

Abstract
Putative transcription factors have only recently been identified in the Plasmodium spp., with the major family of regulators comprising the Apicomplexan AP2 (ApiAP2) proteins. To better understand the DNA-binding mechanisms of these transcriptional regulators, we characterized the structure and in vitro function of an AP2 DNA-binding domain from a prototypical ApiAP2 protein, PF14_0633 from Plasmodium falciparum. The X-ray crystal structure of the PF14_0633 AP2 domain bound to DNA reveals a β-sheet fold that binds the DNA major groove through base-specific and backbone contacts; a prominent α-helix supports the β-sheet structure. Substitution of predicted DNA-binding residues with alanine weakened or eliminated DNA binding in solution. In contrast to plant AP2 domains, the PF14_0633 AP2 domain dimerizes upon binding to DNA through a domain-swapping mechanism in which the α-helices of the AP2 domains pack against the β-sheets of the dimer mates. DNA-induced dimerization of PF14_0633 may be important for tethering two distal DNA loci together in the nucleus and/or for inducing functional rearrangements of its domains to facilitate transcriptional regulation. Consistent with a multi-site binding mode, at least two copies of the consensus sequence recognized by PF14_0633 are present upstream of a previously identified group of sporozoite-stage genes. Taken together, these findings illustrate how Plasmodium has adapted the AP2 DNA-binding domain for genome-wide transcriptional regulation.
Keywords: malaria, Plasmodium falciparum, AP2 domain, x-ray crystallography, domain swapping
Introduction
The malaria parasite Plasmodium falciparum exhibits a complex life cycle that involves infection of both a vertebrate human host and an invertebrate mosquito vector. The various host cell types and morphological transitions involved in the development of the parasite suggest the utilization of fine-tuned regulation of gene expression at all stages 1. However, dissection of transcriptional regulation in P. falciparum and other related apicomplexan parasites has proven difficult because their genomes possesses few proteins with detectable homology to established transcription factors in model eukaryotes. To date, only one major family of transcriptional regulators has been identified, the ApiAP2 family, which has undergone a lineage-specific expansion in the apicomplexan phylum 2.
ApiAP2 proteins vary in size from hundreds to thousands of amino acids and each contains at least one copy of a conserved ~60 residue AP2 domain that is weakly homologous to the plant Apetala2 (AP2) domain. In the plant Arabidopsis thaliana, AP2 domains are the second most common DNA binding domain after the Myb domain and are typically found in small proteins on the order of 300 amino acids3. Plant AP2 domains mediate specific DNA recognition in all members of the AP2/ethylene response factor (ERF) family, which control development and stress responses in species throughout the plant kingdom 3. Typically in plants, the AP2 domains are found as one- or two-domain arrangements in a given protein, and two-domain AP2 proteins have been shown to contact up to 17 contiguous base pairs of DNA 4. An NMR solution structure of a single Arabidopsis thaliana AP2 domain in complex with DNA reveals a mode of DNA binding where three β strands contact the DNA backbone and specific bases within the major groove. The β strands are stabilized by a C-terminal α-helix that does not contact the DNA 5.
Despite having limited homology to plant AP2 domains, apicomplexan AP2 domains bind DNA with high sequence specificity and can function as regulators of transcription 6;7. There are a total of 26 ApiAP2 proteins predicted in the Plasmodium genus, and although a given AP2 DNA binding domain can be highly conserved across related species, there is sparse homology outside of the DNA-binding domain. Presumably, the ApiAP2 proteins must also contain activation domains and additional protein-protein interaction domains, although there are no additional Pfam domains predicted for any of these proteins 8. Furthermore, paralogous AP2 domains from Plasmodium spp. share little sequence identity with each other, in contrast to those from Arabidopsis where paralogous AP2/ERF proteins can be functionally grouped on the basis of their primary sequence identity as well as their DNA-binding preferences 3; 9; 10. This divergence of AP2 domains in the Apicomplexans is reflected in the diversity of DNA sequences to which these domains can bind 11.
Recently, an ApiAP2 protein from the murine P. berghei species was shown to be essential in vivo for the activation of a set of ookinete-specific genes required during the parasite’s replicative stage in infected mosquitoes 7. This protein, named AP2-O, possesses only a single AP2 DNA-binding domain but appears to require two copies of a TAGCTA DNA sequence for high-affinity binding. Furthermore, all experimentally-determined downstream target genes of AP2-O contain 2 or more copies of the high-affinity site (TAGCTA) in their upstream regions. These data raise the possibility that AP2-O, and other ApiAP2 transcriptional regulators, may dimerize when bound to their target sequences. Dimerization could influence regulation of gene expression, as has been well-documented with other eukaryotic specific transcription factors 12. Although dimerization of plant AP2 domains has not been demonstrated, AP2 domains have been shown to contact other transcription factors and mediate protein-protein interactions through the AP2 domain 13; 14.
The AP2-containing protein PF14_0633 from P. falciparum is an 813 amino acid protein that includes a single, highly-conserved 60 amino acid AP2 domain and only one other identifiable domain, a short AT-hook DNA-binding motif that directly precedes the AP2 domain 15. We previously demonstrated that the AP2 domain of PF14_0633 binds a TGCATGCA DNA sequence with high sequence specificity 6. In this work we present the crystal structure of a domain-swapped dimer of the AP2 domain from PF14_0633 in complex with double-stranded (ds) DNA containing its consensus binding sequence (“cognate DNA”), as well as solution studies that examine DNA binding and the mechanism of dimer formation. Strikingly, specific binding of the PF14_0633 AP2 domain to its consensus DNA sequence was found to stimulate domain-swapped dimerization. We propose a model whereby DNA-induced dimerization of the AP2 domain of PF14_0633 facilitates the conformational rearrangements of the remainder of the protein or its interaction partners, while simultaneously looping out intervening DNA between pairs of binding sites enriched in the upstream regions of a set of sporozoite-specific genes.
