

Abstract
Aortic valve leaflets experience varying applied loads during the cardiac cycle. These varying loads act on both cells types of the leaflets, endothelial and interstitial cells, and cause molecular signaling events that are required for repairing the leaflet tissue, which is continually damaged from the applied loads. However, with increasing age, this reparative mechanism appears to go awry as valve interstitial cells continue to remain in their ‘remodeling’ phenotype and subsequently cause the tissue to become stiff, which results in heart valve disease. The etiology of this disease remains elusive; however, multiple clues are beginning to coalesce and mechanical cues are turning out to be large predicators of cellular function in the aortic valve leaflets, when compared to the cells from pulmonary valve, which is under a significantly less demanding mechanical loading regime. Finally this paper discusses the mechanical environment of the constitutive cell populations, mechanobiological processes that are currently unclear, and a mechano-potential etiology of aortic disease will be presented.
2. Introduction
In the burgeoning field of mechanobiology, heart valves (HVs) have a unique position. The leaflets that make up the HVs are relatively ‘simple’ in architecture, and additionally, their biology is not excessively complicated due to significant innervations or vasculature. Both these characteristics are akin to articular cartilage and musculoskeletal tendons and ligaments, but HVs are unique in that they are always under significant forces due to the cardiac cycle. When a HV is open, the surface facing the passing blood is exposed to significant shear stress, while the other side experiences disturbed flow due to blood recirculation and eddy formation (Fig. 1A). When the HV is closed, blood pressure imposes a force normal on the leaflets preventing retrograde blood flow with a distinct orthogonal mechanical tissue response, largely due to an evolved and highly aligned collagen architecture (Fig. 1B).
Figure 1.
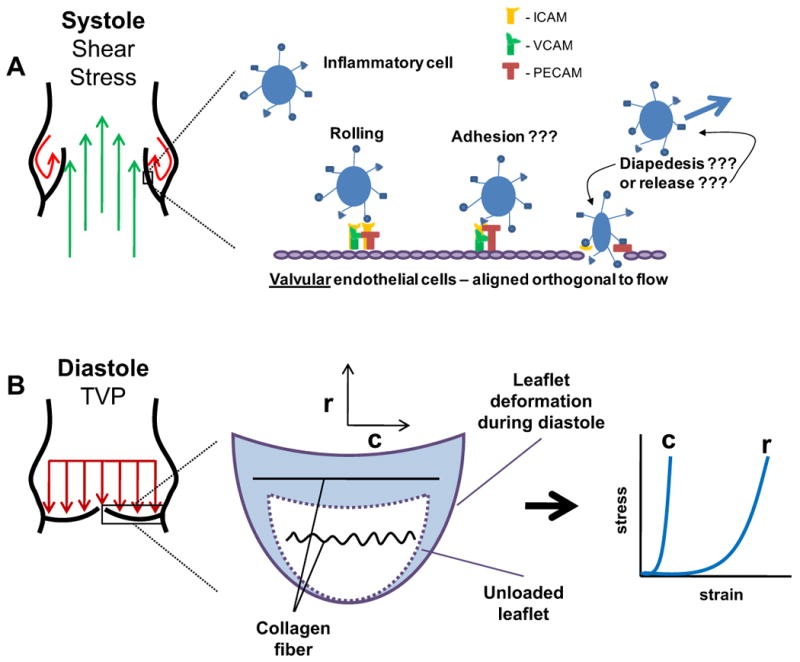
Distinct stress modes applied to AV leaflets during the cardiac cycle. A: Shear stress is applied to both sides of the leaflet during systole. On the side of flowing blood (ventricularis), the leaflets experience laminar shear stress. On the side of recirculation and eddy formation (fibrosa), the leaflets experience disturbed, oscillatory shear stress. This oscillatory shear stress is thought to activate cell adhesion molecules (i.e. ICAM, VCAM, and PECAM), which may or may not recruit inflammatory cells into the leaflet. B: Diastolic pressure results in biaxial planar stretch of the leaflets. The leaflets exhibit an orthogonal stress-strain response due to the aligned collagen architecture in the circumferential direction. Under normal diastolic pressure (∼80 mmHg), AV leaflets are strained to ∼15% in the circumferential direction (c) (Thubrikar 1990), due to the straitening of the collagen fibers, and ∼50% in the radial direction (r) (Christie and Barratt-Boyes 1995).
