

Abstract
Perry syndrome is characterized clinically by autosomal dominantly inherited, rapidly progressive parkinsonism, depression, weight loss and hypoventilation. In the seven families reported previously and the two new families presented herein (the Hawaii family and the Fukuoka-4 Japanese family), the mean disease onset age is 48 years (range: 35-61) and the mean disease duration five years (range: 2-10). Histology and immunohistochemistry show severe neuronal loss in the substantia nigra and locus coeruleus, with TDP-43-positive pathology in neurons (intranuclear and cytoplasmic inclusions, dystrophic neurites, axonal spheroids) and glial cells (glial cytoplasmic inclusions). Compared with other TDP-43-proteinopathies (amyotrophic lateral sclerosis and ubiquitin-positive frontotemporal lobar degeneration), the distribution is unique in Perry syndrome with pallidonigral distribution and sparing of the cortex, hippocampus and motor neurons. The genetic cause of Perry syndrome was recently identified with five mutations in the dynactin gene (DCTN1) segregating with disease in eight families. DCTN1 encodes p150glued, the major subunit of the dynactin protein complex, which plays a crucial role in retrograde axonal and cytoplasmic transport of various cargoes. Evidence suggests the Perry mutations alter the binding of p150glued to microtubules. Further studies will examine reasons for the vulnerability of selected neuronal populations in Perry syndrome, and the link between the genetic defect and TDP-43 pathology.
Keywords: Perry syndrome, parkinsonism, depression, hypoventilation, dynactin, DCTN1, p150glued, TDP-43
Introduction
In 1975, Perry and collaborators reported a Canadian family with rapidly progressive autosomal dominant parkinsonism, depression, weight loss, sleep difficulties and central hypoventilation [1, 2]. Over the next 30 years six additional families have been reported from Canada, the U.S., the U.K., France, Turkey and Japan (reviewed in [3]) [4-10]. Depression, apathy, weight loss and parkinsonism are early symptoms while central hypoventilation develops in later stages. The mean onset age is 48 years (range: 35-61, including two unpublished families, see below) and the mean disease duration five years (range: 2-10). Patients die of respiratory complications, sudden unexplained death or by suicide. Initial reports highlighted reduced taurine levels in CSF from patients with Perry syndrome, however this finding was not replicated in subsequent studies [1, 2, 4, 8, 9].
An international consortium was established in 2001 by ZKW and YT to expand the clinical, pathological and genetic characterization of Perry syndrome. In so doing, we have been able to reactivate seven of the eight previously published families and to identify two additional unreported kindreds from Hawaii and Japan (Fukuoka-4 family). Material collected included detailed clinical information (eight families), brain tissue (eight patients from five families) and DNA samples (17 affected individuals from eight families and 74 unaffected family members).
Herein we present clinical data on two unpublished families (the Hawaiian and Fukuoka-4 families) with Perry syndrome, and review our recent discoveries in the pathological and genetic characterization of the disease.
Unpublished families
Patients were examined by movement disorders neurologists, and signed IRB-approved informed consent.
The Hawaiian family originates from Japan. There are six affected individuals (two men) including two with clinical information and one with pathology (Fig. 1). The proband and her second cousin presented at age 47 with parkinsonism and later developed respiratory symptoms. The proband had partial benefit from levodopa. In contrast to previously reported families, symptoms did not include depression and weight loss. Neuropathologic examination of the proband's second cousin showed neuronal loss in the substantia nigra (SN) and the locus coeruleus, with no Lewy bodies.
Figure 1.
