

Abstract
Background and Aims
Necrotizing enterocolitis (NEC), the leading cause of gastrointestinal death from gastrointestinal disease in preterm infants, is characterized by exaggerated TLR4 signaling and decreased enterocyte proliferation through unknown mechanisms. Given the importance of β-catenin in regulating proliferation of many cell types, we hypothesize that TLR4 impairs enterocyte proliferation in NEC via impaired β-catenin signaling.
Methods
Enterocyte proliferation was detected in IEC-6 cells or in ileum or colon from wild-type, TLR4-mutant or TLR4−/− mice after induction of NEC or endotoxemia. β-catenin signaling was assessed by cell fractionation or immunoconfocal microscopy to detect its nuclear translocation. Activation and inhibition of β-catenin were achieved via cDNA or siRNA respectively. TLR4 in the intestinal mucosa was inhibited using adenoviruses expressing dominant-negative TLR4.
Results
TLR4 activation significantly impaired enterocyte proliferation in the ileum but not colon in newborn but not adult mice, and in IEC-6 enterocytes. β-catenin activation reversed these effects in vitro. To determine the mechanisms involved, TLR4 activation phosphorylated the upstream inhibitory kinase GSK3β, causing β-catenin degradation. NEC in both mouse and humans was associated with decreased β-catenin and increased mucosal GSK3β expression. Strikingly, the inhibition of enterocyte β-catenin signaling in NEC could be reversed, and enterocyte proliferation restored, through adenoviral-mediated inhibition of TLR4 signaling in the intestinal mucosa.
Conclusion
We now report a novel pathway linking TLR4 with inhibition of β-catenin signaling via GSK3β activation, leading to reduced enterocyte proliferation in vitro and in vivo. These data provide additional insights into the pathogenesis of diseases of intestinal inflammation like NEC.
INTRODUCTION
Necrotizing enterocolitis (NEC) is the leading cause of death from gastrointestinal disease in preterm infants, and remains a major cause of long term disability in those who survive the disease1, 2. Despite the increasing frequency with which NEC is diagnosed, its pathogenesis remains largely uncertain, although there is general acceptance that its development reflects persistent intestinal mucosal injury3. In seeking to understand how mucosal injury in NEC occurs, we have recently shown that NEC development requires activation of the innate immune receptor Toll like receptor 4 (TLR4) on enterocytes, and that TLR4 activation by bacterial endotoxin (lipopolysaccharide, LPS) inhibits enterocyte proliferation and leads to disease progression4, 5. Importantly, the mechanisms by which TLR4 activation leads to an inhibition in enterocyte proliferation in the pathogenesis of NEC remain largely unknown. To investigate this further, we now focus on the canonical β-catenin signaling pathway, a series of protein-protein interactions with an established role in the regulation of enterocyte proliferation via the upstream inhibitor GSK3β6. Although a link between TLR4 and β-catenin remains to be established, such a link could provide a novel explanation for the initiation ad propagation of the mucosal injury seen in NEC.
We now report the novel finding that TLR4 activation inhibits enterocyte proliferation in vitro and in vivo via impaired β-catenin signaling after activation of the AKT-GSK3β signaling pathway. These effects were restricted to the small intestine of newborn mice, suggesting relevance in the development of NEC. Strikingly, the TLR4-mediated inhibition of β-catenin signaling could be partially restored in mice with NEC through adenoviral-mediated inhibition of TLR4 signaling in the intestinal mucosa. The current findings support a novel linkage between TLR4 and inhibitory β-catenin signaling in enterocytes, suggest an important physiological context where such signaling may be important, and provide additional insights into the development of intestinal mucosal injury in the pathogenesis of NEC.
METHODS
Cell culture, mice and reagents
IEC-enterocytes were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and maintained as described7. Immortalized primary murine embryonic fibroblasts (MEF) were isolated from 13 day old TLR4-wild-type and TLR4-mutant embryos and cultured in DMEM supplemented with 10% fetal bovine serum, 2mM L-glutamine and 10µg/ml blasticidin (InvivoGen, San Diego, CA). Lipopolysaccharide (LPS, 99% pure) was purified from E. coli O127:B8 by gel filtration chromatography (Sigma-Aldrich, Saint Louis, MO). In all cases, the concentration of LPS used was based upon the measured concentration of LPS found in the stools of mice with experimental NEC as we recently reported (50µg/ml)8. Antibodies were as follows: β-catenin, tubulin, and histone H1, pCNA from Santa Cruz (Santa Cruz, CA); phospho-GSK-3β and phospho-AKT from Cell Signaling (Danvers, MA); phospho-β-catenin from Sigma. LiCl from Sigma. SiRNA to GSK3β, β-catenin and TLR4 from Dharmacon (Lafayette, CO, 100 nM final concentration), and were used as described8. TLR4−/− mice were generated by first generating a floxp-TLR4 mice, in collaboration with Ozgene (Bentley, Australia), which we then bred with the EIIa-Cre mouse (Jackson Labs) to generate the global TLR4−/− mouse. These mice were genetically TLR4 deficient, and failed to mount a septic response to the intraperitoneal injection of LPS.
