

Abstract
Obesity and mild hyperglycemia are characteristic of early or “prediabetes”. The associated increase in fatty acid flux is posited to enhance substrate delivery to mitochondria leading to enhanced superoxide production resulting in mitochondrial dysfunction and progressive worsening of the hyperglycemic state. We quantified superoxide production by gastrocnemius muscle, heart, and liver mitochondria in a rodent model that mimics the pathophysiology of prediabetes by administering low dose streptozotocin (LD-STZ) to rats fed high fat (HF). Superoxide was rigorously determined indirectly as H2O2 largely released from the matrix and by electron paramagnetic resonance spectroscopy which directly detects superoxide released externally. Both HF and LD-STZ mildly increased glycemia (p < 0.05 by 2-way ANOVA). Matrix and external superoxide production by gastrocnemius mitochondria respiring on the complex II substrate, succinate, and matrix superoxide production by liver mitochondria respiring on the complex I substrates, glutamate plus malate, were significantly reduced by HF feeding but not affected by mild hyperglycemia. Superoxide production was not significantly altered by either treatment in heart mitochondria fueled by either complex I or II substrates. The functional status of the mitochondria were assayed as simultaneous respiration and membrane potential which were not affected by HF or mild hyperglycemia. Comparison of substrate and inhibitor effects on superoxide release implied marked differences in the redox mechanism(s) regulating mitochondrial superoxide production from liver mitochondria compared to muscle and heart. In summary, superoxide production from mitochondria of different insulin sensitive tissues differs mechanistically. But, in any case, excess superoxide production as an intrinsic property of mitochondria of insulin sensitive tissues does not result from conditions mimicking the pathophysiology of pre- or early diabetes.
Keywords: Reactive oxygen species, high-fat feeding, diabetes, glucose, respiratory coupling
Introduction
Elevated circulating free fatty acids, as seen in obesity, and hyperglycemia are thought to increase myocyte and hepatocyte lipid content and induce mitochondrial ROS, thereby interfering with insulin signaling leading to insulin resistance (1-4). Type 2 diabetes is known to be a progressive disorder characterized by both insulin resistance and impaired insulin secretion. Both defects develop very early in the course; at the stage of prediabetes (5) and mild type 2 diabetes (6). Muscle and liver represent the major insulin-sensitive tissues, accounting, respectively, for glucose disposal and hepatic glucose output. Hence, it is plausible that excess mitochondrial ROS production in these tissues could contribute to worsening insulin resistance and progression from prediabetes to early diabetes or from mild to more severe type 2 diabetes.
We hypothesized that liver, skeletal muscle and heart mitochondria are intrinsically altered by insulin deficiency, high fat feeding, and obesity that characterize early diabetes such that these organelles become programmed towards excess superoxide production. If this hypothesis is correct, then mitochondria of these tissues should manifest elevated superoxide production when isolated and incubated in vitro. To assess this, we created a rodent model that mimics the pathophysiology or prediabetes. We did this by feeding a high-fat diet to normal rodents followed by repeated low doses of the β-cell toxin, streptozotocin (STZ) resulting in a state of mild hyperglycemia, obesity, and insulin resistance.
Mitochondrial superoxide arises primarily from complex I or complex III. To assess superoxide production in a robust and site specific fashion, we used a combination of electron paramagnetic spectroscopy and fluorescence spectroscopy. As we have documented in prior studies of endothelial cell (7) and muscle mitochondria (8), these methods are, relative to each other, specific for complex I or complex III superoxide. Hence, this rigorous approach allows assessment of mitochondrial superoxide that might be generated by different mechanisms.
Superoxide production may be mitigated by respiratory uncoupling (9). For this reason, and to document the functional status of the mitochondria, we also assessed mitochondrial respiration and membrane potential and calculated proton conductance. In muscle, we assayed the expression of proteins with uncoupling activity including uncoupling protein 3 (UCP3) and the adenine nucleotide translocator-1 (ANT1).
The results we describe herein run counter to our hypothesis, implying the need to reconsider the role of excess mitochondrial superoxide in the progression of diabetes, at least as an intrinsic characteristic of mitochondria. Our results also imply marked tissue specific differences in the mechanisms controlling mitochondrial superoxide production.
Materials and Methods
Experimental Animals
Male Sprague-Dawley rats were obtained from Harlan, Inc. (Indianapolis, IN). Animals were fed and maintained according to standard NIH guidelines. The protocol was approved by our institutional Animal Care Committee.
We studied a rodent model characterized by insulin resistance, impaired insulin release, and mild hyperglycemia; thus resembling human early or prediabetes. To create this models, rats were exposed to high-fat feeding and/or to three low doses of STZ. Four groups of rats were studied. All were generated from normal male Sprague-Dawley rats fed standard rat chow (Harlan Tekland #7001) until age 53 days. High-fat-fed STZ rats (HF-STZ) and high-fat-fed vehicle (HF-Veh) groups were fed a HF diet consisting of 60% of kcals as fat, 20% protein, 20% CHO (Research Diets, Inc., #D12492) from age 53 days until sacrifice between 99 and 135 days later. The HF-STZ group received injections of STZ (15 mg/kg) IP on days 56, 70, and 84 after initiation of the diet, while the HF-Veh group received an equal volume of saline on the same days. Likewise, at age 53 days, normal-fat-fed-STZ rats (NF-STZ) and normal-fat-fed-vehicle (NF-Veh) groups were continued on the standard diet consisting of 4.25% fat, 25% protein until sacrifice between 99 and 135 days later. The NF-STZ group received injections of STZ (15 mg/kg) IP on days 56, 70, and 84 after initiation of the diet, while the NF-Veh group received an equal volume of saline on the same days.
On the day of study rats were euthanized 4 hours after food removal by IP injection of 25 mg/kg pentobarbital followed by incision of the left ventricle. Data show that doses of pentobarbital up to 4-fold more than we used did not affect mitochondrial respiration or potential (10). Moreover, euthanasia was carried out in the same way for all groups. Glucose values were determined just before injection of pentobarbital on a drop of tail vein blood using a reagent strip and meter (OneTouch Ultra).
