

Abstract
The influence of diabetes on brain glutamate (GLU) uptake was studied in insulinopenic (streptozotocin (STZ)) and insulin-resistant (diet-induced obesity (DIO)) rat models of diabetes. In the STZ study, adult male Sprague-Dawley rats were treated with STZ (65 mg/kg iv) or vehicle and studied 3 weeks later. STZ rats had elevated plasma levels of glucose, ketone bodies and branched-chain amino acids; brain uptake of GLU was very low in both STZ and control rats, examined under conditions of normal and greatly elevated (by iv infusion) plasma GLU concentrations. In the DIO study, rats ingested a palatable, high-energy diet for 2 weeks, and were then divided into weight tertiles: rats in the heaviest tertile were designated DIO, rats in lightest tertile diet-resistant (DR), and rats in the intermediate tertile controls. DIO and DR rats continued to consume the high-energy diet for 4 more weeks, while control rats were switched to standard rat chow. All rats were studied at 6 weeks (subgroups were examined under conditions of normal or elevated plasma GLU concentrations). DIO rats ate more food and were heavier than DR or control rats, and had higher plasma leptin levels and insulin:glucose ratios. In all diet groups, the BBB showed very low GLU penetration, and was unaffected by plasma GLU concentration. Brain GLU uptake also did not differ among the diet groups. Together, the results indicate that the BBB remains intact to the penetration of GLU in two models of diabetes under the conditions examined.
Keywords: insulin, brain capillaries, endothelial cells, dicarboxylic amino acids, leptin, diabetes, streptozotocin, diet-induced obesity
1. Introduction
The movement of nutrients into brain from the circulation involves specific transport carriers, located on the membranes of capillary endothelial cells, which form the “blood-brain barrier” (BBB) (1). For amino acids (AA), a number of such carriers exist, that are differentiated by the size and charge of the AA transported (2). For example, separate carriers are found for large, neutral AA (e.g., aromatic and branched-chain AA), basic AA (lysine, arginine), and the acidic AA (glutamic (GLU) and aspartic acids). The large neutral and basic AA carriers generally facilitate the uptake of these AA into brain, while the acidic AA carriers restrict entry (2). Acidic AA are excluded from brain, because they function directly at neuronal synapses as excitatory neurotransmitters. Indeed, roughly two-thirds of all synapses in cerebral cortex utilize GLU as a neurotransmitter (3), making it the dominant brain transmitter. GLU compartmentation in brain is thus an important physiological issue, because the presence of high amounts of GLU in the extracellular fluid (ECF) surrounding neurons can cause them to be overexcited, leading ultimately to their death (3-6). The brain compartmentalizes GLU by synthesizing all that it needs, and keeping it carefully sequestered within neurons and glial cells. It maintains ECF GLU concentrations at very low levels, except during the brief periods when the AA is released from nerve endings to excite GLU receptors on adjacent neurons. And even when GLU concentrations rise in the local ECF milieu of the synapse following its release, powerful transporters on neuronal and glial cells rapidly remove it from the synaptic ECF (3). The GLU absorbed by glial cells is converted to glutamine, and recycled to neurons, where it is deaminated to GLU and reused (3). Such GLU transporters (EAAT-1, EAAT-2, EAAT-3) are also located on the abluminal membranes of capillary endothelial cells (the BBB), and function to remove GLU from ECF, from which it enters the general circulation (7).
Over the past two decades, interest has developed in evaluating the possibility that BBB defects are associated with chronic diseases that affect cognition. One example is diabetes; in this case, interest in BBB integrity derives from prior observations that kidney and retinal capillary functions degrade as the disease progresses (8-10). Indeed, some data suggest that the barrier function of the BBB falters in diabetes (11-13). However, it is not clear that such changes, if prevalent, compromise the effectiveness of the nutrient transporters of brain capillary endothelial cells. The transporters for the large neutral AA and basic AA may (14) or may not (15,16) be modified, and, no data appear to be available for the acidic AA. Because of the importance of the compartmentalization of GLU within brain, which includes normal functioning of GLU transport activities at the BBB, we have examined net GLU uptake into brain in insulinopenic (STZ-induced) and insulin-resistant (DIO obesity) animal models of diabetes.
2. Materials and Methods
2.1 Animals
Male Sprague-Dawley rats (300-325 g initial weight (Charles River Laboratories, Wilmington, MA) were housed individually at 23-24 °C (humidity 40-50%), with a daily lighting cycle of 14 h light: 10 h dark (6:00 h lights on; 10:00 h lights off). They had free access to food (Rodent Diet #8640, Harlan Teklad, Madison WI) and water.
All rats were acquired, cared for and handled in conformance with: the “Public Health Service Policy on Humane Care and Use of Laboratory animals.” (Office of Laboratory Animal Welfare, National Institutes of Health), the “Guide for the care and Use of Laboratory Animal Animals (Institute of Laboratory Animal Resources, Commission on Live Sciences, National Research Council), and the “Guiding Principles for Research Involving Animals and Humans” (recommendations from the declaration of Helsinki) approved by the Council of the American Physiological Society. The Institutional Animal Care and Use Committee of Rosalind Franklin University approved all experimental protocols.
2.2. Streptozotocin (STZ)-induced diabetes
Rats were anesthetized with halothane, and injected with STZ (65 mg/kg) (Sigma-Aldrich, St. Louis, MO) or vehicle (154 mM NaCl) through a tail vein (n = 12/group). This dose of STZ reliably produces diabetes, with rats becoming hyperglycemic and ketotic within 24 hours (17). Following injection, STZ-treated rats had access for 24 hr to water bottles containing 10% glucose; all rats continued to have free access to food and water throughout the study. All rats were weighed 3 times per week and STZ-treated rats were monitored for the presence of glucose and ketone bodies in the urine using Uriscan 2 gluketo strips (YD Diagnostics Corp. Seoul, Korea). BBB GLU transport measurements were conducted three weeks after induction of diabetes.