Plasmids expressing constitutively active β-catenin were generously provided by Dr. S. P. Monga, University of Pittsburgh9 and were transfected into IEC-6 cells using Lipofectamine 2000.
Details of RT-PCR and primers are included in Online Supplementary Material.
Determination of enterocyte proliferation
Enterocyte proliferation in vitro was measured using the colorimetric XTT(2,3-bis[2-Methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide assay (Sigma-Aldrich). 5000 cells were plated to 50% confluence without serum and the extent of proliferation was expressed as a percent of maximum proliferation rate i.e. the rate of untreated cells. Wild-type, TLR4-mutant or TLR4−/− mice were injected with either saline or LPS (5 mg/kg) for 3h. The terminal ileum was then freshly harvested, fixed in formalin and immunostained with antibodies to the proliferation marker pCNA as described10, or subjected to analysis by RT-PCR using mouse specific primers. To quantify pCNA staining in the intestinal crypts, images of pCNA were digitized, regions of interest were drawn around the crypts, and the extent of pCNA expression was evaluated using ImageJ software(NIH).
Generation of adenoviruses bearing wild-type and mutant TLR4
Replication-deficient recombinant adenoviruses that express GFP-tagged mouse wild-type or dominant negative TLR4-cDNA and control adenovirus expressing GFP, were prepared with the Adeno-X-Expression System2 kit (Clontech), and verified for their ability to inhibit TLR4 signaling in vivo as we recently described4.
Immunohistochemistry, cell fractionation and SDS-PAGE
Immunostaining of IEC-6, mouse/human intestine was performed as described11, 12. Nuclear and cytoplasmic fragments in IEC-6 cells were separated according to the methods of Mei et al13, and were probed with antibodies against β-catenin, α-tubulin (cytoplasm) and histone-H1 (nucleus). SDS-PAGE was performed as described5.
Induction of experimental NEC and endotoxemia
All experiments were approved by the Children's Hospital of Pittsburgh Animal Care Committee and the Institutional Review Board of the University of Pittsburgh. C3H/HeOUJ (i.e. TLR4 wild-type) and C3H/HeJ (i.e. TLR4 mutant) strains were obtained from Jackson Laboratories (Bar Harbor, ME). Endotoxemia was induced in 2 week old mice by the intraperitoneal injection of LPS (Escherichia coli 0111:B4 purified by gel-filtration chromatography, >99% pure, 5 mg/kg, Sigma-Aldrich, St. Louis, MO). 2h later, animals were euthanized, and samples of the terminal ileum were obtained 1cm proximal to the ileocecal valve. Experimental NEC was induced in 10–14 day old mice as we described12 using formula gavage (Similac Advanced infant formula (Ross Pediatrics):Esbilac canine milk replacer 2:1) five times/day, and hypoxia (5%O2, 95%N2) for 10min in a hypoxic chamber (Billups-Rothenberg, DelMar, CA) twice daily for 4d. Where indicated, mice were enterally administered adenoviral GFP, GFP-wild-type TLR4, or GFP-dominant-negative TLR4 twice daily for three days (240uL, 1012 PFU). The expression of GFP in mucosal scrapings of the intestine, lung and liver were assessed by RT-PCR for GFP; intestines were studied for GFP expression by confocal microscopy, and confirmed to be expressed in the intestinal mucosa with no detectable expression in the liver or lung4. Intestinal samples were obtained from human neonates undergoing intestinal resection for NEC, for unrelated indications (control), or at time of stoma closure were processed as we described4.
Statistical Analysis
Data presented are mean +/− SEM, and comparisons are by two-tailed Student’s t test or ANOVA, with statistical significance accepted for p<0.05. All experiments were repeated in at least triplicate, with at least five mice per group where indicated.