This biomechanical response is of great significance for the HVs to function properly. Specifically, the leaflets are required to achieve very large strains with low stress in the radial direction in order to co-apt and close the orifice area; however, they must simultaneously withstand significant pressure from the blood to prevent retrograde flow. Thus, if one were to design a homogenous and isotropic material to function as a replacement valve leaflet, it may be able to achieve the high strain needed in the radial direction; however, there would not be ample strength to withstand the pressure and the leaflets would ultimately pull apart during closure, leading to regurgitation. To prevent this, HVs have developed an aligned collagen architecture in the circumferential direction that responds biomechanically with a sharp rise in stress with minimal strain.
The mechanobiologic consequence of the various stress modes experienced by the HV leaflets provides a platform to examine cellular response to mechanical stimuli. However, there are also confounding factors that make teasing out specific responses difficult. For instance, uncoupling effects of fluid shear stress and tissue deformation resulting from in plane stress is largely impossible. Additionally, the molecules that are produced in response to these stresses, which in turn act on the cells that are not under said deformation, offer further challenges (i.e. cytokines from endothelial cells that act on interstitial cells).
Here, I will focus on aortic valve (AV) leaflets specifically, and their low-pressure counterparts, the pulmonary valve (PV) leaflets, when appropriate. I will begin by moving from the outside of the tissue into the interstitial space and discuss recent advances in our understanding of how mechanical forces acting on the leaflets lead to biological changes in the cell population and subsequently leaflet tissue architecture and functional properties. The impetus for focusing on the AV is that it makes up ∼63% of valve disease mortality numbers and ∼53% of the 93,000 valve procedures performed per year in the U.S. (Lloyd-Jones, Adams et al. 2009). These numbers will increase substantially in the coming years as our population continues to live longer.
Over the next 50 years, the population of Americans age 65+ will more than double – from 34 to 79 million (www.bioethics.gov 2005). In fact, the oldest of the old (85+) are currently the fastest growing segment of the population and will more than quadruple by 2050. These numbers are alarming in light of what is known of HV disease, particularly in the AV. AV disease develops in an escalating fashion after 65 years of age such that 48% of AVs are sclerotic by the age 85 (Stewart, Siscovick et al. 1997). Multiple studies demonstrate that sclerosis leads to stenosis and surgery 4-8 years after initial diagnosis (Cosmi, Kort et al. 2002; Faggiano, Antonini-Canterin et al. 2003). Together, the aging trends in the U.S. combined with the progression of AV disease indicate an increasing trend in the number of patients affected and dollars spent on this pathology. In light of these projections, there is considerable interest in developing novel techniques to prevent early stage AV disease (before calcification) in the aging population, and developing strategies that utilize the AVs' inherent mechanobiological processes will likely be critical in achieving this goal. Thus, I will attempt to connect our current understanding of AV mechanobiology, including gaps that exist in our knowledge, with subsequent speculative directives toward research that will hopefully lead to novel preventative or treatment strategies in the coming years.
3. Shear stress and valve endothelial cells
The leaflets of all four HVs are sheathed by a single layer of valve endothelial cells (VECs) that have been shown to be morphologically different from vascular endothelial cells (Butcher and Nerem 2004). Specifically, VECs are aligned perpendicularly to the direction of blood flow (Deck 1986), whereas vascular endothelial cells are aligned parallel. This directionality may be programmed into the VECs as porcine VECs grown on collagen are reported to align perpendicular to flow in vitro (Butcher, Penrod et al. 2004). VECs are believed to regulate tone, inflammation, thrombosis, and remodeling, and their dysfunction has been linked with multiple valve disorders (Leask, Jain et al. 2003). VECs have also been found to provide protection for valve interstitial cells (VICs) in a collagen gel/tissue engineering model by preventing a shift of the VICs to the myofibroblast phenotype (to be discussed in the next section) (Butcher and Nerem 2006). However, this finding has not been substantiated in a native leaflet.