2.3. Diet-induced obesity
A group of 36 rats was given free access to water and a high-energy diet (D12266B: 16.8 % protein, 51.4 % carbohydrate, 31.8% fat (%-energy); 4.41 kcal/gram; Research Diets, New Brunswick NJ). Food intakes and body weights were measured daily. After 2 weeks, the rats were divided into 3 groups (n = 12/group): those that gained the most weight were designated the diet-induced obese (DIO) group, and those that gained the least weight the diet-resistant group (DR); these two groups continued to consume the same high-energy diet for an additional four weeks. The intermediate-weight group was designated the control group, and switched to standard rodent diet (Rodent Diet #8640: 23.6 % protein, 64.1 % carbohydrate, 12.3% fat (%-energy); 3.82 kcal/gram; Harlan Teklad, Madison WI) for the subsequent four-week period (18). BBB GLU transport measurements were conducted at the end of this period, 6 weeks after initiating the study.
2.4. Study subgroups
All rats were studied in the morning. They were not fasted. Each group (STZ, control; DIO, DR, control) was divided into two sub-groups. Following anesthetization, one subgroup received an infusion of saline, while the other received an infusion of L-GLU (100 mM L-GLU infused at 1ml•kg-1•min-1 for 3 min and then at 0.26 ml•kg-1•min-1 until the end of the experiment). The intent in infusing GLU was to test the integrity of the BBB to GLU under conditions of very high circulating GLU concentrations.
2.5. Outline of the basic experimental procedure
The animal model was a rat fitted with an arterial and a venous catheter such that L-[14C]GLU could be introduced into the venous system and samples of arterial blood taken continuously throughout the experimental period. At the end of the experiment the rats were killed and their brains removed. The measurements necessary for the calculation of GLU clearance are the dpm per g tissue (obtained by autoradiography of brain sections) and the average plasma L-[14C]GLU content over the 1 min experimental period.
2.6. Surgery
Rats were anesthetized with a mixture of ketamine and xylazine (90mg ketamine, 10 mg xylazine per kg body weight) (Lloyd Laboratories Shenandoah, Iowa). Catheters were then introduced into the left femoral artery and left femoral vein, and Lidocaine (Sigma-Aldrich, St. Louis, MO) was applied to all incisions. Infusions (saline or 100 mM L-GLU) were then initiated. Three minutes later, 50 μCi of L-[14C]GLU (GE Healthcare/Amersham, Piscataway NJ) was injected through the venous catheter over 45seconds. Blood was withdrawn continuously from the arterial catheter into heparinized syringes, and stored in heparinized tubes. This sample was counted to determine the average dpm/μl in plasma for calculating the rate of clearance Sixty seconds after initiating the injection of L-[14C]GLU, the rats were killed by an intravenous injection of sodium pentobarbital (150 mg in 1 ml of 0.15 M NaCl; Sigma-Aldrich, St. Louis, MO) and decapitated. This dose of pentobarbital stopped the heart within 3 s; the rats did not recover consciousness. Plasma was frozen for later analyses of [14C]GLU, GLU, glucose, leptin, insulin, β-hydroxybutyrate, acetoacetate and branched-chain AA.
2.7. Autoradiography
Rats were decapitated within 6-8 seconds after death. The brains were removed from the skull in less than 3 minutes and frozen between layers of bromobutane and methylbutane chilled to −30°C (19). Care was taken not to dislodge the pineal or pituitary glands. The brains were sectioned at 50 μm in a precision cryostat (Microm International, Richard Allen Scientific, Kalamazoo, MI) maintained between 8 and 12 °C. Every fourth section was fixed to a slide and packed with Biomax® light autoradiography film (Kodak, Rochester, NY) with a set of [14C] standards (GE Healthcare/Amersham, Piscataway NJ) that were calibrated to relate optical density to dpm/g. After 10 days the films were developed and the optical density in specific areas was measured using a densitometer (Tobias, Ivyland, PA) with an aperture of 300μm diameter. The optical densities were converted to dpm/g by comparison with the [14C] standards.
2.8. Plasma measurements
For the assays of β-hydroxybutyrate and acetoacetate an aliquot of plasma was deproteinized in 0.5 M HClO4, followed by neutralization with 20 % KOH (w/v). GLU, glucose, β-hydroxybutyrate and acetoacetate were measured in deproteinized extract by specific enzymatic assays (20). Enzymes and cofactors were purchased from Boehringer Mannheim (Indianapolis, IN).
2.9. Plasma radioactivity
The [14C] activity in plasma samples was measured by liquid scintillation spectroscopy. An aliquot of plasma, 10μl, was placed in 500 μl of H2O in a scintillation vial, counting fluid added (Ready Safe®, Beckman Coulter, Inc., Fullerton, CA) and the samples counted in a LS6500 liquid scintillation counter (Beckman Coulter, Inc., Fullerton, CA). It has previously been shown (21) that ∼97% of the radioactivity in plasma was in the form of L-[14C]GLU under the experimental conditions used.
2.9.1 Hormones
Leptin and insulin were measured in untreated plasma by specific radioimmunoassay (rat insulin kit RI-13K; rat leptin kit RL-83K; Millipore Corp, Billerica MA).
2.9.1 Amino acids
Plasma concentrations of the branched-chain amino acid were determined using HPLC separation and electrochemical detection, following reaction of samples with o-phthaldialdehyde (Sigma-Aldrich, St. Louis MO) (22).
2.9.2 Calculations
Glutamate transport into brain was calculated according to the equation
These calculations assume that there was negligible loss of [14C] from brain during the 1 min experimental period and that virtually all the plasma radioactivity was in L-[14C]GLU (21). The background contamination due to radioactivity in blood was taken to be homogeneous and equivalent to the amount of radioactivity in 5 μl of plasma (21).
2.9.3 Statistical analysis
Differences between groups were detected by t-test and ANOVA (Tukey test) (SigmaPlot 11, Systat Software, San Jose CA). Values were considered significant at P < 0.05.