Materials
Rabbit polyclonal anti-UCP3 (#UCP32-A) was purchased from Alpha Diagnostics International (San Antonio, TX). Goat polyclonal anti-ANT1 (#SC-9299) was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Rabbit polyclonal anti-MnSOD (#06-984) was purchased from Upstate Cell Signaling Solutions (Lake Placid, NY). Rabbit polyclonal anti-GPX (#LF-PA0019) was purchased from Lab Frontier (Seoul, South Korea). Goat anti-rabbit IgG-HRP and donkey anti-goat IgG-HRP were purchased from Santa Cruz Biotechnologies.
Isolation of mitochondria
Mitochondria were isolated by differential centrifugation and washed three times as previously described (11; 12). Muscle and heart tissues were minced for one minute prior to homogenization. As we previously reported (8), mitochondria were highly pure as indicated by distribution of GAPDH and porin in whole tissue and mitochondrial extracts.
Mitochondrial ROS production by fluorescent measurement
H2O2 production was assessed using the fluorescent probe, 10-acetyl-3,7-dihydroxyphenoxazine (DHPA or Amplex Red, Invitrogen) as previously described (7). Samples were prepared in 96-well plates containing 0.06 mL per well and incubated in respiratory buffer [120 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 1 mM EGTA, 3 mM HEPES, pH 7.2 with 0.3% fatty acid free BSA, 2 μM oligomycin to inhibit ATP synthase]. Fluorescence was measured as we previously described (7) once every 44 seconds for 50 cycles during which time the signal increased in linear fashion. For quantification, a H2O2 standard curve ranging from 0-12μM was prepared and included on each plate. Addition of catalase, 500 units/ml, reduced fluorescence to below the detectable limit indicating specificity for H2O2. Addition of substrates to respiratory buffer without mitochondria did not affect fluorescence. Addition of the ETS inhibitor rotenone to mitochondria in the absence of substrate altered fluorescence only about 5%.
Electron paramagnetic resonance (EPR) spectroscopy
ROS were also measured by EPR as we previously described (7; 11). Mitochondria were studied during state 4 respiration in 0.2mL of respiratory buffer with 0.069 M 5,5-dimethyl-l-pyrroline-N-oxide (DMPO), and 0.09 mg mitochondria. Respiration was initiated with the addition of 5 mM succinate and samples incubated for 5 minutes at 37°C before transfer to a flat aqueous EPR cell. Spectra were then recorded at room temperature using the following instrument settings: microwave power 40 mW, modulation amplitude 2G, receiver gain 2×105, conversion time 40.96 ms, time constant 81.92 ms, and scan rate 80G/41.92 s. Spectra represented the average of 7 scans.
Site specificity of superoxide production
As we previously described for endothelial cell (7) and muscle (8) mitochondria (7), our fluorescent and EPR studies measure superoxide differently. In combination, these techniques impart a degree of specificity for complex I or III superoxide. DHPA detects superoxide indirectly. When added to isolated mitochondria, the probe detects H2O2 generated from superoxide by matrix MnSOD. H2O2 so generated diffuses outward from mitochondria and reacts with horseradish peroxidase in the incubation medium to trigger fluorescence. H2O2 produced in this way derives largely from matrix superoxide released at complex I (13). In contrast, our EPR spin trap, DMPO, detects superoxide directly after efflux outward from mitochondria. Superoxide produced in this way should largely derive from the Q cycle at complex III (13). Since DMPO will not easily penetrate mitochondria and because matrix superoxide is rapidly converted to H2O2, our spin trap should not detect superoxide released to the matrix from either complex III or complex I. To further document the site specificity of our ROS detection methods, we have carried out these studies in the presence or absence of ETS inhibitors (7; 8).
Mitochondrial respiration and membrane potential
Respiration and mitochondrial inner membrane potential were determined simultaneously as we previously described (7; 14). Potential was calculated using the Nernst equation based on the distribution (inside and external to the mitochondrial matrix), of the lipophilic cation tetraphenylphosphonium (TPP+). The proton leak was assessed in succinate fueled mitochondria as the kinetic relationship of H+ transfer to mitochondrial membrane potential. Mitochondria were incubated in ionic respiratory buffer with 5 μM rotenone to inhibit electron entry at Complex I, and 0.1 μM nigericin to abolish the ΔpH (15) across the mitochondrial membrane, as previously described (14). Under these conditions, H+ transfer is proton leak dependent and follows a 6:1 stoichiometry (H+/O) with oxygen consumed (16). Malonate was added in incremental amounts to final concentrations ranging from 0.5 to 6.0 mM to inhibit succinate dehydrogenase, creating a range of membrane potentials. For any value of hydrogen transfer and membrane potential, proton conductance is given by the product (nmol H/min/mg/mV). To calculate membrane potential, it is necessary to include a value for mitochondrial matrix volume. We used a value of 1.35 μl for gastrocnemius and heart and 1.8 for BAT mitochondria based on past studies in our laboratory (8; 14). We found that this value did not differ between normal and STZ-diabetic or between normal and HF-fed rodents (8; 14). We used a value of 1.0 for liver based on past reports (17). We assumed a TPP+ binding correction of 0.25 based on literature values (18-20) as there is no reason to suspect these should differ between the treatment conditions we examined. Importantly, due to the exponential nature of the Nernst equation, differences in matrix volumes and binding corrections actually have very little effect on calculated potential.
Immunoblotting
Mitochondrial proteins were separated on 12.5% polyacrylamide gels for immunoblotting as we previously described (12; 14). Primary antibody incubation conditions for UCP3 were 0.2μg/ml overnight at 4°C in TTBS (tris buffered saline, pH 7.6 with 1ml/L TWEEN 20)/BSA; for ANT-1, 1:250 overnight at 4°C in TTBS/BSA; for manganese superoxide dismutase (MnSOD), 1:2500 overnight at 4°C in TTBS/BSA; and for glutathione peroxidase (GPX), 1:2500 overnight at 4°C in TTBS/BSA. Secondary antibody conditions for UCP3, MnSOD and GPX consisted of goat anti-rabbit IgG-HRP, 1:20,000 for 1h at room temperature in TTBS/milk and for ANT-1, donkey anti-goat IgG-HRP, 1:20,000 for 1h at room temperature in TTBS/milk. Densitometry data were normalized to pooled controls included in duplicate on each blot.
Primary antibody specificity was confirmed by signal ablation by competing peptide (UCP3), expected relative tissue distribution (UCP3, ANT1, and GPX), strong signal generation at the expected kD by adenoviral overexpression (UCP3, MnSOD), and lack of cross reactivity with UCP2 (UCP3) (12).