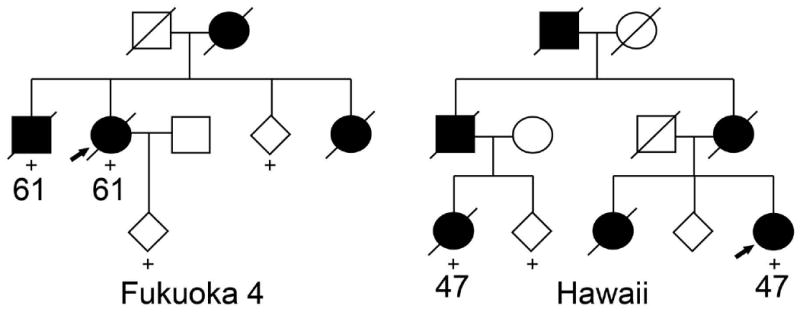
Pedigrees of two new families with Perry syndrome. Circles, women. Squares, men. Diamonds, gender disguised. Diagonal lines, deceased. Black symbols, affected individuals. Numbers under symbols, ages of onset. “+”, DNA sample available. Arrows, probands.
The Fukuoka-4 family has four affected individuals (one man) over two generations (Fig. 1). Disease onset (61 years) and duration (three years) were similar in the proband and her brother. The initial symptom was resting tremor in the proband and weight loss in her brother; both later developed levodopa-responsive parkinsonism, weight loss and hypoventilation. Response to levodopa was transient and did not cause motor complications. The proband died of sudden death and her brother of respiratory failure. Of notice, these two patients did not develop depression or apathy, a feature found in all previously published families with Perry syndrome. Further, onset age in this family (61 years) was five years older than the upper range of onset age in other families. No autopsy was performed on these patients.
Genetic analysis showed that although these two families are of Japanese descent, they harbor distinct mutations from two different founders (see below).
Neuropathology
Previous reports have highlighted severe neuronal loss in the SN and locus coeruleus, with few to no Lewy bodies [3]. We examined autopsy material from eight patients with Perry syndrome (fixed and frozen tissue from two, paraffin-embedded fixed tissue from five, unstained tissue on glass slides from one). All patients had severe SN neuronal loss. Surviving SN neurons had intranuclear (NII) and cytoplasmic (NCI) inclusions that stained positive for transactive-response (TAR) DNA-binding protein of 43 kD (TDP-43) [11]. TDP-43 pathology also included dystrophic neurites, axonal spheroids and glial cytoplasmic inclusions (Fig. 2). Immunohistochemistry for tau and α-synuclein was negative. These findings show TDP-43-positive inclusions are characteristic of Perry syndrome and establish this condition as a TDP-43-proteinopathy.
Figure 2.
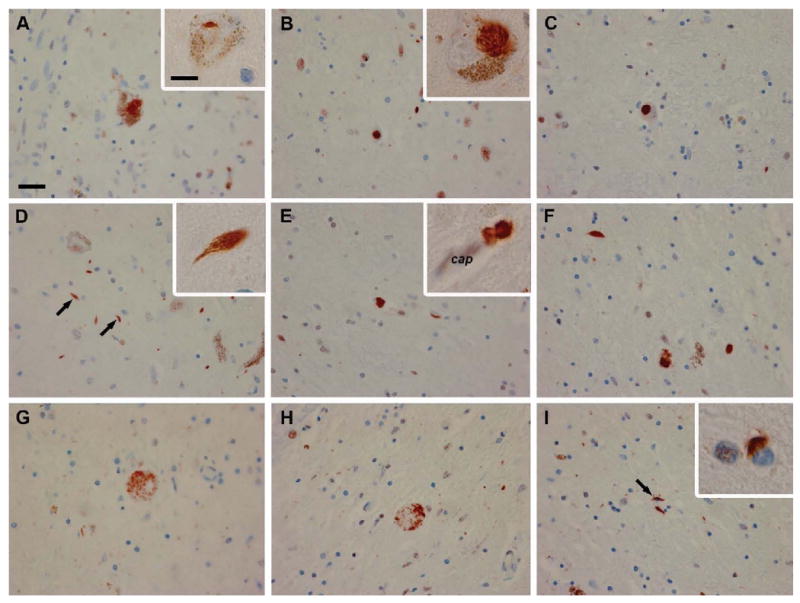
TDP-43 immunohistochemistry of Perry patients shows NCI (A-C; higher magnification in inset of B), NII (inset in A), dystrophic neurites (D arrows, E and F; higher magnification in inset of D), axonal spheroids (G, H), glial cytoplasmic inclusions (I, arrow; higher magnification in inset of I), and a perivascular astrocytic inclusions (inset of E; cap= capillary). Scale bar: 25 μm (A-I), 10 μm (insets of A, B, D, E and I). Reproduced from [11], with permission from Elsevier Inc. ©2008.