Within the proximal third of the leaflets, where they are innervated, there is believed to be a feedback mechanism between the VECs and VICs wherein the nerves transmit information regarding released substances from the VECs (Marron, Yacoub et al. 1996). While it has not been demonstrated to date for HVs, mechano-sensitive release of cytokines from vascular endothelial cells has been shown to cause changes in vascular smooth muscle cell structure and function (Davies 1997); thus, there is a likelihood that this occurs in valvular tissue. There has been speculation that there exists some physical communication between the VECs and VICs via gap junction. However, to date none have been observed between the two cell populations (Filip, Radu et al. 1986), indicating that signaling between the two is likely biochemical. A very recent publication supports this notion, as treatments of intact VECs on porcine leaflets with various neurohumoral stimulants (endothelin 1, serotonin, etc.) resulted in contraction or relaxation of the tissue in both the circumferential and radial directions (El-Hamamsy, Balachandran et al. 2009). Previously, multiple biochemicals have been found to alter AV leaflet stiffness via VIC contraction (Kershaw, Misfeld et al. 2004; Merryman, Huang et al. 2006); however, this is the first evidence that VECs directly communicate with the VICs to alter leaflet mechanical properties.
Some of the most compelling work in the area of VEC mechanobiology in recent years is by Simmons et al. (Simmons, Grant et al. 2005). Here they examined the differentially expressed genes of VECs from both the inflow and outflow surfaces of healthy porcine AVs. Briefly, they demonstrated that VECs on the aorta side, which is much more prone to disease, were permissive to calcification but were protective against oxidative and inflammatory molecules, such as endothelial nitric oxide synthase, vascular adhesion molecule 1, and intracellular adhesion molecule 1. Interestingly, genes expressed for a cytokine implicated in fibrotic remodeling, transforming growth factor-β1 (TGF-β1) (Blobe, Schiemann et al. 2000), was significantly greater on the aorta side of the AV. These results indicate that as a potential mechanism for initiating AV disease, fibrotic and calcific molecules may be readily available from VECs on the aorta side of the leaflets.
The mechanobiological implications for these side-specific differences are significant. In a recent review on the mechano-regulation of VECs, Butcher and Nerem (Butcher and Nerem 2007) discuss potential mechano-dependent molecular mechanisms that may lead to VEC dysfunction and AV disease. Specifically, the ventricular surface of the AV, which is exposed to pulsatile shear stress (average of 26 dynes/cm2 (Weston, LaBorde et al. 1999)), is prone to endocarditis and vegetative bacterial growth, while the aorta surface of the AV, which experiences disturbed, oscillatory shear stress, is prone to sclerotic or fibrotic tissue formation. Hence, there appears to be distinct differences between valve surfaces that are under unique shear stress profiles, in turn leading to distinct pathologies.
Currently, there are multiple unknowns for VEC mechanobiological function that are in serious need of examination. Specifically, how do circulating inflammatory cells ‘stick’ to the VECs, given that their orientation is orthogonal to flow? What are the biophysics of inflammatory cell adhesion on heart valves given that they do not provide a large surface for the paradigm of tethering, rolling, adhesion, and diapedesis found in the vasculature (Fig. 1A)? If most AV diseases are found in the tissue on the aorta side, where VECs are under disturbed oscillatory shear stress, is the effect of the oscillatory shear stress on the VECs or the no-flow condition during diastole more important for inflammatory cell recruitment? Can it be shown that inflammatory cell invasion occurs in the leaflets independent of interstitial fibrosis or calcification? Or, more to the point, does inflammatory cell recruitment via the VECs lead to AV disease, or is their recruitment a byproduct of the disease after the onset of interstitial fibrosis? Finally, how do soluble molecules produced from an activated endothelium interact with the VICs?