Plasma insulin
Insulin concentrations were determined on plasma removed from the left ventricle upon euthanasia by radioimmunoassay for rat insulin (Linco Research, Inc., St. Charles, MO).
Statistics
Differences between groups were determined by one way ANOVA or by two-factor ANOVA with factors consisting of diet (high-fat or normal-fat) and glycemic status (streptozotocin treatment to impair islet function or vehicle control).
Results
Early or prediabetes model
Table 1 lists the characteristics of the four groups of rats. Two rats in the HF-STZ group and one rat in the NF-STZ group died during over the course of the diet exposures leaving the final numbers as indicated. As expected, the HF-fed rats gained more weight. Both HF feeding and low dose STZ significantly increased circulating glucose. Although insulin concentrations trended to be higher in the HF groups the differences were not significant. However, the insulin × glucose product, which is directly proportional to the HOMA measure of insulin resistance as used in humans (21), was significantly increased by HF feeding.
Table 1.
Characteristics of the animal groups. Data represent mean ± SEM. Data were analyzed by 2-factor ANOVA with Bonferroni post-tests. Factors were diet (NF or HF) and glycemia status (STZ-treated or vehicle). NF = normal-fat diet, HF = high-fat diet, Veh = vehicle-treated, STZ = streptozotocin-treated. NS = non-significant. * p < 0.05, ** p < 0.01 compared to NF at same glucose status.
| FACTOR EFFECT (p) | |||||||
|---|---|---|---|---|---|---|---|
| Parameter | NF Veh | HF Veh | NF STZ | HF STZ | Diet | Glucose status | Interaction |
| Age at sacrifice (days) | 168 ± 4 | 166 ± 3 | 167 ± 4 | 162 ± 1 | NS | NS | NS |
| Days on diet | 115 ± 4 | 113 ± 3 | 114 ± 4 | 109 ± 1 | NS | NS | NS |
| Weight at sacrifice (g) | 575 ± 21 | 673 ± 21 ** | 570 ± 15 | 647 ± 19 * | < 0.001 | NS | NS |
| Weight gain on diet (g) | 312 ± 16 | 404 ± 16 ** | 306 ± 15 | 387 ± 18 ** | < 0.001 | NS | NS |
| Glucose (mM) | 82 ± 2 | 89 ± 3 | 87 ± 4 | 99 ± 4 * | 0.004 | 0.025 | NS |
| Insulin (ng/ml) | 6.1 ± 1.3 | 8.7 ± 1.2 | 6.0 ± 1.1 | 7.4 ± 0.5 | NS | NS | NS |
| Insulin × Glucose | 502 ± 115 | 799 ± 127 | 533 ± 103 | 728 ± 60 | 0.03 | NS | NS |
| n | 9 | 11 | 8 | 8 | |||
Superoxide production indirectly assessed as H2O2
Superoxide production indirectly assessed as H2O2, by mitochondria respiring on succinate was significantly decreased in muscle and unchanged in heart (p = 0.06) and liver by high-fat feeding and unaffected by mild hyperglycemia (STZ) (figures 1A, 1E, 1I). Superoxide production assessed in this way in muscle and heart mitochondria is largely through reverse electron transport to complex I (22) as evident by marked inhibition in the presence of rotenone (figures 1B and 1F). Superoxide production was unchanged by high-fat or mild hyperglycemia in muscle and heart mitochondria respiring on the complex I fuels glutamate and malate (figures 1C and 1G) but reduced by HF feeding in liver mitochondria (figure 1K). Rotenone increased superoxide during respiration on the complex I fuels (figures 1D, 1H, 1L) consistent with the known effect of rotenone to block reduction of the semiquinone generated by electron transfer to coenzyme Q in complex I (7; 22; 23).
Figure 1.
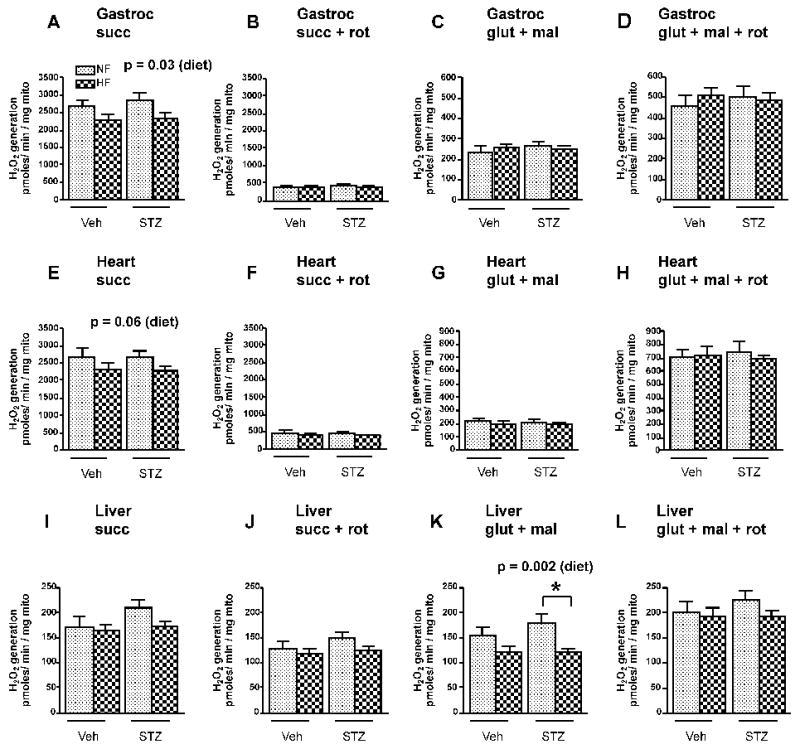
H2O2 release from gastrocnemius (A-D), heart (E-H), and liver (I-L) mitochondria isolated from rats exposed to high-fat (HF, dark stippled bars) or normal-fat (NF, light stippled bars) feeding either treated (STZ) or untreated (Veh) with STZ. Mitochondria were fueled with the substrate indicated with or without rotenone. Data were analyzed by 2-factor ANOVA with Bonferroni post-tests. Factors were diet (HF or LF) and STZ (Veh or STZ). Values represent mean ± SEM, n = 8-11 rats per group. p values indicate effect of diet. Other values for diet effect, STZ effect, or interaction were non-significant. Substrate concentrations were 5 mM glutamate + 1 mM malate (glut + mal) and 5 mM succinate (succ). 5 μM rotenone (rot) was added as indicated. * p < 0.01 compared to STZ group. p values above panels represent significance of diet factor.