RESULTS
TLR4 activation inhibits enterocyte proliferation in vitro and in vivo
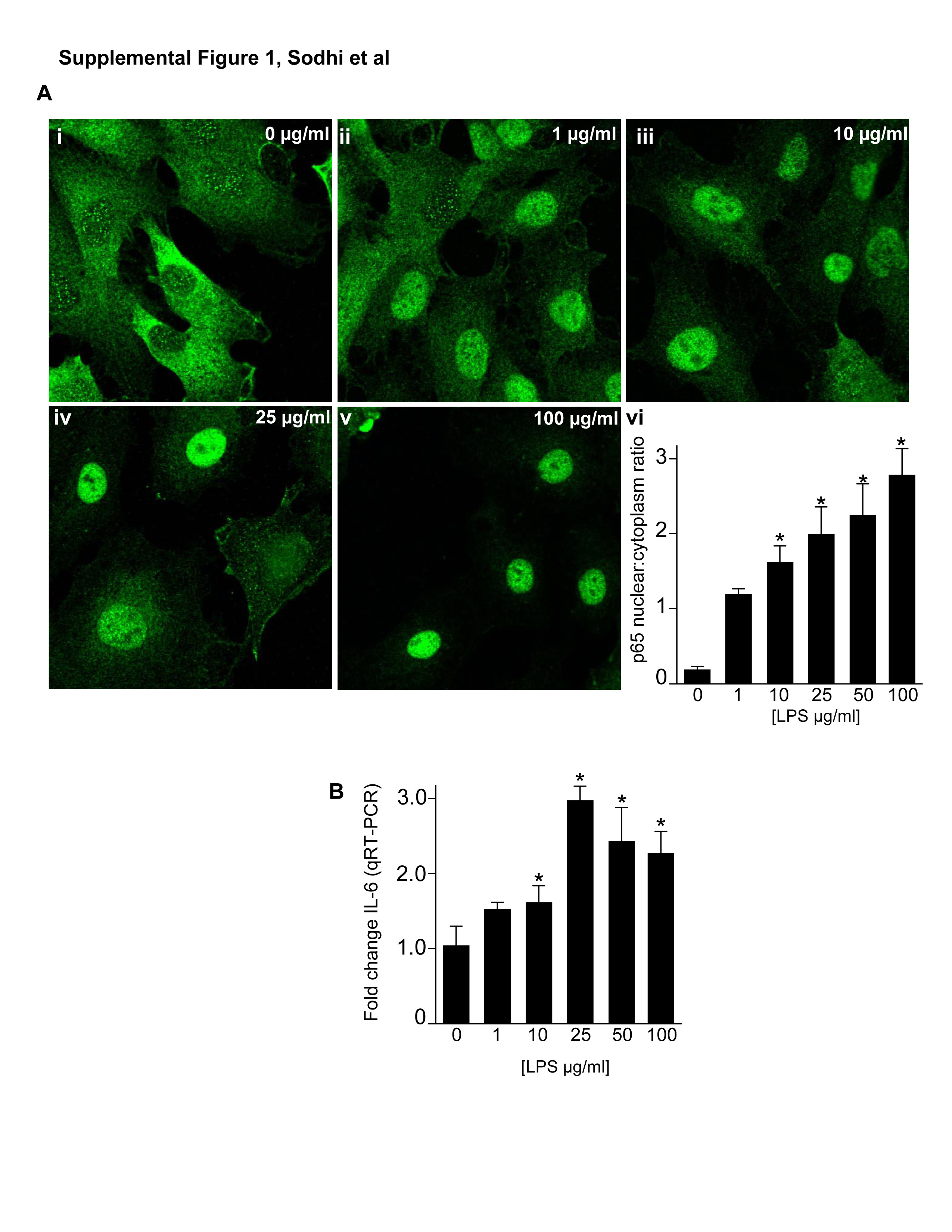
We first sought to assess the effects of TLR4 activation with lipopolysaccharide (LPS) on enterocyte proliferation in vitro and in vivo. As shown in Figure 1, LPS caused a dose-dependent reduction in the extent of enterocyte proliferation, as determined by colorimetric XTT assay (panel A) and reduced expression of the cell proliferation marker pCNA by SDS-PAGE (panels B) and confocal microscopy (note the loss of green pCNA staining with increased concentration of LPS in panel C). There was no effect of LPS on viability as assessed by Trypan blue exclusion (not shown), and LPS caused a dose-dependent increase in NFkB activation and IL-6 expression (Supplemental Figure 1). We next sought to confirm a specific role for TLR4 activation in inhibition of enterocyte proliferation by LPS. Knockdown of TLR4 with TLR4-specific siRNA - which resulted in a >90% reduction in TLR4 expression compared to scrambled siRNA-transfected cells (panel D upper) – reversed the inhibitory effects of LPS on enterocyte proliferation (panel D lower). Importantly, LPS also significantly reduced proliferation in murine embryonic fibroblasts obtained from TLR4 wild-type mice but not TLR4-mutant mice, suggesting that the inhibitory affects of TLR4 are not confined to IEC-6 cells (Panel E). Taken together, these findings strongly indicate that TLR4 activation with LPS significantly inhibits enterocyte proliferation in vitro.
Figure 1. TLR4 activation inhibits enterocyte proliferation in vitro.

A: Percent proliferation in serum-starved IEC-6 cells treated with LPS (3h) at the indicated dosage as determined by XTT assay. B: SDS-PAGE showing pCNA and actin expression and their quantified expression ratio, in IEC-6 cells treated with LPS. *p<0.01 by ANOVA. C: Confocal micrographs showing expression of pCNA (green) and actin (red) treated with LPS for 3h at the indicated concentration; quantification of pCNA pixel density is shown, size bar=10µm. p<0.05 vs. untreated cells, ANOVA. D: RT-PCR showing expression of TLR4 and GAPDH in IEC-6 cells that were either untreated (“mock”), or treated with siRNA to TLR4 (“TLR4”) or to no known targets (“ctrl”), and percent proliferation in IEC-6 cells treated as indicated. E: Effect of inhibition of TLR4 by siRNA on proliferation in embryonic fibroblasts from TLR4-wild-type (C3H/HeOUJ) or TLR4-mutant (C3H/HeJ) mice treated with LPS for 16h as indicated. p<0.05 vs. untreated cells, ANOVA
To define whether TLR4 activation would inhibit enterocyte proliferation in vivo, TLR4 wild-type (C3H/HeOUJ) and TLR4-mutant (C3H/HeJ) mice were injected with either saline or LPS, and the extent of enterocyte proliferation was measured within the enterocytes of the terminal ileum. As shown in Figure 2, LPS significantly reduced enterocyte proliferation in wild-type mice as compared to saline injected animals, yet had no effect in TLR4-mutant mice as shown by immunostaining (panels A–B) and RT-PCR for the proliferation marker pCNA in the intestinal mucosa (panel D). The inhibitory effects of LPS on enterocyte proliferation in vivo were further demonstrated by SDS-PAGE showing a reduction in pCNA expression compared to saline injected animals (panel C). As shown in panel E, LPS did not decrease the rate of enterocyte proliferation in the ileum of adult mice (checkered bars) or in the colon of newborn mice (grey bars), suggesting that the effect may be specific for the small intestine of newborns. We next sought to determine the molecular mechanisms involved.