3. Results
3.1 STZ induced diabetes
Of the twelve rats injected with STZ (65 mg/kg iv), three lost considerable weight over the subsequent three-week period, and were not used in the study. The remaining 9 maintained stable weight (table 1); five were subsequently tested at normal plasma GLU concentrations (saline infusion), and 4 at elevated plasma GLU concentrations (infused with GLU). Of the 12 control rats, three were lost during the surgical protocol; 5 were tested at normal plasma GLU concentrations, and 4 at increased plasma GLU concentrations. All STZ rats became glucosuric and slightly ketonuric (tested with Uriscan 2 gluketo strips) within 24 hr of STZ injection, and remained so throughout the next 3 weeks. At the end of the three-week period, plasma concentrations of the ketone bodies and the branched-chain AA in STZ-treated rats were substantially elevated over values in control rats (table 1).
Table 1.
Body weights and plasma chemistry in control and STZ-diabetic rats
| Variable | Control | STZ-Diabetic |
|---|---|---|
| Group size | 9 | 9 |
| Initial Body weight (grams) | 300 ± 5 | 307 ± 2 |
| Final Body weight (grams) | 447 ± 11 | 305 ± 9* |
| Plasma Glucose (mmol/L) | 10.1 ± 0.4 | 16.9 ± 0.9* |
| Plasma β-hydroxybutyrate (mmol/L) | nd | 2.45 ± 0.36* |
| Plasma Acetoacetate (mmol/L) | nd | 0.91 ± 0.14* |
| Plasma ΣBCAA (nmol/ml) | 455 ± 29 | 1434 ± 92* |
Data are means ± SEM. Plasma samples were collected in the non-fasted state in the morning at the end of the study. STZ = streptozotocin; ΣBCAA = leucine + isoleucine + valine
nd = below limit of assay detection.
P<0.01, t-test.
In rats infused with unlabeled GLU prior to 14C-GLU infusion, plasma GLU concentrations were 30-75-times higher than those in rats infused with saline (table 2). Generally speaking, BBB GLU clearance was very low in all animals (from 2 to 8 μl•min-1•g-1), except in those areas that have fenestrated capillaries (circumventricular organs; see Fig. 1). No statistically-significant differences in GLU uptake were observed in whole brain or in most of the brain regions examined (striatum, hippocampus, midbrain), comparing normal with STZ-treated rats, or within each treatment group, animals with normal or extremely high plasma GLU concentrations. A statistically-significant main effect (STZ, vehicle) was noted in cerebral cortex and in cerebellum (table 2).
Table 2.
Brain glutamate clearance in STZ-diabetic and control rats.
| Variable | Control | Control + GLU | STZ-Diabetic | STZ-Diabetic + GLU |
|---|---|---|---|---|
| Group size | 5 | 4 | 5 | 4 |
| Plasma GLU (mmol/L)** | 0.02 ± 0.03 | 1.51 ± 0.2† | 0.06 ± 0.02 | 1.77 ± 0.31† |
| GLU Clearance (μl·min-1·g brain-1) | ||||
| Whole Brainns | 4.4 ± 0.8 | 2.0 ± 1.0 | 6.2 ± 2.0 | 4.3 ± 2.0 |
| Brain Region | ||||
| Cerebral Cortex* | 2.3 ± 0.6 | 0.4 ± 0.2 | 6.8 ± 2.1 | 3.7 ± 2.8 |
| Striatumns | 4.8 ± 1.1 | 3.0 ± 1.4 | 5.6 ± 2.4 | 3.7 ± 2.8 |
| Hippocampusns | 3.5 ± 1.0 | 3.2 ± 1.7 | 5.9 ± 2.6 | 2.0 ± 1.7 |
| Midbrainns | 6.6 ± 1.5 | 1.8 ± 1.4 | 4.0 ± 2.0 | 4.8 ± 2.4 |
| Cerebellum* | 4.7 ± 2.1 | 1.8 ± 1.4 | 8.5 ± 1.9 | 7.3 ± 1.7 |
Data are means ± sem. Within a treatment group, “+GLU” indicates the subgroup with elevated plasma GLU concentrations during the BBB GLU uptake procedure (iv GLU infusion); the other subgroup had normal plasma GLU concentrations (iv saline infusion).
P<0.05, main effect of treatment (streptozotocin, vehicle), but not infusion (vehicle, glutamate)
no significant main effect of treatment (streptozotocin, vehicle) or infusion (vehicle, glutamate)
P<0.05, main effect of infusion (vehicle, glutamate), but not treatment (streptozotocin, vehicle)
P< 0.05, differs significantly from corresponding non-infusion group; ANOVA (Tukey test).
Fig. 1.
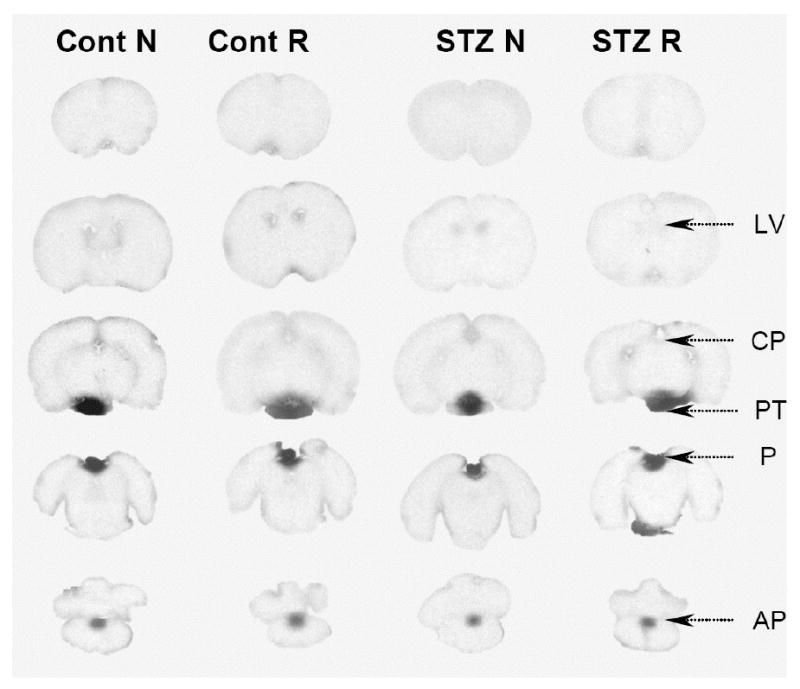
Autoradiographs of brain from STZ-treated and control rats. Representative coronal sections through the brains of rats infused with [14C]glutamate at normal concentrations (N) and raised glutamate concentrations (R) are shown. AP, area postrema, CP, choroid plexus, LV, lateral ventricles, P, pineal gland, PT, pituitary gland. The only areas of brain where 14C was notably visible were the circumventricular organs (e.g., AP, CP). The circumventricular organs have fenestrated capillaries, however, the epithelial lining of the ventricles surrounding these areas are tightly connected by zonula occludens thereby preventing the spread of 14C into adjacent parenchyma of brain (29,41). The pineal and anterior pituitary glands are not part of the brain.