Results and Discussion
Structure of the AP2 domain of PF14_0633 specifically bound to dsDNA
To elucidate the mechanism of DNA binding of an apicomplexan AP2 domain, we determined the 2.2 Å-resolution crystal structure of the AP2 domain of PF14_0633 bound to an 8bp dsDNA comprising its consensus binding sequence TGCATGCA (Figure 1 and Supplemental Figure 1). Single wavelength-anomalous dispersion phasing of a crystal of selenomethionine-incorporated AP2 domain bound to DNA produced a high quality experimental electron density map into which a partial molecular model was built. This model was used as a molecular replacement search model to phase the higher-resolution diffraction data from a native complex, which yielded electron density maps that allowed building of the final model (Figure 1, Table 1). The asymmetric unit contains two molecules of the AP2 domain of PF14_0633, with each monomer bound to an individual dsDNA molecule.
Figure 1. Structure of the AP2 domain of PF14_0633 specifically bound to dsDNA.

(A) Secondary structural elements of the AP2 domain of PF14_0633 are depicted above the primary sequence as arrows (β-strands) and a rectangle (α-helix). Residues that make base-specific contacts to dsDNA are underlined. (B) (Top) Ribbon diagram of the crystal structure of a domain-swapped dimer of the AP2 domain of PF14_0633 (one monomer shaded blue, another red) bound to independent dsDNA molecules (shaded green). Disulfide bonded Cys76 residues are indicated and shaded yellow. (Bottom) Orthogonal view with protein electrostatic surface potentials depicted (blue = electropositive, red = electronegative). (C) DNA contact map depicting direct base-specific contacts between the Asn72, Arg74, Arg88, or Ser90 side chains and the indicated DNA bases.
Table 1. Data collection, phasing and refinement statistics of the AP2 domain of PF14_0633 specifically bound to dsDNA.
| SeMet Anomalous Peak | Native | |
|---|---|---|
| Data Collection a | ||
| Space group | C2221 | C2221 |
| Cell dimensions a, b, c (Å) |
43.65, 59.18, 173.93 | 43.81, 58.82, 177.30 |
| Wavelength | 0.97886 Å | 0. 97886 Å |
| Resolution (Å) b | 50.0 – 2.40 (2.44–2.40) | 50.0–2.20 (2.26–2.20) |
| Rsym c | 0.107 (0.477) | 0.075 (0.381) |
| I / σI | 21.9 (2.5) | 32.1 (4.5) |
| Completeness (%) | 96.7 (71.3) | 91.9 (72.4) |
| Redundancy | 11.4 (4.2) | 11.0 (9.0) |
| Phasing | ||
| Figure of Merit (before/after solvent flattening) |
0.35/0.70 | |
| Refinement | ||
| Resolution (Å) | 29.55 – 2.20 (2.26–2.20) | |
| Rwork / Rfree | 0.230 / 0.279 | |
| No. atoms | 1691 | |
| Protein | 971 | |
| DNA | 623 | |
| Water | 97 | |
| <B-factors> | ||
| Protein | 59.1 | |
| DNA | 65.4 | |
| Water | 61.0 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.009 | |
| Bond angles (°) | 1.37 |
Data statistics from one crystal of Native or SeMet PF14_0633.
Values in parentheses are for highest-resolution shell.
Rsym = ΣΣj| Ij - <I> I/SIj, where Ij is the intensity measurement for reflection j and <I> is the mean intensity for multiply-recorded reflections.
The overall structure of the DNA-bound AP2 domain of PF14_0633 retains many of the canonical features of similar DNA-binding domains for which structures have been determined, such as the Arabidopsis thaliana ethylene response factor-1 (ATERF1) 5 and the yeast PI-SceI homing endonuclease 16. Three anti-parallel β strands from the AP2 domain of PF14_0633 wrap into the major groove of the bound dsDNA, and contain all residues making base-specific contacts. A flexible loop connects these strands with an extended α-helix, which supports the opposing face of the β-sheet. However our structure demonstrates that PF14_0633 dimerizes through a three dimensional domain-swapping mechanism in which the α-helix of one protomer is packed against the β-sheet of its dimer mate (Figure 1B). Similar dimerization was not previously observed in the NMR-solution structure of the related plant AP2 protein ATERF1 5. The interface derived from domain swapping results in burial of a large surface area (~3140Å2) of primarily hydrophobic residues. Dimerization of the PF14_0633 AP2 domain also aligns Cys76 residues of each monomer with one another with sufficient proximity to allow disulfide bond formation (Figure 1B). We note that the Cys76 residue is conserved in all orthologues of PF14_0633 in Plasmodium spp. but is not conserved in other related apicomplexan species.