Figure 2. TLR4 activation leads to an inhibition of enterocyte proliferation in vivo.

A: Confocal micrographs showing sections of terminal ileum from TLR4-wild-type (i–ii) and TLR4-mutant mice (iii–iv) that were injected with LPS or saline and immunostained with pCNA. Size bar = 250 µm. B: Quantification of pCNA staining in crypts in A, *p<0.01 vs. saline C3H/HeOUJ by ANOVA. Representative of 3 separate experiments with at least 5 mice per group. C: SDS-PAGE and quantification showing the expression of pCNA and actin in terminal ileal mucosal scrapings from TLR4-wild-type mice injected with either saline (lanes i–ii) or LPS (lanes iii–iv) for 3h,*p<0.05. Representative of 3 separate experiments with at least 5 mice per group. D: Quantitative RT-PCR showing pCNA expression in terminal ileal mucosal scrapings from TLR4-wild-type and TLR4-mutant mice injected with saline (black bars) or LPS (white bars) for 3h,*p<0.005 vs. saline in C3H/HeOUJ by ANOVA. Representative of 3 separate experiments with at least 5 mice per group. E: RT-PCR expression of pCNA in the terminal ileal mucosa of newborn (white bars) and adult mice (checkered bars), and the colon of newborn mice (grey bars) after injection with LPS. *p<0.005 by ANOVA. Representative of 3 separate experiments with at least 5 mice per group.
TLR4 activation inhibits enterocyte proliferation via the canonical beta-catenin signaling pathway
Given the established role of the canonical β-catenin signaling pathway in the regulation of the proliferation of enterocytes and other cells, we next sought to determine whether β-catenin signaling may mediate the inhibitory effects of TLR4 signaling on enterocyte proliferation. Signaling via the canonical β-catenin pathway results in the translocation of β-catenin from the cytoplasm to the nucleus, which results in the transcription of a variety of genes that induce cell proliferation14. As shown in Figure 3 (A i–iii and B), under control conditions, β-catenin could be detected in IEC-6 cells in both nuclear and cytosolic locations. By contrast, treatment with LPS led to a re-distribution of β-catenin into a predominantly cytoplasmic location, suggesting an inhibition of nuclear translocation in response to LPS (Figure 3A iv–vi, B). LPS also caused a dose-dependent decrease in β-catenin expression in nuclear as compared with cytosolic fractions purified from IEC-6 cells (Figure 3 panel C). To determine directly whether TLR4 activation may inhibit enterocyte proliferation via effects on β-catenin signaling, three specific siRNA's against β-catenin - individually and in combination (“pooled”) - were transfected in IEC-6 enterocytes, and the pooled siRNA resulted in a greater than 95% reduction in β-catenin expression as compared with control siRNA (Figure 3D i–ii). Inhibition of β-catenin prevented further decreases in enterocyte proliferation after the subsequent addition of LPS (Figure 3D iii). Strikingly, overexpression of active β-catenin reversed the inhibitory effects of LPS on enterocyte proliferation as measured by expression of pCNA (Figure 3D iv). Taken together, these findings demonstrate that TLR4 inhibits enterocyte proliferation via effects on the pathways that regulate β-catenin signaling.
Figure 3. TLR4 activation inhibits enterocyte proliferation via inhibition of β-catenin signaling.

A: Confocal images of IEC-6 cells showing β-catenin (red, i, iv) and the nuclear marker Draq-5 (green, ii, v). Size bar=10 µm. B: Extent of colocalization (in iii and vi) was calculated using ImageJ,*p<0.05, representative of 5 separate experiments. C: SDS-PAGE of β-catenin in nuclear and cytoplasmic fractions obtained from IEC-6 cells treated with LPS as indicated, along with the nuclear marker histone and the cytoplasmic marker α-tubulin. Representative of 3 separate experiments. D: SDS-PAGE (i) and quantification of β-catenin:actin expression (ii) in IEC-6 cells transfected with siRNA to no known target (“Ctrl”) or to β-catenin. Three siRNA's (#1, #2, #3) were used either individually or in combination (“pooled”), as indicated. iii: Proliferation in IEC-6 cells treated with the indicated siRNA (XTT assay). iv: RT-PCR showing fold change in pCNA:actin in IEC-6 cells transfected with either GFP or constitutively active β-catenin and treated with LPS. *p<0.05 vs. control, representative of 3 separate experiments.