Again, uncoupling shear and planar stress imposed on the leaflets makes teasing out their effects difficult. If some of these posed questions were readily answerable, it would provide guidance for 1) elucidating the initiating mechanism of AV disease, and 2) development of potential therapeutics prior to end-stage AV disease which requires valve replacement surgery.
4. Sub-endothelial matrix components, valve interstitial cells, & their evolution over a lifetime
As stated above, the VECs form a single cell layer on the surface of the leaflets. Below the VEC layer is a basal membrane and then the interstitial extracellular matrix (ECM) that varies in protein composition throughout the thickness. In the adult AV, the ECM nearest the aorta side of the leaflet is composed mostly of type I collagen that is oriented as the VECs are, in the circumferential direction. This layer spans ∼1/3 the thickness of the leaflet and is the primary contributor to the planar, orthogonal mechanical response of the leaflet during closure mentioned above. The central portion of the leaflet, termed the spongiosa, is made up of sulfated glycosaminoglycans (GAGs), and is likewise ∼1/3 of the leaflet thickness. The biomechanical function of this central region is thought to be primarily a lubricating or shock absorbing layer (Grande-Allen, Calabro et al. 2004). The tissue layer on the ventricular side of the AV, aptly named the ventricularis, is composed of both collagen and elastin (Schoen 1997), which may aid is early closing mechanisms of the valve.
These morphological characteristics are well known and multiple studies have examined changes to AV tissues and cells due to various mechanical stimuli (Weston and Yoganathan 2001; Balachandran, Konduri et al. 2006; Gupta and Grande-Allen 2006; Gupta, Werdenberg et al. 2007; Merryman, Lukoff et al. 2007; Balachandran, Sucosky et al. 2009). These works have demonstrated that varying tissue stretch levels alter cytokine effects, enzymatic activity, and protein biosynthesis. Clearly, an altered strain field in AV tissue leads to multiple changes at the cellular level, leading to further changes in ECM content and mechanical response to physiologic loading. However, previous studies that have examined these changes have all been conducted in vitro and for relatively short durations (14 days or less), leaving an incomplete picture of precisely how mechanical stimuli alter protein synthesis and degradation over a lifetime.
Alternatively, examining healthy, autopsied valves during fetal development, childhood, and adulthood allows for theorizing into the roles of physiologic stresses on the tissues and cells. Indeed, a seminal work has examined the morphology of AVs and pulmonary valves (PVs) from development (human fetuses) to adulthood (∼50 years old) (Aikawa, Whittaker et al. 2006). From early fetal development (14-19 wks to 20-39 wks), the leaflet composition went from almost complete GAGs with little to no collagen or elastin, to a bilaminar structure with the beginnings of disorganized fibrosa and ventricularis layers. Thus, there is minimal prenatal adaptation of the tissue that is likely due to slight increased pressure on the valves as gestation progresses. However, the prenatal valve is quite distinct from the adult valve. This finding indicates that the closing of the foramen ovale after birth, which allowed equal pressure on both the AV and PV during gestation, provides a high and low pressure comparison. Essentially, the AV and PV are extremely similar with respect to cellular function and ECM composition at birth; however, they diverge over our lifetime to resemble two distinct structures that are parameterized by the mechanical forces on the leaflets. The major mechanical difference between the AV and the PV is the amount of pressure imposed on the leaflets due to transvalvular pressure during closure (Merryman, Youn et al. 2006).