Several contacts between the PF14_0633 AP2 domain and its DNA-binding sequence appear to be important to binding specificity (Figure 1C, summarized in Supplemental Table 1). Residues of particular note are Asn72, Arg74, Arg88, and Ser90, which make base-specific contacts to the consensus binding sequence. Of these, only one residue (Asn72) was previously implicated in DNA binding by computational modeling 2. The two arginine side chains can each contact both strands of DNA (O6 atoms of dG2 and dG6) (Figure 1C). The side chains of Asn72 and Ser90 contact a central AT base pair, each to one strand. All four of these residues are invariant among apicomplexan orthologues of PF14_0633 with BLAST expect scores <1e-05, including AP2 domains from Plasmodium spp., Toxoplasma, Theileria, Babesia and Cryptosporidium genera. The AP2 domain in this protein therefore appears to have conserved specific binding to the TGCATGCA consensus DNA over a large evolutionary distance separating these apicomplexan species by conservation of four key DNA contact residues. Indeed, the distantly related Cryptosporidium PF14_0633 orthologue binds specifically to the same TGCATGCA sequence 6. In addition to these base-specific contacts, several phosphodiester backbone contacts are made, most notably His94, which is located in the hinge region between the α-helix and the β-sheet of the AP2 domain. His94, Pro95, and several adjacent residues are all well conserved among the orthologous PF14_0633 AP2 domains. A common mechanistic model for domain swapping implicates strain in Pro-containing hinge regions, which relies on Pro isomerization to help drive conformational changes during dimerization 17; 18. The interaction of His94 with the phosphodiester backbone may influence the conformation of the adjacent Pro95 side chain and act as a trigger for domain swapping (discussed further below).
Residues making base-specific DNA contacts in the crystal structure are important in solution for DNA binding
In order to verify the determinants of DNA binding specificity as predicted in the crystal structure, we generated four variants of the PF14_0633 AP2 domain by substituting alanine for the residues observed to make base-specific DNA contacts (Asn72Ala, Arg74Ala, Arg88Ala, and Ser90Ala). These variants were subjected to electrophoretic mobility shift assays (EMSAs; Figure 2) and protein binding microarrays 19; 20 (PBMs; Supplemental Figure 4) to assess whether these amino acid substitutions altered DNA binding stability and specificity in solution. All four variants were found to have circular dichroism spectra highly similar to that of the wild-type AP2 domain (Supplemental Figure 2), indicating that functional differences were not due to differences in protein folding.
Figure 2. Electrophoretic mobility shift assay (EMSA) of the AP2 domain of PF14_0633 reveals the DNA binding contributions of several residues.

(A) A representative EMSA gel comparing DNA binding by wild-type (left) and a variant (Arg74Ala) PF14_0633 AP2 domain. Ten fmol of end-labeled 28bp probe containing the consensus binding sequence of the PF14_0633 AP2 domain were incubated in the absence or in the presence of 0.1–50μM AP2 domain. (B) Graph of percent specific DNA binding for the wild-type PF14_0633 AP2 domain, four alanine substitution variants, and a fifth variant that includes a native AT-hook located N-terminal of the AP2 domain, as calculated by quantification of the primary EMSA gel-shift.
We assessed the affinity of the purified wild-type PF14_0633 AP2 domain for its consensus sequence using EMSA (Supplementary Figure 3). Binding of this domain to two probes (28 bp and 122 bp in length), each containing a core 28 bp region derived from the upstream region of the pfi0540w gene of the P. falciparum 3D7 genome, as used previously 6 was measured (see Materials and Methods). The Kd, app of the wild-type domain for its TGCATGCA target sequence was determined to be 0.5–1.0 M. Mutation of the central two AT base pairs of the consensus sequence to GC significantly reduced the binding affinity of PF14_0633, confirming that these bases are critical for specific DNA recognition.
The ability of the variant AP2 domains (Asn72Ala, Arg74Ala, Arg88Ala, and Ser90Ala) to bind specifically to dsDNA was evaluated next. The 28bp dsDNA probe (described above) was incubated with increasing concentrations of the wild-type protein or its variants. A representative gel image for wild-type and Arg74Ala AP2 domains is shown in Figure 2A, where the wild-type protein forms a specific complex with dsDNA with a Kd, app of 0.5μM (primary shift). A slower-mobility form of the wild-type protein is observed at concentrations of 2.5μM and above (secondary shift). Quantification of the percent shifted probe for each Ala-substituted variant AP2 domain demonstrated that all bind dsDNA very poorly, binding specifically only at concentrations just below those where non-specific binding to both ssDNA and dsDNA occurs (Figure 2B). This is consistent with the protein-DNA interface observed in the crystal structure being used in solution. We also assessed the contribution of the AT-hook element (residues 39–50) to DNA binding by extending the expressed portion of PF14_0633 to include residues 38–123. AT-hooks are short auxiliary elements that contact DNA in a non-sequence specific manner, and are commonly found adjacent to specific DNA-binding domains 15. The addition of the AT-hook motif did not significantly affect the binding affinity of wild-type PF14_0633 to the 28bp probe, (Figure 2B). These results demonstrate that specific DNA binding was weakened in the variant AP2 domains, consistent with elimination of base-specific contacts provided by each side chain.
We reasoned that alteration of the individual residues that make base-specific contacts to the DNA-binding site might alter the sequence specificity of the PF14_0633 AP2 domain. To address this, GST-tagged forms of these variants of PF14_0633 were assayed using protein binding microarrays (PBMs) and sequence-specific binding was determined as previously described 19. As expected, the wild-type AP2 domain bound the consensus binding sequence TGCATGCA. Surprisingly, two of the four variants, (Asn72Ala and Ser90Ala) also bound this same sequence, albeit more weakly (Supplemental Figure 4). This result implies that these residues are not necessary for DNA binding specificity. Furthermore, it also suggests that the PBM methodology is far more sensitive for detecting lower affinity DNA-binding events which we could not detect by EMSA. In contrast to the Asn and Ser variants, DNA binding was completely abrogated with both Arg variants.