A novel link between TLR4 and AKT-GSK3β signaling mediates the inhibitory effects of LPS on enterocyte proliferation
It has been previously demonstrated that the ability of β-catenin to translocate into the nucleus is dependent upon its stability within the cytoplasm, which is governed by the activity of the upstream serine/threonine kinase glycogen-synthase 3-beta (GSK3β). GSK-3β negatively regulates β-catenin by phosphorylation of Ser33/37/Thr41 - leading to its degradation - while AKT, a critical upstream negative regulator of GSK3β, inactivates GSK-3β by phosphorylation of its Ser9 residue15. We therefore next tested whether the TLR4-induced β-catenin-mediated inhibition of enterocyte proliferation required GSK3β. As shown in Figure 4 panel A, LPS dose-dependently increased the phosphorylation of β-catenin, which was associated with reduced total β-catenin expression, consistent with its proteosomal degradation. LPS also caused a dose-(Figure 4, panel B) and time- (Figure 4, panel C) dependent decrease in the phosphorylation of GSK3β in enterocytes, a finding consistent with an expected increase in GSK3β kinase activity as demonstrated by the increased phosphorylation of β-catenin. Since phosphorylation of GSK3β is known to be mediated by AKT16, we next assessed the effects of LPS on AKT phosphorylation and expression. As shown in Figure 4 panels B–C, LPS caused a dose- and time-dependent decrease in phosphorylation of AKT and a decrease in overall AKT expression (consistent with the observed decrease in phosphorylation of GSK3β over time). Inhibition of p-AKT with LY290043 prevented the LPS-induced reduction in phosphorylation of GSK3β, confirming the link between AKT and GSK3β signaling (Figure 4 panel D, schematic panel F). In support of the physiological relevance of these findings, LPS caused an increase in the expression of phospho-β-catenin and a corresponding increase in the expression of GSK3β in TLR4-expressing mice that were injected with LPS Figure 4 panel E (Quantification and statistical analysis of all blots is given in Supplementary Figure 2).
Figure 4. LPS activates the AKT-GSK3β signaling pathway in enterocytes.

A–E: SDS-PAGE showing expression of the indicated protein in IEC-6 cells treated with LPS at the concentrations and durations indicated (A–D) or mucosal scrapings from the terminal ileum of mice injected with saline or LPS for the durations indicated (E). All blots are representative of 3 separate experiments; over 5 mice per group in E. F: Schematic summarizing panels A–D showing mechanism by which TLR4 inhibits enterocyte proliferation.
To test directly therefore whether GSK3β signaling mediates the inhibitory effects of LPS on enterocyte proliferation, IEC-6 cells were treated with lithium chloride, a remarkably specific GSK3β inhibitor17, which significantly reversed the LPS-induced inhibition of nuclear translocation of β-catenin in IEC-6 cells (Figure 5 panels A–B), and also reversed the LPS-mediated inhibition in enterocyte proliferation (Figure 5 panel C). To confirm this finding, siRNA knockdown of GSK3β completely reversed the inhibitory effects of LPS on enterocyte proliferation (Figure 5 panels D). Taken together, we conclude that TLR4-mediated inhibition in enterocyte proliferation is mediated via GSK3β signaling and correspondingly decreased β-catenin activity.
Figure 5. TLR4 activation inhibits enterocyte proliferation via GSK3β.

A: Confocal images showing immunolocalization of β-catenin (red, i, v, ix, xiii), the nuclear marker draq-5 (ii, vi, x, xiv), merged images (iii, vii, xi, xv) and colocalization of nuclear and β-catenin (iv, viii, xii, xvi) using ImageJ in IEC-6 cells treated with media (control), or the indicated treatment (LPS and/or the GSK3β inhibitor LiCl). Size bar=10µm. B: Quantification of colocalization between the nucleus and β-catenin using ImageJ.*p<0.05 vs. control cells, ANOVA. C: Proliferation of IEC-6 cells treated as indicated (XTT assay) *p<0.05 vs. control cells, ANOVA. D: upper: SDS-PAGE showing GSK3β in IEC-6 cells transfected with indicated siRNA's to GSK3β, either alone (#1,#2,#3) or in combination (“pool”). lower: proliferation of IEC-6 cells (XTT) after treatment with indicated siRNA and LPS or media (“ctrl”)*p<0.01 vs. control cells, ANOVA. Representative of 3 separate experiments.
NEC is associated with TLR4-mediated induction of β-catenin-GSK3β signaling in enterocytes and an inhibition in enterocyte proliferation in mice; similar changes occur in human infants with NEC
We next sought to determine the physiological relevance of these findings in the pathogenesis of NEC, a disease that we have shown to be characterized by TLR4-mediated reduced enterocyte proliferation8. As is shown in Figure 6 panels A–C, NEC in TLR4-expressing mice was associated with a striking reduction in β-catenin and increase in GSK3β within terminal ileal enterocytes as demonstrated by microscopy and RT-PCR compared to breast-fed controls; these changes were not observed in TLR4-mutant mice. Of interest, exposure of mice to formula feeding alone led to a 50% reduction in enterocyte proliferation as measured by intestinal mucosal RT-PCR and β-catenin compared to breast fed animals before evidence of histological injury was observed, suggesting that the reduction in enterocyte proliferation may be an early event contributing to NEC pathogenesis (p<0.05).