TDP-43 was recently identified as the major ubiquitinated component of NCI and NII in ubiquitin-positive frontotemporal lobar degeneration (FTLD-U), frontotemporal dementia – motor neuron disease (FTD-MND) and amyotrophic lateral sclerosis (ALS) [12]. Interestingly, NCI and NII in Perry syndrome are similar to those found in FTLD-U and ALS. However, the distribution differs in Perry syndrome with pallidonigral predominance and sparing of the cortex, hippocampus and motor neurons. This accounts for the lack of dementia and motor neuron disease in Perry syndrome, in contrast with FTLD-U and ALS. The initial hypothesis that TDP-43-positive inclusions were specific to a small number of diseases such as FTLD-U, FTD-MND and ALS has been challenged by the identification of abnormal TDP-43 in Perry syndrome, Lewy body disease, Guam Parkinson dementia complex, Alzheimer's disease, and hippocampal sclerosis [3, 13-15]. Such findings suggest TDP-43 pathology may not be disease-specific but rather represent a more general cellular response to a variety of genetic defects and environmental insults. However, it should be emphasized that in Perry syndrome, FTLD-U, FTD-MND and ALS, TDP-43 immunoreactive inclusions are the dominant pathologic finding, supporting a role in pathogenesis. This contrasts with other conditions such as Alzheimer's disease and Lewy body disease where the main finding is α-synuclein and tau-positive inclusions, TDP-43 pathology being less prominent. Further supporting a more direct implication of TDP-43 in a selected number of diseases, mutations in the TDP-43 gene (TARDBP) were recently shown to cause ALS [16, 17].
While SN neuronal loss explains parkinsonism in Perry syndrome, identifying the pathological basis of depression, weight loss and hypoventilation has proven more challenging [11]. An autopsy study of one patient from the first Japanese family found selective loss of putative respiratory neurons in the ventrolateral medulla (neurokinin-1 receptor / tyrosine hydroxylase-immunopositive neurons) and in the raphe nucleus (serotonergic neurons), which most probably accounts for central-type hypoventilation [18]. Loss of aminergic neurons in the locus ceruleus and the ventral tegmental area may contribute to depression and apathy. No specific pathological abnormality (e.g. hypothalamic) was found that would explain weight loss.
Genetics
Our group recently established mutations in the dynactin gene (DCTN1) as the genetic cause of Perry syndrome [19]. Each of the eight families examined harbored one of five mutations in exon 2 of DCTN1 (p.G71A/E/R, p.T72P, and p.Q74P), all of which segregated with disease. None of the mutations were identified in 949 control individuals and in 475 patients with familial parkinsonism. Another mutation in DCTN1 (p.G59S) was previously reported in a family with a rare form of lower motor neuron disease (comparison with Perry syndrome is presented in the Table) [20, 21]. Additional mutations have been identified in ALS patients and in a family with ALS and FTLD (clinically of the behavioral type), however evidence of pathogenicity is lacking [22, 23].