Taken together with the results from the EMSAs comparing wild-type and mutant DNA probes (Figure 2B, Supplemental Figure 3), the observation that Asn72Ala and Ser90Ala variants still recognize the consensus binding sequence with high statistical significance indicates that base-specific contacts to the central AT base pairs are not necessary for specific DNA binding. Instead, an appropriate balance of DNA rigidity/flexibility provided by central AT base pairs in the palindromic sequence may be important for specific binding. In contrast, Arg74 and Arg88 are required for sequence-specific DNA binding. These data imply that the identified consensus binding sequence represents the minimal binding sequence for the AP2 domain of PF14_0633, which is largely determined by the base-specific contacts of both Arg74 and Arg88.
DNA binding stimulates dimerization by domain swapping within the PF14_0633 AP2 domain
Two striking features of the structure of the AP2 domain of PF14_0633 bound to dsDNA are that the two monomers have domain swapped α-helices to form a dimer, and that the dimeric structure aligns the single native Cys residues within proximity to form a disulfide bond. To explore whether these observations are representative of the behavior of this domain in non-crystallographic conditions, we employed several solution-based experiments to examine the DNA dependence of disulfide bond formation, dimerization, and domain swapping.
We first addressed these questions by assessing whether a disulfide bond is formed at the dimerization interface upon binding to dsDNA. Under non-reducing conditions, no significant difference in disulfide bond formation was observed between the apo and dsDNA-bound forms of the AP2 domain of PF14_0633 (Figure 3A, non-reducing gel: lanes 1, 3, and 5). As an alternate approach, we tested whether the homobifunctional crosslinker BM(PEG)3, which can covalently react with free thiols on exposed Cys residues, has a differential effect on the apo and DNA-bound forms of the AP2 domain. As shown in Figure 3A, the presence of cognate dsDNA (containing the consensus binding sequence of PF14_0633’s AP2 domain, see Materials and Methods for details) stimulated crosslinking of two monomers to form a dimer approximately 50-fold relative to the apo domain (Figure 3A, reducing gel: lane 4 vs. lane 2). This effect is likely due to the proximal localization of the Cys residues by dimerization upon DNA binding. Incubation with non-cognate dsDNA also stimulated crosslinking (Figure 3A, reducing gel: lane 6), but with a reduced efficiency relative to cognate dsDNA. In agreement with the observation that these Cys residues are buried and/or disulfide bonded upon DNA binding, accessibility of these Cys residues was stimulated 9- to 13-fold by the presence of dsDNA as observed by an iodoacetamide protection/2-nitro-5-thiocyanatobenzoic acid (NTCB) cleavage assay (Supplemental Figure 5). These data indicate that DNA binding induces dimerization of the PF14_0633 AP2 domain.
Figure 3. DNA binding stimulates dimerization of the AP2 domain in solution.

(A) PF14_0633 AP2 domain was incubated in the absence or presence of cognate or non-cognate dsDNA, and subjected to crosslinking with BM(PEG)3 or vehicle alone. Samples were analyzed by non-reducing (left) or reducing (right) 20% SDS-PAGE. Monomer and dimer forms of the AP2 domain are indicated at the right. The average percent of total protein crosslinked and standard deviation from three replicates is given below each respective lane. (B) The wild-type AP2 domain of PF14_0633 was incubated in the absence (apo) or presence of cognate or non-cognate dsDNA and applied to a calibrated S100 size exclusion column. The elution volumes of standard marker proteins of masses 44, 17, and 1.35kDa were used to interpolate the molecular mass of resultant elution peaks. The peak containing proteins that eluted in the void volume (~35–40ml) was excluded from this analysis. The elution peak for the apo AP2 domain is indicated due to its low UV absorbance value. Samples from the two elution peaks of the wild-type AP2 domain bound to cognate dsDNA or from the apo AP2 domain were analyzed by non-reducing (left) and reducing (right) 20% SDS-PAGE (inset). The more quickly eluting complex (marked as “*”, elution peak at 60.0ml, “Peak 1”) contains a disulfide bonded dimer that is resolved to a monomer upon inclusion of reducing agent. The sample from the more slowly eluting complex (marked as “**”, elution peak at 67.5ml, “Peak 2”) contains no disulfide bonded complexes. (C) Ribbon diagram of the PF14_0633 AP2/DNA complex indicating the positions of Cys76, Ala86 (teal) and Asn118 (orange). A triple substitution variant (Cys76Ala, Ala86Cys, Asn118Cys) is also used in subsequent experiments. (D) The triple substitution variant (depicted in C) was analyzed as was the wild-type AP2 domain in (A).