Figure 6. Necrotizing enterocolitis is associated with decreased β-catenin and increased GSK3β expression in enterocytes in mice and humans.

A: Immunostaining of β-catenin (brown staining, i, iii) and GSK3β (brown staining, ii, iv) in either breast fed or mice with NEC. Representative of 4 separate experiments with at least 5 mice per group. B–C: Quantitative RT-PCR showing β-catenin (B) and GSK3β (C) in wild-type and TLR4-mutant mice with (white bars) and without (black bars) NEC. Representative of 3 separate experiments with at least 5 mice per group. D: RT-PCR showing the intestinal mucosal expression of pCNA, GSK3β and β-catenin in wild-type (C57Bl-6) and TLR4−/− mice after injection of saline (black bars) or LPS (white bars). *p<0.05, ANOVA. Representative of 3 separate experiments with at least 5 mice per group. E: SDS-PAGE showing β-catenin in mucosal scrapings obtained from two infants without (lanes i, ii) and two infants with (lanes iii, iv) NEC. Blots were stripped and re-probed for GSK3β, pCNA and actin.
To determine whether these effects were seen in other strains, we generated TLR4−/− mice and injected them with LPS(1mg/kg), which significantly decreased β-catenin and pCNA expression and increased GSK3β; these changes were not seen in TLR4−/− mice (Figure 6 panel D). Finally, in human infants with NEC, there was a dramatic decrease in β-catenin and pCNA and increase in GSK3β expression compared with the terminal ileum from infants without NEC (Figure 6 panel E). We note that the control infants were on average 6 to 8 weeks older than the infants with NEC, corresponding to the time at which stoma reversal was performed. Taken together, these findings provide a strong link between TLR4 and β-catenin-GSK3β signaling in enterocytes in the pathogenesis of NEC, and provide direct physiological relevance of the novel pathway identified in Figure 3–Figure 5.
Inhibition of enterocyte TLR4 signaling in mice with NEC restores enterocyte proliferation and reverses the changes in β-catenin and GSK3β expression
We next sought to determine whether we could reverse the inhibitory effects of TLR4 activation on enterocyte proliferation by blocking TLR4 signaling, and therefore reverse the inhibition in enterocyte proliferation in mice with NEC. We have recently demonstrated that GFP-tagged dominant negative TLR4 bearing the P710H mutation that occurs in the C3H/HeJ mouse inhibits LPS-induced TLR4 signaling in enterocytes and protects against experimental NEC4, and that enteral administration of adenoviral GFP, GFP-wild-type TLR4 and GFP-dominant negative TLR4 lead to expression that is restricted to the enterocytes4. As shown in Figure 7, mice with NEC that were enterally administered adenoviral-GFP-TLR4 or -GFP demonstrated reduced enterocyte proliferation as manifest by decreased pCNA (panel A), decreased β-catenin (panel B) and increased GSK3β (panel C) compared with breast fed controls, consistent with the findings shown in Figure 6, and providing further evidence for a link between TLR4-mediated β-catenin-GSK3β signaling and enterocyte proliferation in vivo. Strikingly, the administration of adenoviral-dominant-negative TLR4 significantly reversed the expression of β-catenin and pCNA, and reduced expression of the inhibitory molecule GSK3β, to levels observed in breast fed mice (grey bars in Figure 7 panels A–C). As an important control, in TLR4 mutant mice, adding back wild-type-TLR4 decreased enterocyte proliferation, compared to TLR4 mutant mice that were administered GFP or dominant negative TLR4 (Figure 7 panel D). The distribution of the GFP-tagged proteins were each localized predominantly to the enterocytes (Panel D i–iv). These findings indicate a critical role for TLR4 activation in the inhibition of enterocyte proliferation in vivo in the pathogenesis of NEC, in part via effects on the β-catenin-GSK3β signaling pathway.
Figure 7. Inhibition of enterocyte TLR4 signaling in mice with NEC restores enterocyte proliferation and reverses the changes in β-catenin and GSK3β expression.

A–C: Newborn mice were either breast fed (black bars) or were induced to develop NEC and administered adenoviruses expressing GFP (checkered bars), GFP-wild-type TLR4 (white bars), or GFP—dominant-negative TLR4 by enteral gavage (grey bars). The expression by quantitative RT-PCR of pCNA (A), β-catenin (B) and GSK3β (C) in mucosal scrapings in each group is shown. *p<0.005 vs. breast fed, **p<0.001 vs. both NEC mice administered adeno-GFP and adeno-GFP-wild-type-TLR4, ANOVA. Representative of 5 separate experiments with over 5 mice per group D: i–v: Confocal micrographs showing GFP (green), actin (red) and nuclei (blue) in untreated (“control”) mice, or mice treated by gavage with the indicated adenovirus. v: RT-PCR of pCNA in TLR4 mutant mice that were administered GFP (checkered bars), GFP-wild-type-TLR4 (white bars), GFP-dominant-negative-TLR4 (grey bars) or control (black bars) and LPS as indicated. Representative of 3 separate experiments with over 10 mice per group.