Juxtaposing valve development and valve degeneration with increasing age, there appears to be mirrored characteristics of divergent processes (Fig. 2). During development, VIC density, proliferation, and remodeling markers are high compared to adult valves (from 20-50 years of age). The reason for this VIC activation is the need to constantly remodel the tissue while increasing both in area and volume. Additionally, VIC density is 6-fold higher during fetal development compared to healthy adult valves. This is likely because the ECM of the developing valve is unorganized and unaligned, thus the VICs are responsible for remodeling their surrounding tissue ‘locally’, leading to highly activated VICs that do not work in coordinated fashion. Conversely, VICs from both AV and PV adult sheep demonstrate migration into an aligned pattern on collagen gels prior to contraction, indicating some innate or programmed level of organization and cooperation (Merryman, Liao et al. 2007). This migration prior to contraction takes ∼30 hours more in the VICs from the PV; however, once organized they contract the compliant collagen gels similarly to VICs from the AV. As an aside, dermal fibroblasts exposed to the same treatment as the VICs contracted collagen gels very rapidly and locally further indicating an additional and interesting adaptive mechanobiologic response. Specifically, DFs are exposed to mainly isotropic forces over their lifetime and do not appear to have a coordinate system programmed that dictates their function, while the VICs experience repeated and directional forces in the circumferential direction.
Figure 2.

Coupled response of AVIC phenotype and ECM stiffness with increasing age. In utero, the AVIC population is highly activated and remodeling the very compliant tissue in order to develop a functional AV. The phenotype shifts slowly during childhood to quiescence as stiffness increases. During adulthood the ECM compliance is largely constant, as is the AVIC phenotype, but begins to increase around the age of 50. Ages 60 and up see progressive myofibroblast phenotypic AVICs with increasing stiffness. In essence, the process of valve degeneration mirrors valve development.
Therefore, from birth, alignment of type I collagen and elastin organization continues with age, leading to an increase in ECM stiffness (blue region, Fig. 2) to support the transvalvular pressure of the systemic blood in the adult heart. During adulthood, this ECM stiffness modulates in a healthy range and the VICs of the tissue are responsible for maintaining the ECM proteins in a homeostatic remodeling dynamic. Examining the mechanical properties of VICs from healthy porcine valves indicates the difference between this homeostatic remodeling in the AV and PV. Interestingly, whether assessing by micropipette aspiration (Merryman, Youn et al. 2006) or atomic force microscopy (Merryman, Liao et al. 2007), VICs from the AV are 2× as stiff as those from the PV. Therefore, as VIC stiffness is a function of α-smooth muscle actin (αSMA) (Merryman, Youn et al. 2006) and αSMA is the key indicator of phenotypic change of VICs to the myofibroblast phenotype (Rabkin-Aikawa, Farber et al. 2004), VICs from the AV appear primed and ready to become myofibroblasts while VICs from the PV are not. This could easily explain why PV disease is not prevalent: the VICs of the PV are not under enough stress to become activated myofibroblasts. However, the VICs from the AV are under enough stress to become myofibroblasts, and with age, the leaflets stiffen and AV disease ensues, but why?
5. Cellular deformation with age – extension of stress overload theory
To reiterate, AV leaflets undergo very large deformations during the cardiac cycle, most notably significant strain in both the circumferential and radial directions during diastole to prohibit blood regurgitation. Specifically, the AV exhibits a very high strain-low stress response in the radial direction with much more modest strain and higher stress in the circumferential direction (Billiar and Sacks 2000a). It is estimated that the typical range of circumferential strain in a healthy AV leaflet during diastole is 10-20% (Fig. 1B) (Thubrikar 1990) while radial strains can vary from 40-60% (Christie and Barratt-Boyes 1995). The circumferential response is dictated by the fibrillar type I collagen that becomes taut at very low strains (Sacks, Smith et al. 1998), while the radial response is a result of the elastin fibers (Vesely 1998) and non-collagen fiber components and additionally the dispensability of the basal attachment of the leaflets at the annulus of the aortic wall.