DCTN1 encodes p150glued, the major subunit of the dynactin protein complex (Fig. 3) [24]. Present as a dimer, p150glued is the backbone of the dynactin complex, binding directly to microtubules, the molecular motor dynein, and different dynactin subunits (Fig. 3). Dynactin plays a major role in retrograde axonal and cytoplasmic transport of vesicles, organelles and other cargoes. Its complex multimeric structure allows dynactin to interact with microtubules and dynein (via the p150glued subunit) and various cargoes (via other subunits). In transgenic mice, over-expression of dynamitin (the p50 subunit of dynactin) leads to late-onset progressive motor neuron disease [25]. Interestingly the five DCTN1 mutations which cause Perry syndrome and the previously reported p.G59S mutation are all located within the p150glued highly conserved N-terminal cytoskeleton-associated protein, glycine-rich (CAP-Gly) domain. The CAP-Gly domain is critical for microtubule binding and in vitro assays with Perry (p.G71R and p.Q74P) and p.G59S mutant p150glued demonstrated reduced affinity of dynactin for microtubules [19, 20, 26]. Further, cells transfected with DCTN1 p.G71R, p.Q74P or p.G59S show redistribution of dynactin compared to wild-type protein (Fig. 4) [19]. In vitro and in vivo evidence suggests the p.G59S mutation leads to altered dynactin/dynein function (loss of function) as well as dynactin aggregation (toxic gain of function) [26]. In contrast, experimental data shows that p150glued mutations identified in ALS and FTLD, which lack evidence of pathogenicity, do not impair microtubule binding, alter the dynein-dynactin interaction or confer increased propensity to form aggregates, thus not supporting a direct role in pathogenesis [27].
Figure 3.
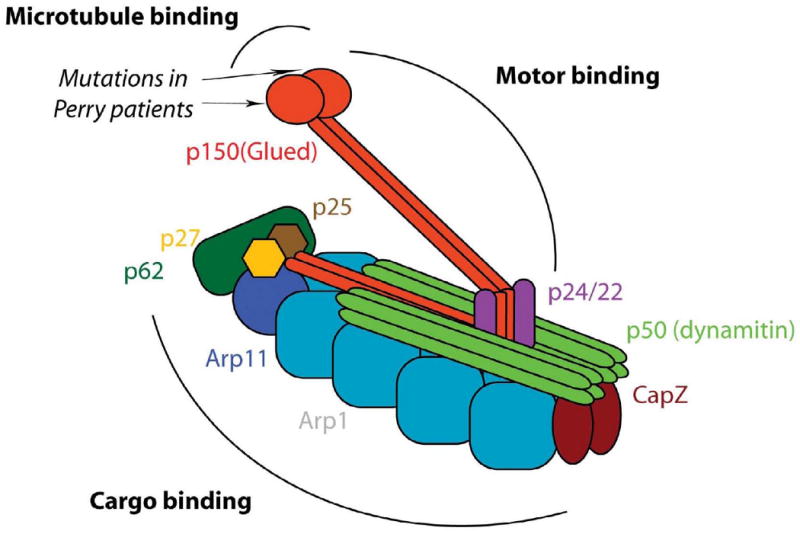
Schematic representation of the dynactin protein complex highlighting the different subunits, the position of Perry mutations in p150glued, and domains binding microtubules, the motor dynein, and various cargoes. Adapted from [24], with permission from the Annual Review of Cell and Developmental Biology Volume 207 ©2004 by Annual Reviews, www.annualreviews.org.
Figure 4.
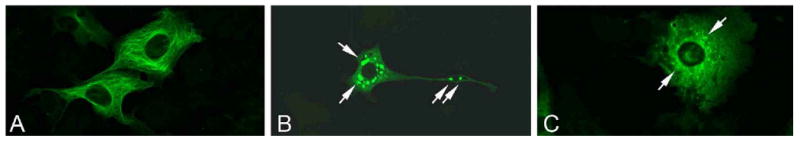
COS7 cells were transiently transfected with plasmids encoding wild-type (A) or mutant (B, p.G59S or C, p.G71R) p150glued protein. One day after transfection cells were fixed in 4% formaldehyde, blocked in 3% BSA and subsequently stained for p150glued (green). A polyclonal goat antibody directed against C′ terminal p150glued was used (1266-1278 aa, Abcam, 1:200 in 1%BSA in PBS) as DCTN1 mutations may affect N′ terminal epitopes and antibody affinity. Confocal pictures were taken using the 40× oil immersion objective of a Zeiss Axivert 200M microscope equipped with LSM510META technology. White arrows indicate representative examples of inclusion bodies.