Size exclusion chromatography of the wild-type AP2 domain in the absence or presence of cognate or non-cognate DNA was used as an independent means of testing whether DNA binding stimulates dimerization in solution (Figure 3B). The wild-type AP2 domain eluted at a volume consistent with the molecular size of a monomer (8.8kDa) in the absence of DNA. In the presence of cognate (8bp, 4.8kDa) or non-cognate (17bp, 10.6kDa) dsDNA, the AP2 domain eluted as two distinct peaks each, at volumes consistent with an AP2 dimer bound to 1 or 2 dsDNA molecules. The presence of DNA in these elution peaks was confirmed by a decrease in the A280:A260 ratio from 1.56 (apo) to 0.56–0.59 (DNA-bound), as well as by the large increases in peak UV absorbance due to the relatively large extinction coefficients of dsDNA. Samples from both peaks of the cognate dsDNA-bound AP2 complexes were compared to the apo form via reducing and non-reducing 20% SDS-PAGE (Figure 3B inset). The faster eluting species (marked as “*”, elution peak at 60.0ml, 7% of total) contained a disulfide-bonded dimer, whereas the slower eluting species (marked as “**”, elution peak at 67.5ml, 93% of total) was composed of a non-covalently bonded dimer (Figure 3B inset). Taken together, these experiments indicate that DNA binding stimulates the dimerization of the wild-type AP2 domain in solution.
In order to “capture” the domain-swapped form of the PF14_0633 dimer in solution, we engineered a triple substitution variant AP2 domain for further cross-linking studies. This protein variant (Cys76Ala Ala86Cys Asn118Cys) lacks the single native Cys residue in the dimerization interface, but introduces one Cys residue on the domain swapped α-helix and another in a spatially proximal loop between β-strands (illustrated in Figure 3C). The introduced Cys residues allow the assessment of domain swapping between dimer mates whereby the β sheet of one monomer can crosslink to the α helix donated by the other monomer. As observed above with the wild-type PF14_0633 AP2 domain, the presence of dsDNA containing the consensus binding sequence stimulated crosslinking of the triple point variant (Figure 3D, reducing gel: lane 4 vs. lane 2). Again, a reduced level of dimerization is seen in the presence of non-specific DNA (Figure 3A, reducing gel: lane 6). These observations imply that domain swapping also occurs in solution conditions. We hypothesize that DNA binding to the beta sheet may stabilize this portion of the domain, thus allowing the helix to adopt the extended conformation and form a domain-swapped dimer.
In light of these data, we propose a model in which the quaternary structure of the AP2 domain is influenced by specific DNA binding (Figure 4). The DNA trigger could act by either stabilizing the dimeric state in a pre-existing monomer-dimer equilibrium of the AP2 domain or, alternatively, DNA binding by an AP2 monomer could induce a conformational change that attracts a second monomer to bind. The AP2 dimer can be stabilized by several factors, including hinge region effects and stabilizing interactions made by the dimer interface or by ligand binding. In the case of the AP2 domain of PF14_0633, both of these factors could be important. First, Pro95 in the hinge region is in a trans conformation in the dimer crystal structure. If the closed monomeric form requires Pro95 to be in a cis conformation, isomerization could both relieve strain and provide energy to produce a conformational change. Notably, other residues in the hinge make backbone contacts with the DNA that may help drive DNA-binding stabilization of the dimeric form of the AP2 domain. Additionally, the structural and biochemical evidence presented herein indicates that the formation of a disulfide bond at the dimerization interface (Cys76) could also increase the stability of the dimer form. Lastly, observations from our three solution-based experiments confirm that binding dsDNA specifically stimulates the formation and/or stabilization of the domain-swapped dimer, as observed in the crystallographic structural model. The influence of these structural features indicates that the composition of the AP2 domain of PF14_0633 is well suited for the formation of a bona fide domain-swapped dimer, which may be relevant to its role in transcriptional regulation.
Figure 4. A model of DNA-induced, domain swapped dimerization and DNA looping.
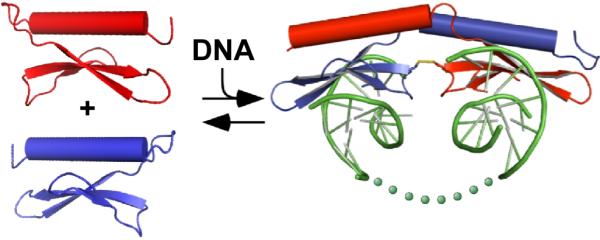
DNA binding stimulates formation and/or stabilization of the domain-swapped dimer, which in turn loops out intervening DNA between the two binding sites (illustrated as dots). Dimerization may allow portions of PF14_0633 not observed in the crystal structure (e.g. the c-terminal ~700 residues) to undergo functional rearrangement of protein interaction/transcriptional regulatory domains.
Many eukaryotic specific transcription factors form requisite dimers or higher-order multimers to assemble into their functional conformation. Some, such as Oct-1, Pit-1, and Ets-1 become competent for transcriptional regulation by differential homodimerization induced by the sequence and architecture of their DNA-binding sites 21; 22. Notably, the human FOXP2 transcription factor forms a domain-swapped dimer, with known disease-associated mutations affecting residues of the domain swapping interface 23. Similarly to the AP2 domain of PF14_0633, the orientation of the DNA-binding region of the domain-swapped FOXP2 dimer would only allow the binding of two distal DNA binding sites, thus looping out intervening DNA. The precedence of such a structural explanation of transcriptional regulation by FOXP2 thus lends credence to a similar mechanism for the function of PF14_0633.