3.2. Diet induced obesity
In this study, all rats (n=36) were initially placed on the high-energy diet. At two weeks, they were weighed, and separated into three groups based on body weight (lowest, highest, intermediate; n = 12/group) (table 3). The lowest third was designated diet-resistant (DR), and the highest third diet-induced-obese (DIO). These groups continued to consume the high-energy diet for the remainder of the experiment (4 additional weeks). The group of intermediate weight was designated the control group, and switched back to standard rat chow (Chow) for the succeeding 4 weeks. All rats continued to gain weight, though at six weeks, chow and DR rats weighed the same, while the DIO rats were significantly heavier than the control or DR rats (table 3). DIO rats consumed significantly more kcal/day than DR and control rats throughout the study (table 3).
Table 3.
Diet-induced obesity study: body weights, daily food intake and plasma chemistries.
| Variable | DR | Control | DIO |
|---|---|---|---|
| Group size | 12 | 11 | 12 |
| Body weight (grams) | |||
| Initial | 297 ± 1 | 300 ± 1 | 302 ± 2 |
| Final | 512 ± 3 | 510 ± 3 | 589 ± 4* |
| Food Consumption (kcal/day) | |||
| Weeks 0-2 | 98 ± 2 | 103 ± 2 | 116 ± 2* |
| Weeks 2-6 | 97 ± 2 | 97 ± 1 | 114 ± 2* |
| Fat Pads (grams)† | 27.8 ± 1.7 | 20.3 ± 1.2 | 36.8 ± 2.3 |
| Plasma Glucose (μmol/ml) | 10.6 ± 0.6 | 10.9 ± 0.4 | 11.2 ± 0.8 |
| Plasma Insulin (ng/ml) | 7.8 ± 1.9 | 7.4 ± 2.3 | 17.3 ± 3.7* |
| Insulin/Glucose Ratio (ng/μmol) | 0.73 ± 0.17 | 0.65 ±018 | 1.63 ± 0.38* |
| Plasma Leptin (ng/ml) | 4.0 ± 0.6 | 1.3 ± 0.2** | 5.1 ± 0.5 |
Data are means ± sem. DR, diet-resistant rats; Control, chow-fed rats; DIO, diet-induced obese rats. Fat pads weighed were: epididymal, mesenteric and retroperitoneal.
P < 0.05 differs significantly from either DR or control values
P < 0.05, any group comparison significantly different
P < 0.05, differs significantly from either DR or DIO values (ANOVA, Tukey test).
Plasma glucose, leptin and insulin were measured in non-fasting samples obtained at the conclusion of the study (6 weeks). No differences in glucose concentrations were noted among the groups, but insulin levels in the DIO group were significantly higher than those in either the DR or chow group. The insulin/glucose ratio, an indicator of insulin resistance, was also higher in the DIO group than in either of the other two groups (see table 3). Retroperitoneal, mesenteric and epididymal fat pads were also removed and weighed and used as an index of total fat mass. These weights were greatest in DIO rats, least in chow rats, and intermediate in DR rats (table 3), and the insulin/glucose ratio was correlated with the combined mass of these fat depots (Fig. 2, top panel). Adipose tissue mass and plasma leptin values correlated extremely well (r2 = 0.92; Fig. 3), consistent with adipose tissue being a major source of circulating leptin (23,24).
Fig. 2.
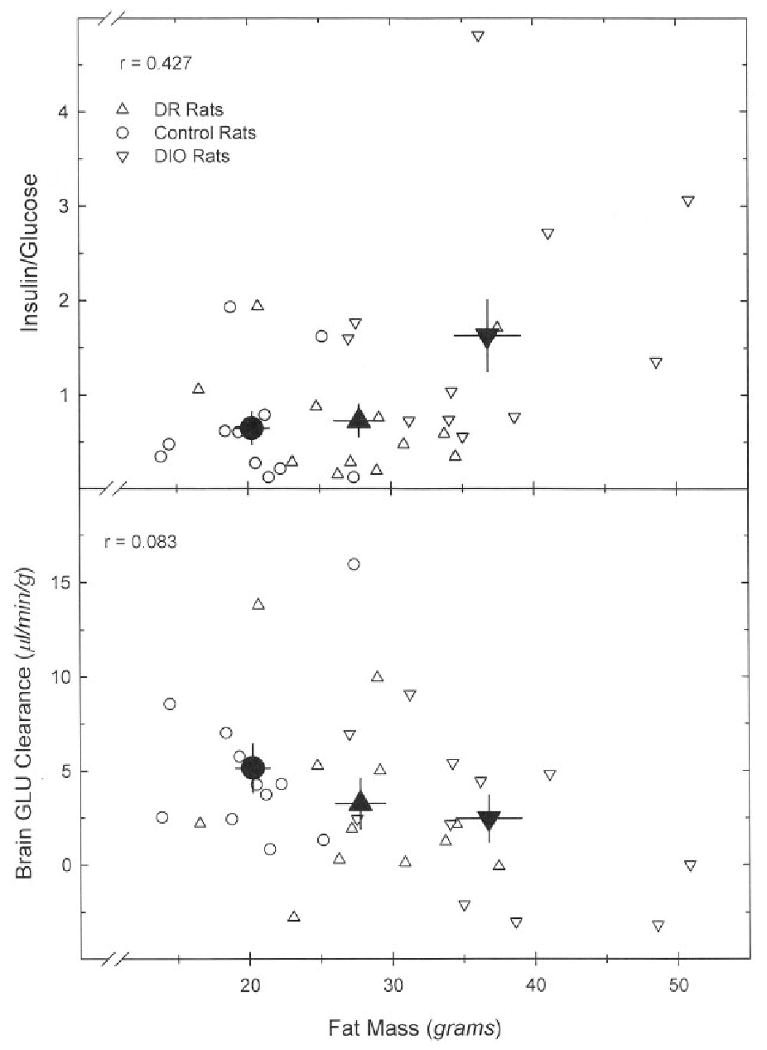
Correlation between fat mass and the plasma insulin/glucose ratio (top panel) and brain GLU clearance (bottom panel). Data points for individual animals are shown as open symbols; filled (black) symbols represent group means; bars are SEM. A linear regression of insuin/glucose vs. fat mass was statistically significant (F = 7.127, P = 0.012), while that of whole brain GLU clearance vs. fat mass was not (F = 2.173, P = 0.150).