Superoxide production by EPR spectroscopy
Figure 2 illustrates representative spectral signals (panels A and B) and quantitative data (panel C) depicting superoxide released from gastrocnemius mitochondria of normal male Sprague-Dawley rats respiring on succinate and detected by EPR. Superoxide detected in this way was not affected by rotenone, which inhibits complex I; decreased by stigmatellin, which blocks electron entry into complex III; and markedly increased by antimycin, which blocks semiquinone reduction in the Q-cycle of complex III. Spectral signals were abolished by MnSOD demonstrating specificity for superoxide. Very similar findings were observed for heart and liver mitochondria (not shown). In contrast to the complete lack of effect of rotenone on superoxide detected by EPR, H2O2 production by muscle mitochondria respiring on succinate detected by fluorescence was markedly reduced by rotenone (figure 1, panels A, B, E, and F).
Figure 2.
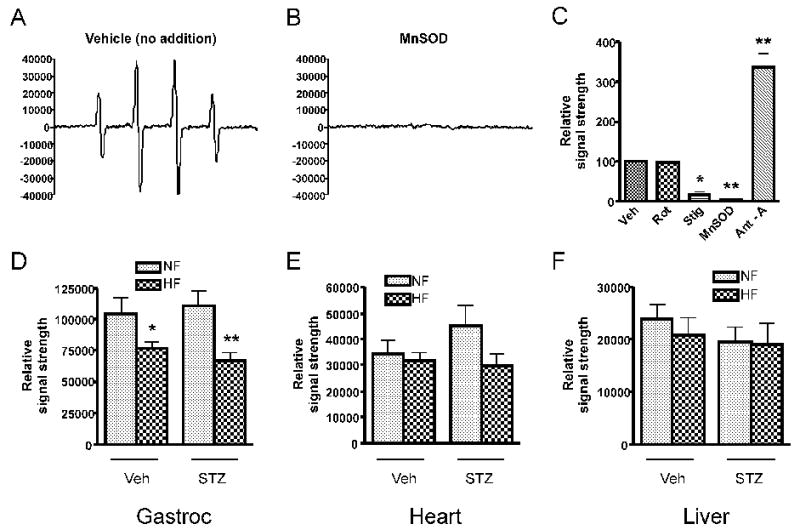
Superoxide production by mitochondria fueled by 5 mM succinate as detected by EPR spectroscopy. Panel A: Representative spectrum generated by gastrocnemius muscle of a NF, vehicle-treated rat generated using the spin trap DMPO which is specific for superoxide or the hydroxyl radical. Panel B. Spectrum generated as in panel A in the presence of 200 μg/ml manganese superoxide dismutase (MnSOD) demonstrating specificity for superoxide. Panel C: EPR signal strength (mean ± SEM) normalized to the vehicle condition (n = 3 repetitions, * p < 0.05, ** p < 0.01, by one-way ANOVA). Additions consisted of 5 μM rotenone, 80 nM stigmatellin, 200 μg/ml manganese superoxide dismutase (SOD), or 1 μM antimycin A. Panels D and F: Quantitative assessment of superoxide release (mean ± SEM) from gastrocnemius (gastroc), heart, and liver mitochondria isolated from rats exposed to high-fat (HF) or normal-fat (NF) feeding either treated (STZ) or untreated (Veh) with STZ. * p < 0.05, ** p < 0.01 compared to NF condition, n=7-11 rats per group. Data were analyzed by 2-factor ANOVA with Bonferroni post-tests. Factors were diet (HF or LF) and STZ (Veh or STZ). For gastrocnemius mitochondria, the effect of diet was significant at p = 0.0002 with no significant effects for diabetes or interaction. Diabetes, diet, and interaction had no significant effects for heart or liver mitochondria.
Superoxide measured by EPR was decreased in gastrocnemius mitochondria and not significantly changed in heart or liver mitochondria by HF feeding and not affected by mild hyperglycemia (figures 2D, 2E, 2F).
Respiration and potential
Table 2 depicts mitochondrial respiration, membrane potential, and calculated proton conductance in mitochondria of muscle, heart, and BAT in the uninhibited state (no added malonate). HF feeding increased respiration by BAT mitochondria, but had no significant effects on gastrocnemius, heart, or liver mitochondria.
Table 2.
Respiration and potential in mitochondria isolated from gastrocnemius muscle, heart, or brown adipose tissue (BAT) incubated in respiratory buffer and fueled by succinate (5 mM) under conditions of uninhibited state 4 respiration. Data represent mean ± SEM, n = 8-11 repetitions per group. Data were analyzed by 2-factor ANOVA with Bonferroni post-tests. Factors were diet (NF or HF) and glycemic status (STZ-treated or vehicle). NF = normal-fat diet, HF = high-fat diet, Veh = vehicle-treated, STZ = streptozotocin-treated. NS = non-significant. * p < 0.01 compared to low fat diet at the same glycemic status.