Multiple PF14_0633 AP2-binding sites are found upstream of a set of sporozoite stage genes
Previous work by Young and colleagues identified a statistically-enriched DNA motif (PfM24.1) located upstream of 30 genes expressed in the mosquito sporozoite stage of the P. falciparum lifecycle 24. This motif is highly similar to the consensus TGCATGCA motif recognized by PF14_0633 6 suggesting that the sporozoite-expressed ApiAP2 protein PF14_0633 25 may be the transcription factor responsible for the transcriptional regulation of these genes. Our structural model implies that binding to multiple TGCATGCA binding sites may facilitate the function of PF14_0633 as a transcriptional regulator by inducing protein dimerization. To test this possibility, we used the PBM-derived position weight matrix for the TGCATGCA consensus sequence to scan the 2kbp upstream regions of these 30 sporozoite genes for secondary occurrences of the TGCATGCA sequence (see Materials and Methods). Of the 30 sporozoite genes, 27 contained two or more matches to the motif (Supplemental Table 2). Furthermore, 24 of these upstream regions contained pairs of sites that are spaced sufficiently distant (>100bp apart) to permit dimer-induced DNA looping (Supplemental Table 2).
The occurrence of multiple DNA-binding sites upstream of this subset of well-established sporozoite genes suggests that dimer binding may be important for the function of PF14_0633 in transcriptional regulation. Co-occurrence of motifs was also reported for the ookinete-specific Plasmodium transcriptional regulator AP2-O 7. One model is that the positioning of multiple binding sites in a promoter mediates a higher affinity interaction of PF14_0633 with DNA, facilitated by dimer formation. It remains to be tested whether higher order structures are essential for function in vivo. Structural rotations of the C-terminal α-helix, upon dimer formation, may expose anticipated additional domains of PF14_0633 that activate/repress transcription or modulate interaction with other auxiliary proteins required for regulation. Activation and repression domains that mediate transcriptional regulation have been defined only for a few examples of AP2/ERF domain proteins in the plant kingdom 26, and to date no such characterization exists for any Apicomplexan ApiAP2 protein. However, the wide range of predicted sizes of ApiAP2 proteins (200 – 4000 amino acids in Plasmodium spp.) presumably speaks to the likely diversity of sequence space available to serve as necessary functional activation and repression domains.
We anticipate that the apicomplexan AP2 DNA-binding domain structure presented herein will serve as a model for assessing the binding interaction of other apicomplexan ApiAP2 proteins. PF14_0633 orthologues from two highly diverged Apicomplexans, Cryptosporidium parvum and P. falciparum, have conserved their DNA-binding preference for the TGCATGCA sequence 6, the most over-represented DNA sequence in all sequenced Apicomplexa 27. Notably, the four key DNA-binding residues identified in the PF14_0633 structure are conserved in the C. parvum orthologue cgd2_3490. This conservation suggests that insights gained on transcription factor domains in one apicomplexan species will be broadly applicable to understanding how related Apicomplexan AP2 domains contact DNA and regulate gene expression. Although it could be possible to model the structure of other AP2 domains upon that of PF14_0633, the prediction of DNA-binding specificity and which residues make base-specific DNA contacts would likely not be similarly predictable. Important differences that should be taken into consideration are that many ApiAP2 domains do not contain conserved cysteine residues that are present at the dimerization interface, nor do they possess appropriately placed proline residues that might facilitate domain swapping identified here. Additional structural and biochemical characterization of these domains will be needed to elucidate these functional details. In addition, apicomplexan AP2 domains possess affinities for a much more diverse set of DNA sequences than in plants where an overwhelming majority of AP2/ERF proteins bind the canonical GCC box (A/GCCGCC) 9; 11. This difference from plant AP2 domains demonstrates the need for structural and biochemical studies tailored towards Apicomplexan-derived proteins. Such studies are of great importance given that the ApiAP2 proteins are key regulators of parasitic developmental progression, and the absence of any homologous proteins in their mammalian hosts make these proteins notable candidate drug targets.
Materials and Methods
Purification of the PF14_0633 AP2 domain and variants
The PF14_0633 AP2 domain (residues 63–123) or its variants were expressed as a GST-fusion protein as previously described 6. Cells were lysed by sonication in GST Lysis Buffer (50mM Tris pH 8.0, 150mM NaCl, 10% glycerol) containing 1mM phenylmethanesulphonylfluoride (PMSF) and 1mM benzamidine. Proteins were purified from the soluble fraction by binding to glutathione sepharose 4B resin (GE Healthcare) and eluting with GST Lysis Buffer containing 20mM reduced L-glutathione (Acros). The GST epitope tag was removed by thrombin cleavage. Proteins were dialyzed exhaustively to remove glutathione, and were further purified by cation exchange and size-exclusion chromatography, which yielded >95% pure samples in monomeric form. Selenomethionine-incorporated PF14_0633 was expressed in E. coli grown in M9 minimal media containing selenomethionine (Acros) and purified as above, except that 2mM fresh dithiothreitol (DTT) was included in buffers. Protein concentrations were determined by measurement of the absorbance at 280nm.
Crystallization and structure determination of DNA-bound PF14_0633 AP2 domain
PF14_0633 AP2 domain (15mg/ml in GST Lysis Buffer) was mixed at a 1:1.2 molar ratio (protein:dsDNA) with a palindromic dsDNA (5′-TGCATGCA-3′, annealed in 10mM Tris pH 8.0, 50mM NaCl). Equivalent volumes of the complex and mother liquor (10mM CoCl2, 100mM sodium acetate pH 5.1, 0.5–1.25M 1,6-hexanediol, 10–20% v/v ethylene glycol) were mixed and crystals of the complex formed in 1–3 days by hanging-drop vapor diffusion. Crystals were cryoprotected in mother liquor adjusted to 25% ethylene glycol prior to being flash-frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source at Argonne National Laboratory (Argonne, IL) at the LS-CAT 21-ID-D beamline (Table 1). Data were indexed and scaled with HKL2000 28 and an experimental electron-density map of the selenomethionine-incorporated AP2 domain of PF14_0633 bound to dsDNA was generated by single-wavelength anomalous dispersion phasing using SOLVE and solvent flattening using DM 29. A partial model of the complex was generated and then used as a molecular replacement search model to determine the structure of the native complex using Phaser 30. The native structure model was improved by rounds of manual building using Coot 31 and refinement with REFMAC 32.