From gene to inclusion: the conundrum of regional specificity
One of the crucial challenges facing researchers in neurodegenerative diseases is to understand what determines selective neuronal vulnerability. The five Perry mutations in DCTN1 lead to a complex albeit uniform clinico-pathological entity, yet the p.G59S mutation located only 12-15 amino acids away is associated with a clearly different disease (Table). The bulk of the pathology in Perry syndrome is found in pallidonigral neurons (accounting for parkinsonism in patients), whereas the p.G59S mutation affects lower motor neurons (clinically manifested by lower motor neuron disease).
In addition to the CAP-Gly domain, a second p150glued microtubule-binding domain was identified which is basic and highly conserved across species [28]. Evidence showed the CAP-Gly domain binds microtubules firmly as an “anchor”, whereas binding of the basic domain displays dynamic properties which allow “skating” along microtubules even in the absence of the motor dynein. This suggests antagonizing effects of the two p150glued microtubule-binding domains on dynein-promoted motility, the CAP-Gly domain acting as a “parking brake”, counteracting the dynamic properties of the basic domain [28]. Contrarily to the p.G59S mutation, Perry mutations are located within or immediately adjacent to the critical CAP-Gly “GKNDG” motif. Therefore it is conceivable that Perry mutations alter microtubule binding qualitatively by destabilizing the progression of dynactin along microtubules, favoring increased but unstable mobility. Differences in morphology, connectivity and function must explain why nigral neurons are more susceptible than motor neurons to such altered mobility. In particular, vesicular transport of potentially toxic dopamine precursors may confer nigral neurons a lower threshold to cell damage. However, patients with familial motor neuron disease and DCTN1 p.G59S, although they have an earlier onset and a more gradual progression, do not develop parkinsonism and nigrostriatal dysfunction (Table). Nor do Perry patients have any signs of motor neuron disease. Of note, the p.G59S is buried within the center of the CAP-Gly domain, whereas the Perry mutations are within a hydrophobic pocket upon the surface. In vitro, all DCTN1 pathogenic mutations in the CAP-Gly domain modestly impair microtubule binding, but there may be qualitative differences. In addition, p150glued CAP-Gly microtubule binding is only one of the many protein interactions whose binding and transport might be differentially perturbed in a cell-specific manner [29].
In physiological conditions, over 90% of the RNA-binding protein TDP-43 is located in the nucleus, where it plays a role in transcription repression, exon skipping and alternative splicing [30]. In the cytosol and in synaptic sites, TDP-43 may function as a translation regulator [30]. Two domains play a critical role in the regulation of TDP-43 trafficking between the cytosol and the nucleus, a nuclear localization signal and a nuclear export signal [31]. Under stress conditions such as starvation, and oxidation, TDP-43 levels are elevated the cytoplasm, and its transport may require fully functional dynactin. This hypothesis is supported by the co-localization of two subunits of dynactin (p50 and p62) and TDP-43 immunohistochemistry in some NCI from patients with Perry syndrome [19]. We should point out that TDP-43 immunostaining has not been reported in the patient with the p.G59S mutation as the publication antedated the identification of TDP-43 in neurodegenerative diseases [21]. Positive TDP-43 pathology would be expected given the underlying genetic defect is similar to that of Perry syndrome. However, negative results could indicate a cell-specific propensity to TDP-43 aggregation that may shed further light on selective vulnerability.
Diagnosis and treatment
To accurately diagnose a disease, to effectively treat it, to halt its progression and to affect a cure, requires an understanding of its molecular pathogenesis. The finding of p150glued mutations in Perry syndrome can now be used to formally diagnose this age-associated disorder in asymptomatic subjects and may help advanced directives, family planning and therapeutic intervention. Depression and suicidal ideation in carriers can be successfully managed. Assuming that lowering the quantitative amount of p150glued may not be detrimental to the stoichiometry and function of the dynactin complex, specific mutant allele silencing in the brain may also prevent the core features of disease and their progression. Several approaches for transcript silencing and delivery are in development for neurodegenerative disorders, including allele-specific targeting, upon which treatments for orphan diseases such as Perry syndrome can be included [32, 33].