| FACTOR EFFECT (p) | |||||||
|---|---|---|---|---|---|---|---|
| Parameter (tissue) | NF Veh | HF Veh | NF STZ | HF STZ | Diet | Glucose status | Interaction |
| Gastrocnemius | |||||||
| Respiration (nmol O/min/mg) | 174 ± 18 | 208 ± 19 | 186 ± 28 | 235 ± 16 | NS | NS | NS |
| Potential (mV) | 215 ± 4 | 217 ± 2 | 213 ± 4 | 222 ± 2 | NS | NS | NS |
| Heart | |||||||
| Respiration (nmol O/min/mg) | 311 ± 25 | 307 ± 23 | 258 ± 31 | 333 ± 21 | NS | NS | NS |
| Potential (mV) | 221 ± 3 | 224 ± 3 | 216 ± 4 | 226 ± 5 | NS | NS | NS |
| Liver | |||||||
| Respiration (nmol O/min/mg) | 75 ± 11 | 82 ± 9 | 64 ± 9 | 81 ± 7 | NS | NS | NS |
| Potential (mV) | 231 ± 3 | 234 ± 3 | 229 ± 4 | 239 ± 5 | NS | NS | NS |
| BAT | |||||||
| Respiration (nmol O/min/mg) | 426 ± 40 | 584 ± 34 * | 452 ± 26 | 626 ± 48 * | < 0.001 | NS | NS |
| Potential (mV) | 199 ± 5 | 199 ± 6 | 203 ± 2 | 197 ± 3 | NS | NS | NS |
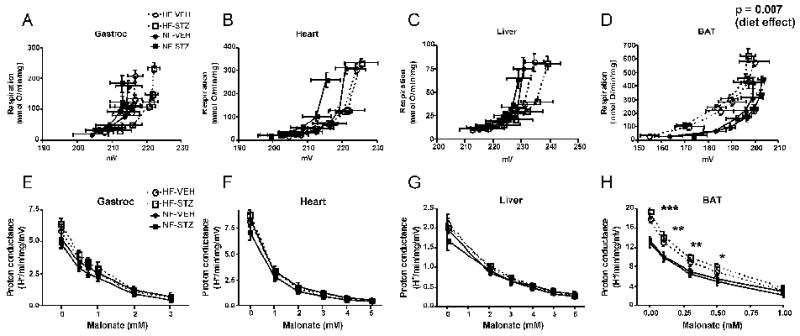
As assessed by the kinetic curves relating respiration to potential (figure 3, panels A-C), we observed essentially no differences in respiratory coupling of gastrocnemius, heart, or liver mitochondria isolated from rats in any of the treatment groups. For contrast and as a positive control to document the validity of our methods, we also examined respiration and potential in BAT mitochondria where uncoupling under conditions of HF feeding is expected (14). As expected, HF feeding resulted in a shift in the kinetic curves upwards and to the left (figure 3, panel D) consistent with enhanced proton leak activity. We also observed significant differences in proton conductance at most malonate concentrations for BAT mitochondria (figure 3, panel H) but not for gastrocnemius, heart, or liver mitochondria (figure 3, panels EG). Low dose STZ treatment did not affect proton conductance by mitochondria from any tissue.
Figure 3.
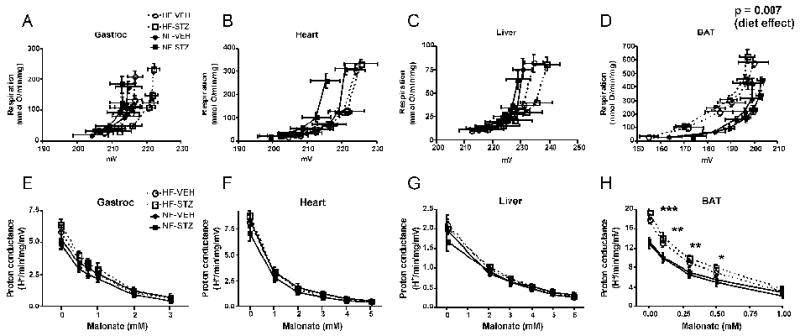
Assessment of the mitochondrial proton leak. Panels A-D: Kinetics of the proton leak in mitochondria isolated from gastrocnemius muscle, heart, liver, or brown adipose tissue (BAT) according to diet and glycemic status. Data points represent mean ± SEM for both respiration (proportional to hydrogen transfer) and membrane potential. Data were analyzed by comparing the proton conductance at the midpoint for individual curves. Analysis was carried out by 2-factor ANOVA (diet and glycemic status). Glycemic state had no significant effect for mitochondria of any tissue and there were no significant differences by diet for gastrocnemius, heart, or liver mitochondria. Panels E-H: Proton conductance of mitochondria isolated from the indicated tissues plotted as a function of the malonate concentration used to titrate potential (generating curves A-D, respectively). *** p < 0.001, ** p < 0.01, * p < 0.05 by 2-factor ANOVA for HF compared to NF at each malonate concentration. There was no effect of glycemic state and no interaction. n = 8-11 rats per group. HF-VEH = high-fat, vehicle-treated; HF-STZ = high-fat, STZ-treated; NF-VEH = normal-fat, vehicle-treated; NF-STZ= normal-fat, STZ-treated.
Uncoupling and antioxidant protein expression
We observed a significant upregulation of UCP3 in muscle mitochondria isolated from the HF groups compared to NF. STZ had no effect on UCP3 expression. We observed no differences in the expression of ANT1, another mitochondrial protein with uncoupling capacity, and no differences in MnSOD or GPX.
Discussion
The model we studied incorporated weight gain generated by high-fat intake with an imposed β-cell defect due to low dose STZ. For cells aside from pancreatic β-cells, this model is closer to human diabetes than genetic rodent models; especially those with defects in leptin or the leptin receptor wherein mitochondrial function would be severely impacted by the gene defect. This model has been characterized by Zhang, et. al. (24) who fed Sprague-Dawley rats with or without high-fat intake for two months followed by STZ 15 mg/kg. In these authors' hands, this resulted in substantial hyperglycemia averaging 16.9 mM two months after STZ compared to 5.2 mM in normal controls. In our work, we observed a significant but much smaller increase in glucose in spite of three injections of low dose STZ. We are not sure why our results differed in this way but we think it likely that the Sprague-Dawley rats we purchased may have been derived from different recent ancestry.
Thus, our HF-low dose STZ model resembled the human prediabetes condition, defined as a fasting glucose between 5.6 and 6.9 mg/100 ml (25). Individuals with prediabetes have a high incidence of progression to overt diabetes. Although the mean blood glucose in our high-fat/STZ group was 5.5 ± 4 (thus, not quite 5.6 mg/dl), that value was significantly increased. Moreover, rat glucose by reagent strip may not be equivalent to human plasma glucose. Further, the normal-fat, vehicle group had an average glucose of 4.6 ± 0.1, which is lower than the mean adult human male fasting glucose; which was reported to be 4.9 ± 0.1 in the middle quintile among 13,163 healthy men age 26-45 years (26).
Mitochondrial dysfunction (27) and/or decreased density per unit muscle tissue (28) are considered important in the pathogenesis of insulin resistance and, thus, type 2 diabetes. Insulin resistance is likely the consequence of increased fatty acid flux to insulin sensitive tissues leading to intracellular fat accumulation and excess ROS production; both of which activate serine kinases which disrupt insulin signaling (3). Moreover, ongoing superoxide production favors lipid peroxidation and oxidized proteins (29; 30) potentially creating a vicious cycle wherein superoxide production impairs mitochondrial function leading to oxidative damage, further inability to oxidize fat, worsening insulin resistance, and consequent progressive worsening of diabetes.