Electrophoretic Mobility Shift Assay (EMSA)
Oligonucleotides containing a 28bp fragment of the genomic sequence from P. falciparum located upstream of pfi0540w that contains the PF14_0633 AP2 consensus binding sequence (GCATGC) was synthesized by the University of Wisconsin Biotechnology Center to create a DNA substrate (“wt”). A mutant substrate with the central AT base pairs of the consensus binding site changed to GC (GCGCGC, “mut”) was also produced. Both substrates were inserted into the StuI site of the pCR-Blunt vector (Invitrogen) to confirm their sequence identity, and to produce 122bp DNA substrates by restriction with HindIII and XhoI and polyacrylamide gel electrophoretic purification (probe sequences listed in Supplemental Table 3). One pmol of each duplex DNA probe was end-labeled by T4 Polynucleotide Kinase (New England Biolabs) with γ32P-ATP. Ten fmol of end-labeled probe was incubated with 0.1–50uM concentrations of the PF14_0633 AP2 domain (or a variant) in 20μl GST Lysis Buffer supplemented with 0.1mg/ml BSA and 5μg/ml poly (dI-dC) (Sigma) for 30 min at room temperature. Samples were electrophoresed through a 12% polyacrylamide gel and exposed to a phosphor screen (Molecular Dynamics). Signals were visualized by a Storm 860 PhosphorImager and band intensities were quantified by ImageQuant (Molecular Dynamics).
Protein Binding Microarrays
Protein binding microarray experiments using the PF14_0633 AP2 variant domains were performed as described previously 33. A minimum of two replicate experiments were performed for each variant.
Cysteine Crosslinking
The PF14_0633 AP2 domain (or triple variant, Cys76Ala Ala86Cys Asn118Cys) were treated with 2mM DTT to reduce disulfide bonds, and subsequently dialyzed exhaustively against GST Lysis Buffer (pH 7.4) to remove the DTT. Individual proteins (60μM) were incubated in the absence of dsDNA, or in the presence of 75μM cognate dsDNA (5′-TGCATGCA-3′) or non-cognate dsDNA (5′-TGTGCATAGTGGTGCGA-3′) for 30min at 4°C. Dimethylformamide (DMF) (0.5μl) or 20mM BM(PEG)3 (a 17.8Å homobifunctional maleimide crosslinker, Thermo Scientific) dissolved in DMF was added to samples for 30min at 4°C. In these conditions, thiol-specific crosslinking proceeds 1000-fold more quickly than non-specific reaction with primary amines. Samples were electrophoresed on reducing or non-reducing 20% SDS-PAGE gels, and visualized by Coomassie Blue. Band intensities were analyzed by ImageQuant.
Calibrated Size Exclusion Chromatography
PF14_0633 AP2 domain (400μM) was incubated with 500μM cognate dsDNA, non-cognate dsDNA (sequences described above), or without DNA for 1–2 hours in GST Lysis Buffer at 4°C. Samples were applied to a GE Healthcare S-100 HR column and elution volumes determined by detection of UV absorbance peaks. Average molecular weights of each peak were determined by comparing the elution volumes to calibrated standard markers (Bio-Rad Gel Filtration Standard) run just prior to and following experimental runs. Peak fractions were also analyzed by reducing and non-reducing SDS-PAGE as above.
Bioinformatic identification of multiple binding sites with transcriptional upstream regions of P. falciparum
ScanACE 34 and the PBM-derived PF14_0633 wild-type position weight matrix were used to scan the 2kb upstream regions of the P. falciparum sporozoite-expressed genes (listed in 24). The 2kb upstream regions of the 30 sporozoite genes were extracted from the Plasmodium genome resource PlasmoDB.org v5.5 35. Scoring cut-offs were set to capture two standard deviations below the mean score in the alignment file.
Supplementary Material
Acknowledgments
We thank Spencer Anderson of LS-CAT for beamline assistance, Ken Satyshur for assistance with crystallographic data collection and model building, Darrell R. McCaslin of the University of Wisconsin-Madison Biophysics Instrumentation Facility, which is supported by the University of Wisconsin-Madison, National Science Foundation grant BIR-9512577, and National Institutes of Health grant S10 RR13790, for assistance with circular dichroism spectroscopy. This investigation was supported by the National Institutes of Health under a Ruth L. Kirschstein National Research Service Award (1F32GM083438) to SEL; a Shaw Foundation award to JLK; and by NIH grant R01 AI076276, the Arnold and Mabel Beckman Foundation, and in part by NIH grant # P50 GM071508 to ML. SEL and EDS performed research; SEL, EDS, JLK and ML designed research, analyzed data, and wrote the paper.
Accession Numbers: The x-ray crystallographic structure and structure factors have been deposited in the protein data bank (PDB) and has been assigned the identifier 3IGM.
Footnotes
Data Deposition: Crystallographic coordinates and structure factors have been deposited in the protein data bank (PDB) and have been assigned the identifier 3IGM.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no conflict of interest.
