDNAs. a Nol, Nephroselmis olivacea; Ota, Ostreococcus tauri; Hsp, Helicosporidium sp.; Pwi, Prototheca wickerhamii; Ovi, Oltmannsiellopsis viridis; Pak, Pseudendoclonium akinetum; Sob, Scenedesmus obliquus; Pmi, Pedinomonas minor; Cre, Chlamydomonas reinhardtii; Ceu, Chlamydomonas eugametos; Cel, Chlorogonium elongatum. Presence/absence of a gene is denoted by +/−. b Me, elongator methionine; Mf, initiator methionine. c Genome specifies a single trnM(cau).
(0.09 MB DOC)
Codon usage in the 32 protein-encoding genes of Helicosporidium mtDNA. a Percentage of each amino acid specified by the specified codon. b Anticodon of Helicosporidium mtDNA-encoded tRNA recognizing the corresponding codon. The following tRNAs with an uracyl in the first position of their anticodon are assumed to decode all four members of the four-codon families: alanine, GCN; glycine, GGN; leucine, CUN; proline, CCN; serine, UCN; threonine, ACN; valine, GUN. c Amino acids are labelled by their one-letter IUPAC code. Termination codons are indicated by asterisks. d In chlorophytes mtDNAs, the gene coding for tRNAThr (ugu) has been found only within the mitochondrial genomes of Helicosporidium, Prototheca and Pseudendoclonium. e The initiator and elongator tRNAMet (cau) are encoded by different genes.
(0.07 MB DOC)