A very interesting theory of stress overload in the AV was proposed by Robicsek et al. (Robicsek, Thubrikar et al. 2002); however, this theory has not been widely explored since. Essentially, they theorized that a likely cause of degenerative AV disease is increased stress on the AV leaflets due to diminished compliance in the aortic wall at the basal attachment. There is very compelling evidence that ascending aorta biaxial compliance near the AV annulus decreases by 60% in humans above the age of 60 (Haskett, Johnson et al. 2009). As compliance decreases in the aortic wall, vessel dilation during diastole decreases, which inhibits stress transfer from the collagen fibers in the circumferential direction to the highly extensible elastin fibers oriented radially (Schoen 1997; Vesely 1998). In the normal AV, the perimeter of the AV annulus increases ∼15% during diastole (Thubrikar 1990); however, above age 60 aorta compliance decreases, prohibiting this increase in perimeter (Fig. 3). Because annular compliance allows the radial deformation of the leaflets to be maximized during diastole, inhibition of this radial strain by a stiffened annular ring implies that the circumferential collagen fibers must bear more load than normal because the load typically supported by the radial deformation is unavailable. Moreover, as the VICs are oriented circumferentially and bound to the type I collagen in the fibrosa layer of the leaflet tissue (Fig. 4), their deformation would likely increase as well with age.
Figure 3.

Aorta and AV annular perimeter changes during the cardiac cycle and with increasing age. In younger people, the aorta is compliant and the perimeter of the AV annulus expands by ∼15% during diastole which accommodates strain in the radial direction. Around the age of 60, aorta and AV annulus compliance decreases by ∼60% (Haskett, Johnson et al. 2009), prohibiting radial strain. Since strain is prohibited in the radial direction of the leaflets, a stress transfer likely occurs wherein the collagen fibers (and VICs) in the circumferential direction are strained more than normal (Robicsek, Thubrikar et al. 2002).
Figure 4.

Transmission electron microscopy images of porcine AVICs fixed in situ at circumferential strains of 0, 10, and 20%. Reprinted with permission from (Merryman 2007). Note that planar strain in the circumferential direction is directly transduced to the AVICs.
The importance of VIC deformation due to applied strain has been indicated previously by multiple studies (Balachandran, Konduri et al. 2006; Balachandran, Sucosky et al. 2009); however, these studies were conducted with mechanical stimulation being the only input. While simply over-stretching the VICs is likely of consequence, a more pressing question is: how are certain biomolecules processed and/or synthesized by the VICs in light of increasing strain? This was addressed cursorily with active TGF-β1 in static (strain = 0%) and 15% circumferential cyclic strain environments for 14 days in vitro (Merryman, Lukoff et al. 2007). Interestingly, it was found that 15% strain and the fibrotic cytokine, TGF-β1, acted synergistically to lead to AV disease-like characteristics (αSMA expression in VICs, collagen production by VICs as quantified by surrogates heat shock protein 47 (Hsp47) and collagen I c-terminal propeptied (CICP), and further synthesis of TGF-β1) (Fig. 5). Not only did they act synergistically, but they generated a feed-forward loop due to the synthesis or activation of excess TGF-β1 (Merryman 2008). Therefore, using the working hypothesis that mechanical strain imposed on VICs later in life, this finding indicates a potential mechanism for VICs to become constitutive myofibroblasts. Further, the work by Simmons et al. discussed above demonstrated that VECs on the aorta surface of AV had significantly higher TGF-β1 gene expression compared to the ventricular surface (Simmons, Grant et al. 2005). This indicates that not only are the VICs likely generating more TGF-β1 due to increased strain, but the VECs nearest them are also providing TGF-β1, further increasing the chance of VIC activation and AV disease.
Figure 5.

Enzyme-linked immunosorbent assay results of αSMA, Hsp47, CICP, and total TGF-β1 from isolated and combined treatments of TGF-β1 and 15% cyclic strain (Tension) on AV leaflets for 14 days. Note synergistic effects of combined treatments (Tension+TGF) on the AVIC phenotype and biosynthesis. This result demonstrates the importance of mechanical strain on the cytokine signaling of AVICs. Reprinted with permission from (Merryman, Lukoff et al. 2007).