Fig. 3.
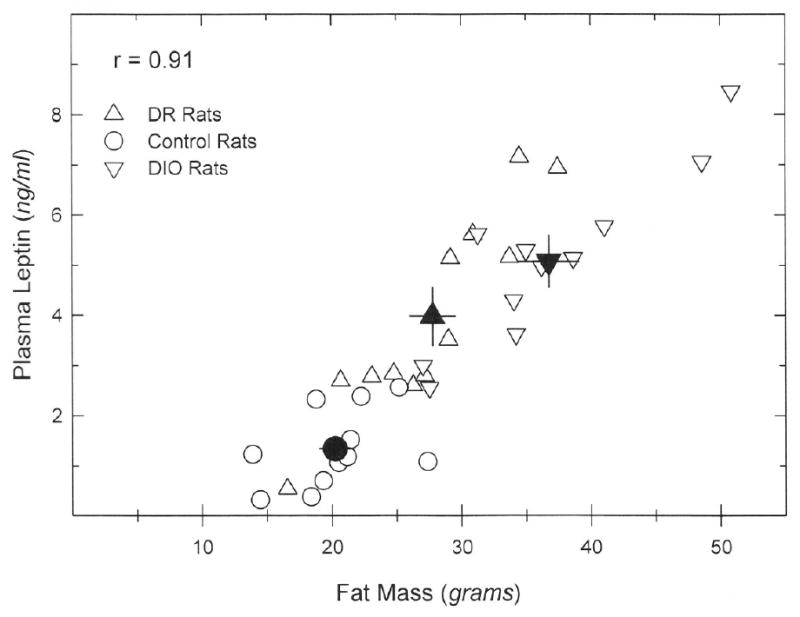
Correlation between fat mass and plasma leptin concentrations. Data points for individual animals are shown as open symbols; filled (black) symbols represent group means; bars are SEM. A linear regression of plasma leptin vs. fat mass was statistically significant (F = 144.583, P < 0.001).
In rats infused with unlabeled GLU prior to 14C-GLU infusion, plasma GLU concentrations were 25-times higher than those in rats infused with saline (table 4). BBB GLU clearance was very low (from 0 to 9 μl•mn-1•g-1), except in those areas with circumventricular organs (i.e., with fenestrated capillaries; Fig. 4). No statistically-significant differences in GLU clearance were observed in any of the brain regions examined, comparing DR, chow and DIO groups (table 4). Elevation of circulating GLU concentrations produced a statistically-significant effect on GLU clearance in whole brain, hippocampus and cerebellum (table 4). Whole brain GLU clearance showed no relationship to fat mass, and was not elevated in animals showing the largest insulin/glucose ratios (i.e., greatest insulin insensitivity) (Fig. 2, bottom panel).
Table 4.
Brain glutamate clearance in diet-induced obesity study.
| Variable | DR | DR + GLU | Control | Control + GLU | DIO | DIO + GLU |
|---|---|---|---|---|---|---|
| Group size | 6 | 6 | 6 | 5 | 6 | 6 |
| Plasma GLU (mmol/L)ns,* | 0.10 ± 0.02 | 2.70 ± 0.10† | 0.10 ± 0.01 | 2.40 ± 0.20† | 0.10 ± 0.02 | 2.40 ± 0.05† |
| GLU Clearance (μl·min-1·g brain-1) | ||||||
| Whole Brainns,* | 5.7 ± 2.2 | 0.8 ± 0.8 | 6.3 ± 2.1 | 3.7 ± 1.3 | 3.6 ± 1.7 | 1.6 ± 1.5 |
| Brain Region | ||||||
| Cerebral Cortex ns | 2.1 ± 1.9 | 1.4 ± 0.5 | 7.8 ± 2.0 | 4.6 ± 2.0 | 3.9 ± 3.6 | 3.0 ± 1.6 |
| Striatum ns | 3.1 ± 2.4 | 0.3 ± 0.8 | 3.4 ± 2.9 | 2.6 ± 2.1 | 0.0 ± 2.1 | 0.0 ± 2.0 |
| Hippocampus ns,* | 8.6 ± 2.9 | 0.0 ± 1.1 | 6.3 ± 2.1 | 3.5 ± 1.4 | 4.0 ± 1.9 | 1.5 ± 2.3 |
| Midbrain ns,* | 6.3 ± 2.5 | 0.6 ± 1.3 | 6.0 ± 2.3 | 3.2 ± 1.9 | 3.1 ± 2.0 | 0.0 ± 2.0 |
| Cerebellum ns,* | 8.4 ± 3.1 | 2.2 ± 1.6 | 8.2 ± 1.8 | 4.7 ± 1.3 | 8.7 ± 2.1 | 3.6 ± 1.4 |
Data are means ± sem. DR, diet-resistant rats; Control, chow-fed rats; DIO, diet-induced obese rats. Within a treatment group, “+GLU” indicates the subgroup with elevated plasma GLU concentrations during the BBB GLU uptake procedure (iv GLU infusion); the other subgroup had normal plasma GLU concentrations (iv saline infusion).
no significant effect of diet (DR, chow, DIO)
P<0.05 significant effect of infusion (vehicle vs GLU)
P < 0.05 vs non-infused value in same diet group (ANOVA, Tukey test).