Given these considerations a largely unanswered question arises; that is, whether mitochondria of insulin sensitive tissues, independent of the in vivo milieu, are altered early in the course of insulin resistance and diabetes in a way leading to intrinsic overproduction of superoxide. If so, then superoxide could be primarily causative of insulin resistance or manifest as an early defect contributing to progression. However, within the confines of our model, our results show that this is not the case. In fact, matrix and external superoxide production by gastrocnemius mitochondria respiring on succinate and matrix superoxide production by liver mitochondria respiring on glutamate plus malate, isolated from the HF and HF-STZ groups, were decreased; an effect attributed to HF-feeding when analyzed by 2-factor ANOVA. Hence, our results show that heart, skeletal muscle, and liver mitochondria of insulin resistant HF and HF-STZ treated rats are not intrinsically altered or programmed towards excess superoxide production. Therefore, if superoxide induces mitochondrial damage and/or cellular insulin resistance, that effect would depend on the high substrate flux imposed upon mitochondria in vivo rather than upon altered mitochondria per se. This concept has important implications suggesting that efforts to mitigate worsening diabetes by protecting cells from oxidative damage might be best directed at substrate flux rather than mitochondrial abnormalities per se.
There is little other information regarding the intrinsic release of mitochondrial superoxide in insulin resistant/mildly hyperglycemic rodents or humans. Boudina et. al. (31) reported an increase in ROS production assessed as H2O2 release (similar to our DHPA method) by heart mitochondria isolated within permeabilized muscle fibers isolated from insulin resistant, obese, and leptin receptor deficient db/db mice. Hence these findings differ from our observations. However, that model was far different form ours consisting of extreme obesity associated with leptin deficiency and diabetes, a condition associated with severe lipid accumulation and cardiac steatosis; so not typical of early diabetes. Our current findings are similar to what this group reported for superoxide production by heart mitochondria isolated from an insulin deficient type 1 model, the Akita mouse (32), and to our past findings for streptozotocin diabetic rats (33); that is no intrinsic mitochondrial increase in superoxide production.
Anderson et. al. (34) recently reported that superoxide production (measured as H2O2 release by DHPA fluorescence) was increased in permeabilized muscle fibers from three week HF-fed rats or morbidly obese humans compared normally fed rodents or lean humans. Hence, these results differ from ours. We are not sure of the reasons for this difference. Conceivably isolated mitochondria differ in intrinsic properties from permeabilized muscle fibers. However, as in isolated mitochondria, the in vitro milieu surrounding the mitochondria within the permeabilized fibers is determined by the incubation medium and not by the usual intracellular fluid.
Our results are compatible with findings of Bonnard, et. al (35) who found that 16 weeks of a high-fat, high-sucrose diet administered to mice led to oxidative damage and mitochondrial respiratory dysfunction, preventable with antioxidant therapy. However, these changes were not evident at 4 weeks of the diet even though the mice did become glucose intolerant. These studies assessed markers of oxidative stress and not ROS production, but agree with our findings that early changes in glucose metabolism did not increase superoxide.
The methods we used for superoxide production were both robust and rigorous and assessed both matrix and external release of the radical thereby adding mechanistic information. H2O2 production was linear over the time period analyzed and care was taken to avoid measuring non-specific fluorescence (see methods). The EPR technique is highly specific and completely blocked by SOD. Figure 5 schematically depicts our representation of the effects of substrates and inhibitors on superoxide production as detected by fluorescence and EPR. The diagram is based on the considerations discussed under methods (“site specificity of superoxide production”) and supported by the rotenone, stigmatellin, and antimycin A effects shown in figures 1 and 2. These events are similar to what we described in past studies of bovine aortic endothelial cell mitochondria (7). Thus, DHPA fluorescence and EPR detect superoxide produced in different fashion; the former representing matrix superoxide released largely from complex I and the latter detecting superoxide released externally, largely from complex III. Although, it is hard to claim that either of these two methods as entirely specific for superoxide at any particular site, when used together these methods impart a degree of specificity, at least relative to each other.
Figure 5.
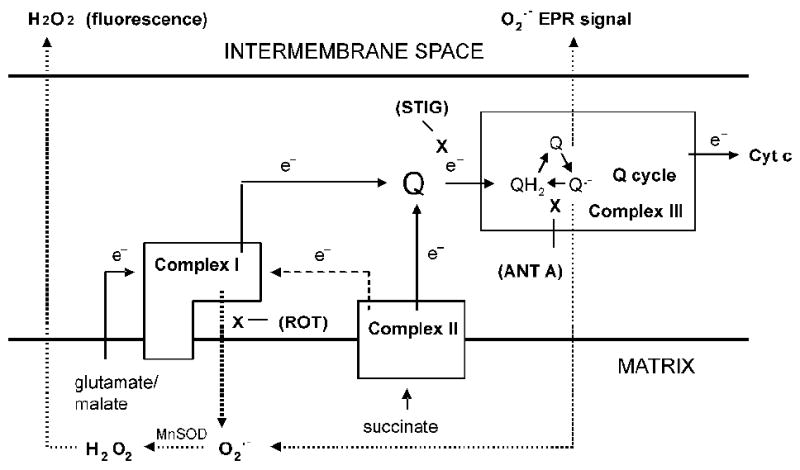
Schematic diagram depicting forward (line arrows with e- symbol) or reverse (dashed line arrow with e- symbol) electron transport, the action of substrates and inhibitors, and sites of ROS production (dotted line arrows). Boxes represent complexes I, II, and III. X depicts sites of inhibition by rotenone (ROT), stigmatellin (STIG), or antimycin A (ANT A).