DISCUSSION
We now describe the identification of a novel and potentially important link between TLR4 signaling and activation of the AKT-GSK3β pathway leading to the inhibition of enterocyte proliferation, an effect that plays a role in the pathogenesis of NEC. The current findings are consistent with previous work indicating that enterocyte proliferation may be inhibited after endotoxin exposure18, 19, yet extend these observations by providing both mechanistic insights and physiological relevance. In support of the relevance of the current findings to the pathogenesis of NEC, we now show that TLR4 activation – which we and others have previously shown to be important in NEC pathogenesis8, 20 - leads to decreased β-catenin and increased GSK3β expression in the intestinal mucosa in both human an murine NEC, while inhibition of TLR4 signaling in enterocytes in vivo reverses these effects and restores levels of enterocyte proliferation in experimental NEC. It is interesting to note that other diseases of intestinal inflammation are associated with impaired enterocyte proliferation, including various models of colitis that mimic inflammatory bowel disease in humans21, 22. Given the observation that TLR4 expression has been shown to be increased in specimens obtained from regions of active ulcerative colitis in humans23, it is tempting to speculate that by inhibition of enterocyte proliferation – perhaps via effects on β-catenin signaling - TLR4 activation could contribute to persistent mucosal injury in these inflammatory intestinal diseases also.
With further respect to NEC pathogenesis, we note that it has recently been reported that in the newborn intestine, vaginal delivery and LPS exposure lead to LPS unresponsiveness by the primary enterocytes24. Based upon our recent discovery that the hypoxia and endotoxemia that characterize NEC leads to persistent upregulation of intestinal TLR48, we now propose that part of the mechanism of NEC reflects the inability of the intestine to down-regulate TLR4 signaling in order to become tolerant to the luminal bacteria. This would be expected to lead to exaggerated TLR4 signaling upon bacterial colonization, resulting in impaired enterocyte proliferation and persistent susceptibility to mucosal injury as seen in NEC. These findings expand the current understanding of the role of TLR4 in intestinal homeostasis, while also providing novel insights into the molecular mechanisms that lead to NEC.
How can the current findings be reconciled with the observation that patients with chronic intestinal inflammation have an increased propensity to develop intestinal tumors, suggesting that enterocyte proliferation may be increased at least transiently during states of chronic intestinal injury? First, we point out that the current findings may be specific for the newborn small intestine, as TLR4 activation did not decrease enterocyte proliferation in either adult intestine or newborn colon. Second, it is well established that downstream mediators of TLR4 signaling – including the transcription factor NFkB and associated molecules - have established roles in the development of tumors, in part via transcription of oncogenes25, which could lead to induction of tumors after chronic intestinal inflammation. And third, while we now demonstrate that TLR4 signaling in enterocytes is responsible for the inhibition of enterocyte proliferation in vivo, it is quite possible that in conditions of colitis that favor the development of tumors, the effects of TLR4 signaling may occur via hematopoietic cells and not on enterocytes. In support of this possibility, Abreu et al has recently used TLR4 wild-type and mutant chimeric mice to show that TLR4 signaling in leukocytes plays a key role in colitis associated tumor formation26. We now propose that in the intestinal inflammatory environment, TLR4 activation in different cell types leads to disparate effects: in newborn enterocytes, an inhibition of cell turnover is observed leading to mucosal injury, while in the setting of chronic NFkB activation – potentially via effects on leukocytes - tumorogenesis may occur. It is also likely that the extent and duration of inflammation, the age of the individual and the location of the inflammation (colon vs. ileum) play important roles in determining whether enterocyte proliferation is impaired, as we now show, as opposed to the more long term models of colitis in which tumors may form27.
In summary, we now provide evidence for a novel link between TLR4 and the GSK3β-β-catenin signaling pathway in enterocytes, and show that the interaction of these signaling pathways plays a key role in the pathogenesis of NEC via impaired enterocyte proliferation. The mechanisms by which this pathway is regulated in vivo may open up future directions of study regarding both the pathogenesis and potential treatment of devastating diseases such as NEC.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
DJH is supported by R01GM078238 and RO1DK08752 from the National Institutes of Health.
Role in the study:
Shi – study concept and design, data acquisition, data analysis; Sodhi – study concept and design, data acquisition, data analysis; Steve Gribar – data acquisition; Ward Richardson – data acquisition, Zachary Grant – data acquisition; Richard A. Shapiro – data acquisition; Thomas Prindle – data acquisition; Maria Branca – data acquisition; Anthony Russo – data Acquisition; David Hackam – study concept and design, data analysis, supervision, obtaining funding, drafting of manuscript.
Abbreviations
- GSK3β
glycogen-synthase 3-beta
- TLR4
Toll like receptor-4
- NEC
necrotizing enterocolitis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest:
Xia-hua Shi – no conflicts exist; Chhinder P. Sodhi - – no conflicts exist ; Ward M. Richardson - – no conflicts exist; Steven C. Gribar - – no conflicts exist; Thomas Prindle Jr - – no conflicts exist; Maria Branca – no conflicts exist; Anthony Russo - – no conflicts exist; Congrong Ma - – no conflicts exist ; Richard Shapiro – no conflicts exist; Zachary Grant – no conflicts exist; David J. Hackam – no conflicts exist.