Fig. 4.
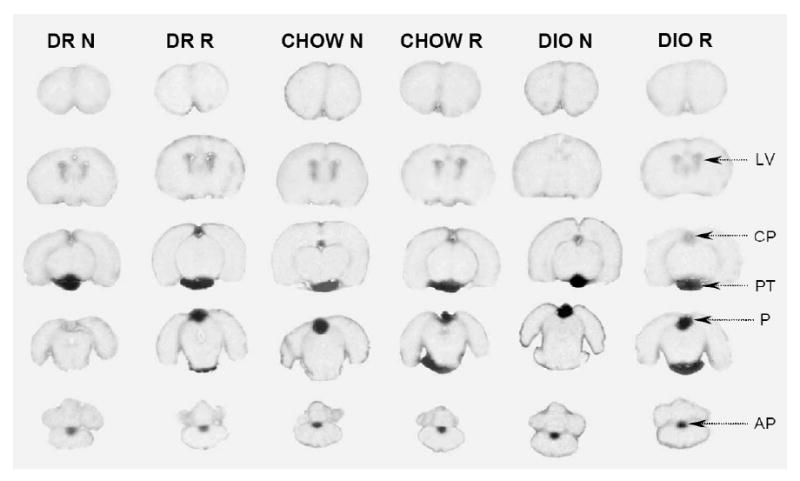
Autoradiographs of brains from diet-induced obesity study. Representative coronal sections through the brains of rats infused with [14C]glutamate at normal concentrations (N) and raised glutamate concentrations (R) are shown. DIO = diet-induced obese rats, DR = diet-resistant rats; chow = rats fed chow from weeks 2-6 of study. The only areas of brain where 14C was visible were the circumventricular organs (see Fig. 1). AP, area postrema, CP, choroid plexus, LV, lateral ventricles, P, pineal gland, PT, pituitary gland.
4. Discussion
To pass the BBB, glutamate must cross both membranes of the capillary endothelial cell. The luminal membrane (blood facing) has a facilitative transporter with a very low capacity (25) and a high affinity (26). This facilitative carrier exists only on the luminal membrane, and cannot be found on the abluminal side (brain facing) (27). Efflux from brain back into plasma appears to be driven in large part by a Na+-dependent active transport system at the capillary abluminal membrane (7). Because the Na+ gradient is about 10 times higher in ECF than in endothelial cells for each GLU molecule that passes into the ECF, it is expected that 10 will be moved back to the endothelial cell whence GLU may diffuse into the plasma (7). This mechanism thus contributes to the maintenance of the GLU concentration in brain ECF at very low concentrations. As such, it also represents one component of a regulatory system that helps maintain the brain interstitial fluid GLU concentration independent of that in the circulation. (7,26,28). Consistent with these known relationships, the present results affirm that GLU uptake into the brain is normally very low. They further demonstrate that brain GLU uptake remains low when rats are made insulinopenic or insulin-resistant, even when blood GLU concentrations are very high.
We employed the STZ-treated rat as the insulinopenic model of diabetes. First, we observed that GLU clearance in this study was 2-8 μl•mn-1•g-1 tissue in normal and STZ-treated rats in brain regions having a BBB (table 2). Such values are normally very low, and similar to those obtained previously (21). In contrast, clearance values for other AA are typically 10-30-fold higher. For example, clearance values are 100-200 μl•mn-1•g-1 tissue for phenylalanine (a large neutral amino acid) and 8 μl•mn-1•g-1tissue for lysine, a basic amino acid (14). These AA are transported at the BBB by carriers different from that for acidic AA (e.g., GLU) (25). Such differences suggest that the BBB limits the access of circulating GLU to most portions of the brain. In several discrete brain areas, the circumventricular organs (CVOs), however, where no BBB is found, GLU is not excluded, and indeed, in the present study and a previous study (21), GLU penetration is readily observed (Fig. 1; e.g., area postrema). Cellular elements in the CVOs are known not to be sensitive to elevated ECF GLU concentrations, and are isolated from adjacent brain regions by tight junctions that exist between the epithelial cells that line the cerebral ventricles (28-30). Second, GLU clearance values were not appreciably different from control values in STZ-treated rats (table 2). In all cases, clearance values were within the normal range (2-8 μl•mn-1•g-1 tissue), though in two regions, a statistically-significant, but materially insignificant, effect of STZ treatment was observed (cerebral cortex, cerebellum). The latter apparent difference is probably due to chance, since in a previous study (21), cerebral cortex and cerebellum GLU clearance values in normal rats were somewhat higher than those seen in STZ rats in the present study (8-11 μl•mn-1•g-1 tissue). And third, GLU clearance values were unaltered in the presence of extremely high circulating GLU concentrations (GLU infusion groups). The GLU infusion group was included, based on the hypothesis that if the BBB to GLU appeared intact in diabetic animals at normal blood GLU concentrations, but was actually weakened, this weakness might emerge in the presence of enormously high circulating GLU levels. Such clearly was not the case, further attesting to the intactness of the BBB under the diabetic conditions examined.
By way of comparison, there is but one study that has assessed GLU clearance across the brains of normal human subjects and subjects having insulin-dependent diabetes of long duration (2 decades) (31). Clearance was estimated from measurements of the arterio-venous difference of amino acid concentrations and the rate of cerebral blood flow. GLU clearance was found to be normal in the diabetic subjects. Our findings are consistent with this result. The results in diabetic subjects of long duration are also helpful in balancing the primary limitation of our study in rats, that of time. It could be argued that had the duration of diabetes been longer in our STZ rats, clearance differences might have been observed. The human study suggests such might not be the case. Indeed, in rats treated with STZ, though some reports indicate physical BBB disruption can occur as early as 14-28 days after STZ injection for small molecules like sucrose (12,32), other studies offer data providing little indication of BBB disruption (for mannitol or sucrose) in rats treated with STZ as long as 9 or 14 months earlier (33,34). It seems that diabetes affects the BBB differently for different molecules (35).