Inspection of figures 1 and 2 shows that liver superoxide production clearly differed from muscle and heart in several ways. First, on a quantitative basis, liver mitochondria released less overall superoxide both to the matrix and externally; consistent with a lower rate of respiration (table 2). Second, the extent of superoxide production by reverse transport (rotenone inhibitable) relative to forward transport on the complex II substrate, succinate, was far less for liver mitochondria (compare figure 1 panels B, F, and J to the adjacent left panels for each tissue). Third, liver mitochondria generated roughly equivalent portions of matrix superoxide on complex I or complex II substrates whereas muscle and heart generated more superoxide on the complex II substrate (figure 1, compare panels A, E, and I to C, G, and K). Hence, the molecular redox mechanisms regulating mitochondrial superoxide differ considerable for liver compared to muscle and fat. We cannot explain why this should be the case except to point out the obvious functional differences between liver, which packages, stores, and releases nutrients and muscle and heart which predominantly oxidize available substrates.
The extent of mitochondrial superoxide is dependent on respiration and mitigated (at any level of respiration) by mild reduction in membrane potential referred to as “mild uncoupling” (9). We did find a marked increase in UCP3 expression, as we and others observed in the past in studies of mice fed HF (14; 35). The mechanism(s) underlying this increase in UCP3 is not clear but it is postulated to be adaptive possibly to increase the mitochondrial export of potentially toxic lipids and lipid peroxides (36) or to represent a means of protection form oxidative stress through reduction in membrane potential induced by “mild uncoupling” (9).
However, in spite of the increase in UCP3, we observed no shift in proton leak kinetics. This lack of change in leak kinetics was not unexpected and, in fact, has been reported before when mitochondria were isolated from muscle tissue under conditions where UCP3 expression is upregulated. In addition to HF feeding (14), these conditions include other states of high fatty acid flux to mitochondria such as fasting (37), lipopolysaccharide-induced free fatty acid release (38), and severe STZ diabetes with insulin deficiency (8). Why this occurs is not clear but it is possible that the lack of UCP3 activated uncoupling ex vivo results from the absence of activation by superoxide. As above, superoxide was reduced under the conditions used to assess respiratory uncoupling (state 4 respiration in mitochondria fueled by succinate).
Another reason we may not have observed a change in leak kinetics of the HF-fed rats may be that uncoupling was assessed in muscle mitochondria respiring on succinate in the presence of rotenone, as is commonly employed, to drive electrons forward from complex II rather than back to complex I through reverse transport. This has the advantage of maintaining a stoichiometric relationship between oxygen consumption and hydrogen transfer. However, reverse transport generates considerable matrix superoxide which has been shown to be a cofactor for activation of UCP3 mediated mitochondrial uncoupling (39). Matrix superoxide might still arise form complex III which is believed to release electrons both to the matrix and externally. However, it is possible that under the conditions of our assay matrix superoxide was not adequate for induction of uncoupling activity.
Our results beg the question of why gastrocnemius mitochondrial superoxide was actually reduced in our ex vivo mitochondrial incubations. Unfortunately our data do not answer this question. This did not result from decreased respiration or potential as evident from the data in table 2 or from detectable alterations in MnSOD or GPx (figure 4). Liver mitochondrial matrix superoxide was also reduced by HF feeding (figure 1) but only on complex I substrates.
Figure 4.
Expression of proteins with uncoupling properties, UCP3 and ANT1, and the antioxidant enzymes, MnSOD and GPX in mitochondria isolated from gastrocnemius muscle of normal-fat-fed, vehicle-treated (NF-VEH); normal-fat-fed, STZ-treated (NF-STZ); high-fat-fed, vehicle-treated (HF-VEH); and high-fat-fed, STZ-treated (HF-STZ) rats. Quantitative protein expression data (arbitrary units) are shown for each protein with representative immunoblots (below in corresponding order). Data were analyzed by 2-factor ANOVA with Bonferroni post-tests. Factors were diet (HF or LF) and glycemic state (VEH or STZ). Values represent mean ± SEM, n = 5-11 per group. p value indicates effect of diet factor. Other significance levels for diet or glycemic status and interaction were non-significant. * p < 0.05 compared to NF treated condition at the same glycemic state.
There are limitations to the current work. We do not exclude the possibility that increased mitochondrial superoxide might manifest under conditions of HF and/or mild hyperglycemia in the in vivo environment. Thus, we only looked for intrinsic mitochondrial changes that manifest impendent of the intracellular milieu. Second, there are limitations to the model employed. Although, we created physiologic conditions in normal rodents that mimic prediabetes, we did not mimic the genetic background of the disorder. So it is possible that genetic susceptibility to diabetes might somehow be required for early changes in mitochondrial superoxide production. On the other, diabetes is felt to be multi-genetic so a specific defect leading to increased mitochondrial superoxide would not likely account for all or most cases of type 2 diabetes. We were not able to measure UCP2 expression in our studies since the level of detectability was too low for immunoblotting. However, we have carried out past quantitative studies measuring UCP3 and UCP2 in molar amounts using purified UCP2 and UCP3 standards expressed in inclusion bodies (11; 14). We have never been able to observe more than borderline detectable amounts of UCP2 in muscle and essentially none in liver. Based on personal communications this is the experience of other investigators as well.
In summary, superoxide production by isolated liver, heart, or gastrocnemius mitochondria of HF-fed rats is unchanged or decreased compared to NF-fed rats. Induction of mild hyperglycemia does not affect superoxide production assessed in this way. These findings are important since they imply that mitochondria of insulin sensitive tissues subject to mildly impaired insulin secretion and insulin action; i.e. conditions that mimic the pathophysiology of pre- or early diabetes, are not intrinsically altered towards overproduction of superoxide. Therefore, if mitochondrial superoxide contributes to the early progression of diabetes, the process either depends on the intracellular milieu or requires genetic or other factors not manifest in our model. Our data also highlight marked differences in the redox mechanisms controlling liver mitochondrial superoxide compared to muscle and heart.
Acknowledgments
Supported by Veterans Affairs Medical Research Funds and grant DK25295 from the National Institutes of Health. We thank Brian L. Dake for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts-of-interest or disclosures to report.