Conclusions
Orphan diseases such as Perry syndrome are difficult to study due to lack of funding and low prevalence of disease. However, clinical, pathological and genetic features of Perry syndrome overlap with those of a number of more common conditions (Fig. 5). The distinct selective vulnerability of neuronal populations (extrapyramidal or motor neurons) is perhaps the most remarkable finding given the position of DCTN1 CAP-Gly mutations. Insights gained from studies of Perry syndrome, and the role of dynactin in cargo trafficking are now likely to shed light on the pathogenesis of a variety of diseases, including Parkinson's disease, multiple system atrophy, depression, FTLD-U, ALS, sleep disorders and metabolic diseases.
Figure 5.
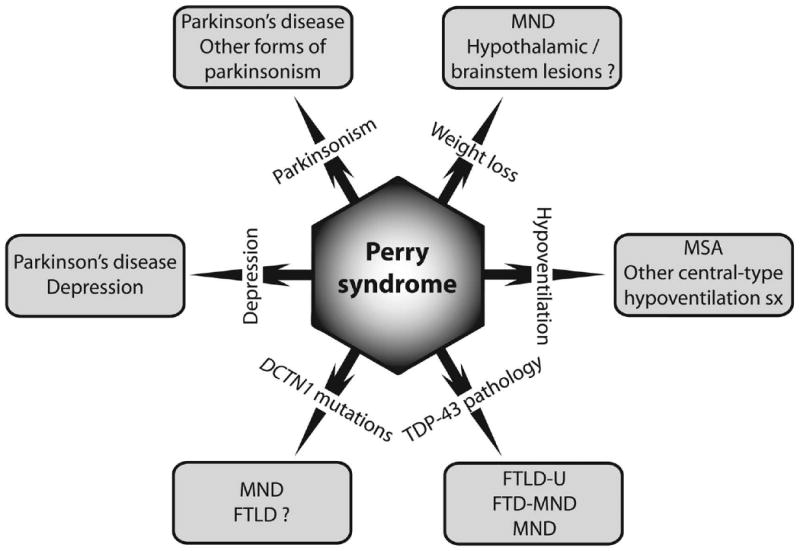
Schematic representation of clinical, pathological and genetic overlap of Perry syndrome with other conditions. FTD-MND, frontotemporal dementia – motor neuron disease. FTLD, frontotemporal lobar degeneration (-U, ubiquitin-positive). MND, motor neuron disease. MSA, multiple system atrophy.
Acknowledgments
We are grateful to the patients and their families for participating in our study. We thank posthumously Dr. Leon Thal for providing clinical material on the Hawaiian family. We are grateful to Sarah Lincoln and Caroline Kent for technical assistance. CW is supported by the Swiss National Science Foundation (PASMP3-123268/1). ZKW, MJF and DWD are supported by the Morris K. Udall NIH/NINDS Parkinson Disease Center of Excellence Grant awarded to the Mayo Clinic Jacksonville P50NS40256, by NIH/NIA P01AG017216 and NIH/NIA R01AG015866 grants, and by the Pacific Alzheimer Research Foundation Centre (PARF) C06-01 grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL. Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol. 1975;32(2):108–13. doi: 10.1001/archneur.1975.00490440058009. [DOI] [PubMed] [Google Scholar]
- 2.Perry TL, Wright JM, Berry K, Hansen S, Perry TL., Jr Dominantly inherited apathy, central hypoventilation, and Parkinson's syndrome: clinical, biochemical, and neuropathologic studies of 2 new cases. Neurology. 1990;40(12):1882–7. doi: 10.1212/wnl.40.12.1882. [DOI] [PubMed] [Google Scholar]
- 3.Wider C, Wszolek ZK. Rapidly progressive familial parkinsonism with central hypoventilation, depression and weight loss (Perry syndrome)--a literature review. Parkinsonism Relat Disord. 2008;14(1):1–7. doi: 10.1016/j.parkreldis.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Purdy A, Hahn A, Barnett HJ, Bratty P, Ahmad D, Lloyd KG, et al. Familial fatal Parkinsonism with alveolar hypoventilation and mental depression. Ann Neurol. 1979;6(6):523–31. doi: 10.1002/ana.410060611. [DOI] [PubMed] [Google Scholar]
- 5.Lechevalier B, Schupp C, Fallet-Bianco C, Viader F, Eustache F, Chapon F, et al. Familial parkinsonian syndrome with athymhormia and hypoventilation. Rev Neurol (Paris) 1992;148(1):39–46. [PubMed] [Google Scholar]
- 6.Lechevalier B, Chapon F, Defer G, Rivrain Y, Le Doze F, Schupp C, et al. Perry and Purdy's syndrome (familial and fatal parkinsonism with hypoventilation and athymhormia) Bull Acad Natl Med. 2005;189(3):481–90. discussion 90-2. [PubMed] [Google Scholar]
- 7.Bhatia KP, Daniel SE, Marsden CD. Familial parkinsonism with depression: a clinicopathological study. Ann Neurol. 1993;34(6):842–7. doi: 10.1002/ana.410340614. [DOI] [PubMed] [Google Scholar]
- 8.Roy EP, 3rd, Riggs JE, Martin JD, Ringel RA, Gutmann L. Familial parkinsonism, apathy, weight loss, and central hypoventilation: successful long-term management. Neurology. 1988;38(4):637–9. doi: 10.1212/wnl.38.4.637. [DOI] [PubMed] [Google Scholar]