Transcript profiling – Not applicable
Writing assistance – Not applicable.
References
- 1.Gribar SCAR, Sodhi CP, Hackam DJ. The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J Leukoc Biol. 2008;83:493–498. doi: 10.1189/jlb.0607358. [DOI] [PubMed] [Google Scholar]
- 2.Anand RJ, Leaphart CL, Mollen KP, Hackam DJ. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27:124–133. doi: 10.1097/01.shk.0000239774.02904.65. [DOI] [PubMed] [Google Scholar]
- 3.Grave GD, Nelson SA, Walker WA, Moss RL, Dvorak B, Hamilton FA, Higgins R, Raju TN. New therapies and preventive approaches for necrotizing enterocolitis: report of a research planning workshop. Pediatr Res. 2007;62:510–514. doi: 10.1203/PDR.0b013e318142580a. [DOI] [PubMed] [Google Scholar]
- 4.Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, Jakub A, Shi XH, Shah S, Ozolek JA, Hackam DJ. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol. 2009;182:636–646. doi: 10.4049/jimmunol.182.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem. 2004;279:24592–24600. doi: 10.1074/jbc.M313620200. [DOI] [PubMed] [Google Scholar]
- 6.Ireland H, Kemp R, Houghton D, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Qureshi FG, Leaphart CL, Cetin S, Jun L, Grishin A, Watkins S, Ford HR, Hackam DJ. Increased expression and function of integrins in enterocytes by endotoxin impairs epithelial restitution. Gastroenterology. 2005;2005 doi: 10.1053/j.gastro.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 8.Leaphart CL, Cavallo JC, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 9.Zeng G, Apte U, Micsenyi A, Bell A, Monga SP. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp Cell Res. 2006;312:3620–3630. doi: 10.1016/j.yexcr.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Hassan GS, Williams TM, Minetti C, Pestell RG, Tanowitz HB, Frank PG, Sotgia F, Lisanti MP. Loss of caveolin-1 causes the hyper-proliferation of intestinal crypt stem cells, with increased sensitivity to whole body gamma-radiation. Cell Cycle. 2005;4:1817–1825. doi: 10.4161/cc.4.12.2199. [DOI] [PubMed] [Google Scholar]
- 11.Cetin S, Leaphart CL, Li J, Ischenko I, Hayman M, Upperman J, Zamora R, Watkins S, Ford HR, Wang J, Hackam DJ. Nitric oxide inhibits enterocyte migration through activation of RhoA-GTPase in a SHP-2-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1347–G1358. doi: 10.1152/ajpgi.00375.2006. [DOI] [PubMed] [Google Scholar]
- 12.Leaphart CL, Qureshi F, Cetin S, Li J, Dubowski T, Batey C, Beer-Stolz D, Guo F, Murray SA, Hackam DJ. Interferon-[gamma] inhibits intestinal restitution by preventing gap junction communication between enterocytes. Gastroenterology. 2007;132:2395–2411. doi: 10.1053/j.gastro.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 13.Mei JM, Hord NG, Winterstein DF, Donald SP, Phang JM. Expression of prostaglandin endoperoxide H synthase-2 induced by nitric oxide in conditionally immortalized murine colonic epithelial cells. FASEB J. 2000;14:1188–1201. doi: 10.1096/fasebj.14.9.1188. [DOI] [PubMed] [Google Scholar]
- 14.Jin T, Fantus I, Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell Signal. 2008;20:1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes K, Mikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sukhotnik I, Agam M, Shamir R, Shehadeh N, Lurie M, Coran AG, Shiloni E, Mogilner J. Oral glutamine prevents gut mucosal injury and improves mucosal recovery following lipopolysaccharide endotoxemia in a rat. J Surg Res. 2007;143:379–384. doi: 10.1016/j.jss.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Kessel A, Toubi E, Pavlotzky E, Mogilner J, Coran AG, Lurie M, Karry R, Sukhotnik I. Treatment with glutamine is associated with down-regulation of Toll-like receptor-4 and myeloid differentiation factor 88 expression and decrease in intestinal mucosal injury caused by lipopolysaccharide endotoxaemia in a rat. Clin Exp Immunol. 2008;151:341–347. doi: 10.1111/j.1365-2249.2007.03571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177:3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiagarajah JR, Zhao D, Verkman AS. Impaired enterocyte proliferation in aquaporin-3 deficiency in mouse models of colitis. Gut. 2007;56:1529–1535. doi: 10.1136/gut.2006.104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renes IB, Verburg M, Van Nispen DJ, Taminiau JA, Büller HA, Dekker J, Einerhand AW. Epithelial proliferation, cell death, and gene expression in experimental colitis: alterations in carbonic anhydrase I, mucin MUC2, and trefoil factor 3 expression. Int J Colorectal Dis. 2002;17:317–326. doi: 10.1007/s00384-002-0409-4. [DOI] [PubMed] [Google Scholar]
- 23.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med. 2006;203:973–984. doi: 10.1084/jem.20050625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukata M, Hernandez Y, Conduah D, Cohen J, Chen A, Breglio K, Goo T, Hsu D, Xu R, Abreu MT. Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.20880. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.