References
- 1.Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53:S110–118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 2.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 3.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 5.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2008;31 1:S55–60. doi: 10.2337/dc08-S055. [DOI] [PubMed] [Google Scholar]
- 6.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 7.O'Malley Y, Fink BD, Ross NC, Prisinzano TE, Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766–39775. doi: 10.1074/jbc.M608268200. [DOI] [PubMed] [Google Scholar]
- 8.Herlein JA, Fink BD, O'Malley Y, Sivitz WI. Superoxide and respiratory coupling in mitochondria of insulin-deficient diabetic rats. Endocrinology. 2009;150:46–55. doi: 10.1210/en.2008-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochimica et Biophysica Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 10.Takaki M, Nakahara H, Kawatani Y, Utsumi K, Suga H. No suppression of respiratory function of mitochondrial isolated from the hearts of anesthetized rats with high-dose pentobarbital sodium. Jpn J Physiol. 1997;47:87–92. doi: 10.2170/jjphysiol.47.87. [DOI] [PubMed] [Google Scholar]
- 11.Fink BD, Reszka KJ, Herlein JA, Mathahs MM, Sivitz WI. Respiratory uncoupling by UCP1 and UCP2 and superoxide generation in endothelial cell mitochondria. American Journal of Physiology - Endocrinology & Metabolism. 2005;288:E71–79. doi: 10.1152/ajpendo.00332.2004. [DOI] [PubMed] [Google Scholar]
- 12.Hong Y, Fink BD, Dillon JS, Sivitz WI. Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142:249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- 13.St-Pierre J, B JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry. 2002;4277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 14.Fink BD, Herlein JA, Almind K, Cinti S, Kahn CR, Sivitz WI. The mitochondrial proton leak in obesity-resistant and obesity-prone mice. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1773–1780. doi: 10.1152/ajpregu.00478.2007. [DOI] [PubMed] [Google Scholar]
- 15.Lionetti L, Iossa S, Liverini G, Brand MD. Changes in the hepatic mitochondrial respiratory system in the transition from weaning to adulthood in rats. Archives of Biochemistry & Biophysics. 1998;352:240–246. doi: 10.1006/abbi.1998.0579. [DOI] [PubMed] [Google Scholar]
- 16.Porter RK, Joyce OJ, Farmer MK, Heneghan R, Tipton KF, Andrews JF, McBennett SM, Lund MD, Jensen CH, Melia HP. Indirect measurement of mitochondrial proton leak and its application. International Journal of Obesity & Related Metabolic Disorders: Journal of the International Association for the Study of Obesity. 1999;23 6:S12–18. doi: 10.1038/sj.ijo.0800937. [DOI] [PubMed] [Google Scholar]
- 17.Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, Walajtys-Rode E, Rashid A, Chen CH, Huang CC, Wu TC, Lane MD, Diehl AM. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. Journal of Biological Chemistry. 1999;274:5692–5700. doi: 10.1074/jbc.274.9.5692. [DOI] [PubMed] [Google Scholar]
- 18.Brown GC, Brand MD. Proton/electron stoichiometry of mitochondrial complex I estimated from the equilibrium thermodynamic force ratio. Biochemical Journal. 1988;252:473–479. doi: 10.1042/bj2520473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannella CA, Pfeiffer DR, Bradshaw PC, Moraru II, Slepchenko B, Loew LM, Hsieh CE, Buttle K, Marko M. Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life. 2001;52:93–100. doi: 10.1080/15216540152845885. [DOI] [PubMed] [Google Scholar]
- 20.Rolfe DF, Hulbert AJ, Brand MD. Characteristics of mitochondrial proton leak and control of oxidative phosphorylation in the major oxygen-consuming tissues of the rat. Biochimica et Biophysica Acta. 1994;1188:405–416. doi: 10.1016/0005-2728(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 21.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 22.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) Journal of Biological Chemistry. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 23.Doughan AK, Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal. 2007;9:1825–1836. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]
- 24.Zhang F, Ye C, Li G, Ding W, Zhou W, Zhu H, Chen G, Luo T, Guang M, Liu Y, Zhang D, Zheng S, Yang J, Gu Y, Xie X, Luo M. The rat model of type 2 diabetic mellitus and its glycometabolism characters. Experimental Animals. 2001;52:401–407. doi: 10.1538/expanim.52.401. [DOI] [PubMed] [Google Scholar]
- 25.Standards of medical care in diabetes--2008. Diabetes Care. 2008;31 1:S12–54. doi: 10.2337/dc08-S012. [DOI] [PubMed] [Google Scholar]
- 26.Tirosh A, Shai I, Tekes-Manova D, Israeli E, Pereg D, Shochat T, Kochba I, Rudich A. Normal fasting plasma glucose levels and type 2 diabetes in young men. N Engl J Med. 2005;353:1454–1462. doi: 10.1056/NEJMoa050080. [DOI] [PubMed] [Google Scholar]
- 27.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 28.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mezzetti A, Cipollone F, Cuccurullo F. Oxidative stress and cardiovascular complications in diabetes: isoprostanes as new markers on an old paradigm. Cardiovasc Res. 2000;47:475–488. doi: 10.1016/s0008-6363(00)00118-8. [DOI] [PubMed] [Google Scholar]
- 30.Piconi L, Quagliaro L, Ceriello A. Oxidative stress in diabetes. Clin Chem Lab Med. 2003;41:1144–1149. doi: 10.1515/CCLM.2003.177. [DOI] [PubMed] [Google Scholar]
- 31.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 32.Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 Diabetic Akita Mouse Hearts are Insulin Sensitive but Manifest Structurally Abnormal Mitochondria that Remain Coupled Despite Increased Uncoupling Protein 3. Diabetes. 2008;57:2924–2932. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herlein JA, F BD, O'Malley Y, Sivitz WI. Superoxide and Respiratory Coupling in Mitochondria of Insulin Deficient Diabetic Rats. Endocrinology. 2009;150:46–55. doi: 10.1210/en.2008-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009 doi: 10.1172/JCI37048. ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hesselink MKC, Mensink M, Schrauwen P. Human uncoupling protein-3 and obesity: An update. Obesity Research. 2003;11:1429–1443. doi: 10.1038/oby.2003.192. [DOI] [PubMed] [Google Scholar]
- 37.Cadenas S, Buckingham JA, Samec S, Seydoux J, Din N, Dulloo AG, Brand MD. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Letters. 1999;462:257–260. doi: 10.1016/s0014-5793(99)01540-9. [DOI] [PubMed] [Google Scholar]
- 38.Yu XX, Barger JL, Boyer BB, Brand MD, Pan G, Adams SH. Impact of endotoxin on UCP homolog mRNA abundance, thermoregulation, and mitochondrial proton leak kinetics. Am J Physiol Endocrinol Metab. 2000;279:E433–446. doi: 10.1152/ajpendo.2000.279.2.E433. [DOI] [PubMed] [Google Scholar]
- 39.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]