- 9.Tsuboi Y, Wszolek ZK, Kusuhara T, Doh-ura K, Yamada T. Japanese family with parkinsonism, depression, weight loss, and central hypoventilation. Neurology. 2002;58(7):1025–30. doi: 10.1212/wnl.58.7.1025. [DOI] [PubMed] [Google Scholar]
- 10.Elibol B, Kobayashi T, Atac FB, Hattori N, Sahin G, Gurer G. Familial parkinsonism with apathy, depression and central hypoventilation (Perry's syndrome) Boston, MA: Kluwer Academic/Plenum Publishers; 2002. [Google Scholar]
- 11.Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord. 2008 doi: 10.1016/j.parkreldis.2008.07.005. Epub August 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114(1):63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- 13.Dickson DW. TDP-43 immunoreactivity in neurodegenerative disorders: disease versus mechanism specificity. Acta Neuropathol. 2008;115(1):147–9. doi: 10.1007/s00401-007-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008 doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435–45. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535–8. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuboi Y, Dickson DW, Nabeshima K, Schmeichel AM, Wszolek ZK, Yamada T, et al. Neurodegeneration involving putative respiratory neurons in Perry syndrome. Acta Neuropathol. 2008;115(2):263–8. doi: 10.1007/s00401-007-0246-1. [DOI] [PubMed] [Google Scholar]
- 19.Farrer M, Hulihan MM, Kachergus JM, Stoessl AJ, Grantier LL, Calne S, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2008 doi: 10.1038/ng.293. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–6. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 21.Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57(5):687–94. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58(5):777–80. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- 23.Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724–6. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- 24.Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–79. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 25.LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34(5):715–27. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 26.Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172(5):733–45. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixit R, Levy JR, Tokito M, Ligon LA, Holzbaur EL. Regulation of dynactin through the differential expression of p150glued isoforms. J Biol Chem. 2008 doi: 10.1074/jbc.M804840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Culver-Hanlon TL, Lex SA, Stephens AD, Quintyne NJ, King SJ. A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat Cell Biol. 2006;8(3):264–70. doi: 10.1038/ncb1370. [DOI] [PubMed] [Google Scholar]
- 29.Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, et al. Structure-function relationship of CAP-Gly domains. Nat Struct Mol Biol. 2007;14(10):959–67. doi: 10.1038/nsmb1291. [DOI] [PubMed] [Google Scholar]
- 30.Wang IF, Wu LS, Shen CK. TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol Med. 2008;14(11):479–85. doi: 10.1016/j.molmed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic Tar DNA binding protein (TDP-43) induces disease-like redistribution, sequestration and aggregate formation. J Biol Chem. 2008 doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431(7006):371–8. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 33.Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. 2008;3(1):19. doi: 10.1186/1750-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]