We used a dietary paradigm to attempt to create insulin-resistant rats (18). Rats were fed a highly-palatable diet for two weeks, and then divided into three groups of equal size, based on body weight gain. The groups gaining the most and the least continued on the same diet, while the group of intermediate weight gain was switched to chow for the remaining experimental period. As described by Levin and associates, the group that showed the greatest initial weight gain ingesting the highly-palatable diet continued to eat more and gained significantly more weight by the end of the experiment than the group initially gaining the least weight on the same diet (DIO vs DR groups, respectively; table 3). By most measures, DR rats were very similar to the rats that had initially gained an intermediate amount of weight, that were switched to the rat chow diet (control group). They differed only in their fat pad mass and serum leptin levels. Indeed, as observed previously in such dietary paradigms, plasma leptin levels were notably different among the groups, and correlated remarkably well with fat mass in individual rats across all groups (Fig. 3) (36,37). In our study, plasma insulin concentrations were also significantly higher in the DIO group, compared to the other two groups, but the effects were much smaller than those seen for leptin. However, since plasma glucose levels did not differ among groups, a significant difference was evident in the insulin/glucose ratio (I/G) (table 3), a measure of insulin resistance (38). To affirm the relationship of fat mass to insulin insensitivity, which should be positive (23,36), data from individual rats were plotted (Fig. 2, top panel), and showed that the I/G ratio tended to rise with increasing fat mass (the effect was statistically significant; linear regression, P = 0.01). In contrast, a plot of brain GLU clearance vs. fat mass (Fig. 2, bottom panel) revealed that, if anything, GLU clearance tended to be lower as fat mass increased, though this effect was not significant (linear regression, P = 0.15). A regression analysis of whole brain GLU clearance vs. the I/G ratio was also not statistically significant (not shown; r = 0.08, F = 0.22, P = 0.64). Hence, rats with higher I/G ratios, indicative of insulin resistance, did not show higher brain clearances for GLU. The conclusion, however, that insulin resistance is not associated with increased GLU penetration into brain, must be tempered by the length of the study: 6 weeks. This time may be insufficient for the development of the metabolic sequelae that accompany insulin resistance of long duration. In humans with type II diabetes (non-insulin-dependent) of long duration, the few data that are available are conflicting regarding the integrity of the BBB: one study indicates it is intact (39), while another indicates it is not (13). A third study, quantitating GLU clearance across the brain of long-term diabetic subjects requiring insulin (discussed above), reports no difference relative to non-diabetic subjects, though these subjects had type I diabetics, a more severe form of diabetes (31). Further work would thus be useful with the DIO model, to assess if it does does develop more marked insulin insensitivity with time, which is at least suggested by some earlier data (40).
In conclusion, the results with STZ suggest only minor, if any, changes in GLU clearance in whole brain and brain regions of severely diabetic rats. In addition, no changes in brain GLU clearance were evident in rats made insulin resistant using a dietary paradigm that promotes obesity. While the findings generally support the opinion, at least in the paradigms employed, that diabetes is not associated with changes in GLU transport across the BBB, further studies in an animal model of prolonged insulin resistance would be useful, because of the growing prevalence of type II (non-insulin dependent) diabetes in the population, particularly that associated with obesity.
Acknowledgments
These studies were supported by a grants from: the International Glutamate Technical Committee, the Ministerio de Ciencia y Tecnología (BFU 2007/62036), the National Institutes of Health (NS31017 and NS 041405).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hawkins RA, Peterson DR, Viña JR. The complementary membranes forming the blood-brain barrier. IUBMB Life. 2002;54:101–107. doi: 10.1080/15216540214541. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins RA, O'Kane R L, Simpson IA, Vina JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 3.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 4.Castillo J, Davalos A, Naveiro J, Noya M. Neuroexcitatory amino acids and their relation to infarct size and neurological deficit in ischemic stroke. Stroke. 1996;27:1060–1065. doi: 10.1161/01.str.27.6.1060. [DOI] [PubMed] [Google Scholar]
- 5.Castillo J, Davalos A, Noya M. Progression of ischaemic stroke and excitotoxic aminoacids. Lancet. 1997;349:79–83. doi: 10.1016/S0140-6736(96)04453-4. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Kane RL, Martinez-Lopez I, DeJoseph MR, Viña JR, Hawkins RA. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J Biol Chem. 1999;274:31891–31895. doi: 10.1074/jbc.274.45.31891. [DOI] [PubMed] [Google Scholar]
- 8.Edwards MS, Wilson DB, Craven TE, Stafford J, Fried LF, Wong TY, Klein R, Burke GL, Hansen KJ. Associations between retinal microvascular abnormalities and declining renal function in the elderly population: the Cardiovascular Health Study. Am J Kidney Dis. 2005;46:214–224. doi: 10.1053/j.ajkd.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Ward JD. Abnormal microvasculature in diabetic neuropathy. EYE. 1993;7:223–226. doi: 10.1038/eye.1993.53. [DOI] [PubMed] [Google Scholar]
- 10.McMillan DE. The microcirculation in diabetes. Microcirc Endothelium Lymphatics. 1984;1:3–24. [PubMed] [Google Scholar]
- 11.Bradbury MW, Lightman SL. The blood-brain interface. Eye. 1990;4(Pt 2):249–254. doi: 10.1038/eye.1990.36. [DOI] [PubMed] [Google Scholar]