References
- 1.Horrocks P, Wong E, Russell K, Emes RD. Control of gene expression in Plasmodium falciparum - ten years on. Mol Biochem Parasitol. 2009;164:9–25. doi: 10.1016/j.molbiopara.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Balaji S, Babu MM, Iyer LM, Aravind L. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005;33:3994–4006. doi: 10.1093/nar/gki709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–46. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- 4.Krizek BA. AINTEGUMENTA utilizes a mode of DNA recognition distinct from that used by proteins containing a single AP2 domain. Nucleic Acids Res. 2003;31:1859–68. doi: 10.1093/nar/gkg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen MD, Yamasaki K, Ohme-Takagi M, Tateno M, Suzuki M. A novel mode of DNA recognition by a beta-sheet revealed by the solution structure of the GCC-box binding domain in complex with DNA. EMBO J. 1998;17:5484–96. doi: 10.1093/emboj/17.18.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Silva EK, Gehrke AR, Olszewski K, Leon I, Chahal JS, Bulyk ML, Llinas M. Specific DNA-binding by apicomplexan AP2 transcription factors. Proc Natl Acad Sci U S A. 2008;105:8393–8. doi: 10.1073/pnas.0801993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuda M, Iwanaga S, Shigenobu S, Mair GR, Janse CJ, Waters AP, Kato T, Kaneko I. Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol Microbiol. 2009;71:1402–14. doi: 10.1111/j.1365-2958.2009.06609.x. [DOI] [PubMed] [Google Scholar]
- 8.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–8. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakuma Y, Liu Q, Dubouzet JG, Abe H, Shinozaki K, Yamaguchi-Shinozaki K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem Biophys Res Commun. 2002;290:998–1009. doi: 10.1006/bbrc.2001.6299. [DOI] [PubMed] [Google Scholar]
- 10.Shigyo M, Hasebe M, Ito M. Molecular evolution of the AP2 subfamily. Gene. 2006;366:256–65. doi: 10.1016/j.gene.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Llinas M, Deitsch KW, Voss TS. Plasmodium gene regulation: far more to factor in. Trends Parasitol. 2008;24:551–6. doi: 10.1016/j.pt.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Amoutzias GD, Robertson DL, Van de Peer Y, Oliver SG. Choose your partners: dimerization in eukaryotic transcription factors. Trends Biochem Sci. 2008;33:220–9. doi: 10.1016/j.tibs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Buttner M, Singh KB. Arabidopsis thaliana ethylene-responsive element binding protein (AtEBP), an ethylene-inducible, GCC box DNA-binding protein interacts with an ocs element binding protein. Proc Natl Acad Sci U S A. 1997;94:5961–6. doi: 10.1073/pnas.94.11.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler JW, Cole M, Flier A, Grewe B, Werr W. The AP2 transcription factors DORNROSCHEN and DORNROSCHEN-LIKE redundantly control Arabidopsis embryo patterning via interaction with PHAVOLUTA. Development. 2007;134:1653–62. doi: 10.1242/dev.001016. [DOI] [PubMed] [Google Scholar]
- 15.Aravind L, Landsman D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;26:4413–21. doi: 10.1093/nar/26.19.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moure CM, Gimble FS, Quiocho FA. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat Struct Biol. 2002;9:764–70. doi: 10.1038/nsb840. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–99. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newcomer ME. Protein folding and three-dimensional domain swapping: a strained relationship? Curr Opin Struct Biol. 2002;12:48–53. doi: 10.1016/s0959-440x(02)00288-9. [DOI] [PubMed] [Google Scholar]
- 19.Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, 3rd, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;24:1429–35. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukherjee S, Berger MF, Jona G, Wang XS, Muzzey D, Snyder M, Young RA, Bulyk ML. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat Genet. 2004;36:1331–9. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamber EP, Vanhille L, Textor LC, Kachalova GS, Sieweke MH, Wilmanns M. Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 2008;27:2006–17. doi: 10.1038/emboj.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remenyi A, Tomilin A, Pohl E, Lins K, Philippsen A, Reinbold R, Scholer HR, Wilmanns M. Differential dimer activities of the transcription factor Oct-1 by DNA-induced interface swapping. Mol Cell. 2001;8:569–80. doi: 10.1016/s1097-2765(01)00336-7. [DOI] [PubMed] [Google Scholar]
- 23.Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L. Structure of the forkhead domain of FOXP2 bound to DNA. Structure. 2006;14:159–66. doi: 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Young JA, Johnson JR, Benner C, Yan SF, Chen K, Le Roch KG, Zhou Y, Winzeler EA. In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genomics. 2008;9:70. doi: 10.1186/1471-2164-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301:1503–8. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- 26.Mitsuda N, Ohme-Takagi M. Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 2009;50:1232–48. doi: 10.1093/pcp/pcp075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ling KH, Rajandream MA, Rivailler P, Ivens A, Yap SJ, Madeira AM, Mungall K, Billington K, Yee WY, Bankier AT, Carroll F, Durham AM, Peters N, Loo SS, Isa MN, Novaes J, Quail M, Rosli R, Nor Shamsudin M, Sobreira TJ, Tivey AR, Wai SF, White S, Wu X, Kerhornou A, Blake D, Mohamed R, Shirley M, Gruber A, Berriman M, Tomley F, Dear PH, Wan KL. Sequencing and analysis of chromosome 1 of Eimeria tenella reveals a unique segmental organization. Genome Res. 2007;17:311–9. doi: 10.1101/gr.5823007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994. [Google Scholar]
- 30.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 33.Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–45. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 35.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Jr., Treatman C, Wang H. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–43. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.