- 12.Huber JD, VanGilder RL, Houser KA. Streptozotocin-induced diabetes progressively increases blood-brain barrier permeability in specific brain regions in rats. Am J Physiol Heart Circ Physiol. 2006;291:H2660–2668. doi: 10.1152/ajpheart.00489.2006. [DOI] [PubMed] [Google Scholar]
- 13.Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74:70–76. doi: 10.1136/jnnp.74.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mans AM, DeJoseph MR, Davis DW, Hawkins RA. Regional amino acid transport into brain during diabetes: effect of plasma amino acids. Am J Physiol. 1987;253:E575–E583. doi: 10.1152/ajpendo.1987.253.5.E575. [DOI] [PubMed] [Google Scholar]
- 15.McCall AL, Millington WR, Wurtman RJ. Metabolic fuel and amino acid transport into the brain in experimental diabetes mellitus. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1982;79:5406–5410. doi: 10.1073/pnas.79.17.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brosnan JT, Forsey RG, Brosnan ME. Uptake of tyrosine and leucine in vivo by brain of diabetic and control rats. Am J Physiol. 1984;247:C450–453. doi: 10.1152/ajpcell.1984.247.5.C450. [DOI] [PubMed] [Google Scholar]
- 17.Junod A, Lambert AE, Stauffacher W, Renold AE. Diabetogenic action of streptozotocin: relationship of dose to metabolic response. The Journal of Clinical Investigation. 1969;48:2129–2139. doi: 10.1172/JCI106180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levin BE, Hogan S, Sullivan AC. Initiation and perpetuation of obesity and obesity resistance in rats. Am J Physiol. 1989;256:R766–771. doi: 10.1152/ajpregu.1989.256.3.R766. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins RA, Mans AM. Determination of cerebral glucose use in rats using [14C] glucose. In: Boulton AA, Baker GB, Butterworth RF, editors. Neuromethods 11 Carbohydrates and Energy Metabolism. Humana Press Inc; Clifton, New Jersey: 1989. pp. 195–230. [Google Scholar]
- 20.Bergmeyer HU, editor. Methods of Enzymatic Analysis. 2nd. Academic Press; New York: 1974. [Google Scholar]
- 21.Hawkins RA, DeJoseph MR, Hawkins PA. Regional brain glutamate transport in rats at normal and raised concentrations of circulating glutamate. Cell & Tiss Res. 1995;281:207–214. doi: 10.1007/BF00583389. [DOI] [PubMed] [Google Scholar]
- 22.Bongiovanni R, Yamamoto BK, Jaskiw GE. Improved method for the measurement of large neutral amino acids in biological matrices. J Chromatogr B Biomed Sci Appl. 2001;754:369–376. doi: 10.1016/s0378-4347(00)00629-0. [DOI] [PubMed] [Google Scholar]
- 23.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 24.Klein S, Coppack SW, Mohamed-Ali V, Landt M. Adipose tissue leptin production and plasma leptin kinetics in humans. Diabetes. 1996;45:984–987. doi: 10.2337/diab.45.7.984. [DOI] [PubMed] [Google Scholar]
- 25.Oldendorf WH, Szabo J. Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol. 1976;230:94–98. doi: 10.1152/ajplegacy.1976.230.1.94. [DOI] [PubMed] [Google Scholar]
- 26.Smith QR. Transport of glutamate and other amino acids at the blood-brain barrier. J Nutr. 2000;130:1016S–1022S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- 27.Lee WJ, Hawkins RA, Viña JR, Peterson DR. Glutamine transport by the blood-brain barrier: a possible mechanism for nitrogen removal. Am J Physiol. 1998;274:c1101–c1107. doi: 10.1152/ajpcell.1998.274.4.C1101. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins RA. The blood-brain barrier and glutamate. American J Clin Nutr. 2009 doi: 10.3945/ajcn.2009.27462BB. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brightman MW, Reese TW. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reese TS, Feder N, Brightman MW. Electron microscopic study of the blood-brain and blood-cerebrospinal fluid. 1971;30:137–138. [PubMed] [Google Scholar]
- 31.Grill V, Bjorkman O, Gutniak M, Lindqvist M. Brain uptake and release of amino acids in nondiabetic and insulin-dependent diabetic subjects: important role of glutamine release for nitrogen balance. Metabolism. 1992;41:28–32. doi: 10.1016/0026-0495(92)90186-e. [DOI] [PubMed] [Google Scholar]
- 32.Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 33.Bradbury MW, Lightman SL, Yuen L, Pinter GG. Permeability of blood-brain and blood-nerve barriers in experimental diabetes mellitus in the anaesthetized rat. Exp Physiol. 1991;76:887–898. doi: 10.1113/expphysiol.1991.sp003551. [DOI] [PubMed] [Google Scholar]
- 34.Rechthand E, Smith QR, Latker CH, Rapoport SI. Altered blood-nerve barrier permeability to small molecules in experimental diabetes mellitus. J Neuropathol Exp Neurol. 1987;46:302–314. doi: 10.1097/00005072-198705000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Hawkins BT, Ocheltree SM, Norwood KM, Egleton RD. Decreased blood-brain barrier permeability to fluorescein in streptozotocin-treated rats. Neurosci Lett. 2007;411:1–5. doi: 10.1016/j.neulet.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 37.Archer ZA, Corneloup J, Rayner DV, Barrett P, Moar KM, Mercer JG. Solid and liquid obesogenic diets induce obesity and counter-regulatory changes in hypothalamic gene expression in juvenile Sprague-Dawley rats. J Nutr. 2007;137:1483–1490. doi: 10.1093/jn/137.6.1483. [DOI] [PubMed] [Google Scholar]
- 38.Buse JB, Polansky KS, Burant CF. Insulin resistance and the risk of type 2 diabetes. 10. Saunders; Philadelphia: 2003. [Google Scholar]
- 39.Dai J, Vrensen GF, Schlingemann RO. Blood-brain barrier integrity is unaltered in human brain cortex with diabetes mellitus. Brain Res. 2002;954:311–316. doi: 10.1016/s0006-8993(02)03294-8. [DOI] [PubMed] [Google Scholar]
- 40.Levin BE, Sullivan AC. Glucose-induced norepinephrine levels and obesity resistance. Am J Physiol. 1987;253:R475–481. doi: 10.1152/ajpregu.1987.253.3.R475. [DOI] [PubMed] [Google Scholar]
- 41.Brightman MW, Reese TS, Feder N. Assesment with the electronmicroscope of the permeability to peroxidase of cerebral endothelium and epithelium in mice and sharks. In: Crone C, Lassen NA, editors. Capillary permeability. Academic Press; New York: 1970. pp. 468–476. [Google Scholar]