Work is currently underway in examining the mechano-dependent processing of multiple cytokines by VICs. It should be mentioned that the focus of this current article was primarily on mechano-dependent myofibroblast activation of the VICs as this is believed to be an early indicator of valve pathogenesis (Jian, Narula et al. 2003), prior to calcification. Further down the disease cascade once calcification has initiated there is a myriad of signaling factors, particularly osteogeneic, that are likely also mechano-dependent (O'Brien 2006; Chen, Yip et al. 2009; Yip, Chen et al. 2009) and these have not been discussed here.
Elucidation of how mechanical strain alters the synthesis or use of various cytokines could ultimately lead to potential preventative therapeutic options for AV disease. Recently, a very interesting study demonstrated that TGF-β1 effects in VICs could be blocked with fibroblast growth factor (FGF) by inhibiting TGF-β1 signaling through the canonical Smad signaling pathway (Cushing, Mariner et al. 2008). While very exciting, this strategy has limitations as it would be extremely difficult to selectively provide FGF to VICs; however, this work demonstrates that it is possible to prevent TGF-β1 mediated activation of VICs, which may be an important aspect of the hopes preventing inevitable fibrotic remodeling by developing preventative strategies against AV disease.
6. Conclusions and future directions
The hemodynamic forces imposed on the AV during the cardiac cycle dictate the passive response of the leaflets, which in turn elicits mechanobiological response from the cell population. Understanding and being able to control the mechanobiological response of VECs and VICs is likely the only means possible for developing preventative strategies toward heart valve disease for elderly adults. Further, while not discussed here, the understanding of cross-talk between the VECs and VICs of the leaflet is of paramount importance. If it was discovered that valve disease does indeed initiate with the VECs, getting pharmaceuticals to ‘stick’ to the valves will certainly not be easy. Similarly, if a drug could be developed that prevented myofibroblast activation of VICs, it would still have to ‘stick to’ and then ‘clear’ the VEC barrier. Finally, targeting of only one of the valve cell populations may not do the trick. For instance, if fibrotic endogenous remodeling could be halted in the VIC population, there may still be no way to prevent VECs, who have short distances to send biomolecules/cytokines via diffusion, from overriding this blockade. Therefore, we must begin to analyze both cell populations in tandem when we experimentally perturb one because developing a strategy that targets VECs or VICs alone may ultimately be futile.
Acknowledgments
W. David Merryman is supported by the American Heart Association (0835496N and 09GRNT2010125), the Wallace H. Coulter Foundation (Early Career Award), and the National Institutes of Health (HL094707). The author would like to thank Joshua D. Hutcheson and Michael P. Nilo for editing prior to submission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aikawa E, Whittaker P, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113(10):1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- Balachandran K, Konduri S, et al. An ex vivo study of the biological properties of porcine aortic valves in response to circumferential cyclic stretch. Ann Biomed Eng. 2006;34(11):1655–65. doi: 10.1007/s10439-006-9167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran K, Sucosky P, et al. Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: implications for degenerative aortic valve disease. Am J Physiol Heart Circ Physiol. 2009;296(3):H756–64. doi: 10.1152/ajpheart.00900.2008. [DOI] [PubMed] [Google Scholar]
- Billiar KL, Sacks MS. Biaxial mechanical properties of the natural and glutaraldehyde treated aortic valve cusp--Part I: Experimental results. Journal of Biomechanical Engineering. 2000a;122(1):23–30. doi: 10.1115/1.429624. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, et al. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells. J Heart Valve Dis. 2004;13(3):478–85. discussion 485-6. [PubMed] [Google Scholar]
- Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. 2006;12(4):905–15. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1445–57. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher JT, Penrod AM, et al. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. 2004;24(8):1429–34. doi: 10.1161/01.ATV.0000130462.50769.5a. [DOI] [PubMed] [Google Scholar]
- Chen JH, Yip CY, et al. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol. 2009;174(3):1109–19. doi: 10.2353/ajpath.2009.080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie GW, Barratt-Boyes BG. Age-dependent changes in the radial stretch of human aortic valve leaflets determined by biaxial testing. Ann Thorac Surg. 1995;60(2 Suppl):S156–8. doi: 10.1016/0003-4975(95)00219-b. discussion S159. [DOI] [PubMed] [Google Scholar]
- Cosmi JE, Kort S, et al. The risk of the development of aortic stenosis in patients with “benign” aortic valve thickening. Arch Intern Med. 2002;162(20):2345–7. doi: 10.1001/archinte.162.20.2345. [DOI] [PubMed] [Google Scholar]
- Cushing MC, Mariner PD, et al. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22(6):1769–77. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PF. Mechanisms involved in endothelial responses to hemodynamic forces. Atherosclerosis. 1997;131 Suppl:S15–7. doi: 10.1016/s0021-9150(97)06118-2. [DOI] [PubMed] [Google Scholar]
- Deck JD. Endothelial cell orientation on aortic valve leaflets. Cardiovasc Res. 1986;20(10):760–7. doi: 10.1093/cvr/20.10.760. [DOI] [PubMed] [Google Scholar]
- El-Hamamsy I, Balachandran K, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. 2009;53(16):1448–55. doi: 10.1016/j.jacc.2008.11.056. [DOI] [PubMed] [Google Scholar]
- Faggiano P, Antonini-Canterin F, et al. Progression of aortic valve sclerosis to aortic stenosis. Am J Cardiol. 2003;91(1):99–101. doi: 10.1016/s0002-9149(02)03011-4. [DOI] [PubMed] [Google Scholar]
- Filip DA, Radu A, et al. Interstitial cells of the heart valve possess characteristics similar to smooth muscle cells. Circulation Research. 1986;59(3):310–320. doi: 10.1161/01.res.59.3.310. [DOI] [PubMed] [Google Scholar]
- Grande-Allen KJ, Calabro A, et al. Glycosaminoglycans and proteoglycans in normal mitral valve leaflets and chordae: association with regions of tensile and compressive loading. Glycobiology. 2004;14(7):621–33. doi: 10.1093/glycob/cwh076. [DOI] [PubMed] [Google Scholar]
- Gupta V, Grande-Allen KJ. Effects of static and cyclic loading in regulating extracellular matrix synthesis by cardiovascular cells. Cardiovasc Res. 2006;72(3):375–83. doi: 10.1016/j.cardiores.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Gupta V, Werdenberg JA, et al. Synthesis of glycosaminoglycans in differently loaded regions of collagen gels seeded with valvular interstitial cells. Tissue Eng. 2007;13(1):41–9. doi: 10.1089/ten.2006.0091. [DOI] [PubMed] [Google Scholar]
- Haskett D, Johnson G, et al. Age related and location dependent microstructural and biomechanical characterization of human aortas. Proceedings of the ASME 2009 Summer Bioengineering Conference; Lake Tahoe, CA. 2009. [Google Scholar]
- Jian B, Narula N, et al. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75(2):457–65. doi: 10.1016/s0003-4975(02)04312-6. discussion 465-6. [DOI] [PubMed] [Google Scholar]
- Kershaw JD, Misfeld M, et al. Specific regional and directional contractile responses of aortic cusp tissue. J Heart Valve Dis. 2004;13(5):798–803. [PubMed] [Google Scholar]
- Leask RL, Jain N, et al. Endothelium and valvular diseases of the heart. Microsc Res Tech. 2003;60(2):129–37. doi: 10.1002/jemt.10251. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams R, et al. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- Marron K, Yacoub MH, et al. Innervation of human atrioventricular and arterial valves. Circulation. 1996;94(3):368–75. doi: 10.1161/01.cir.94.3.368. [DOI] [PubMed] [Google Scholar]
- Merryman WD. Bioengineering Doctoral Dissertation, University of Pittsburgh PhD. 2007. Mechanobiology of the aortic valve interstitial cell; p. 172. [Google Scholar]
- Merryman WD. Insights into (the interstitium of) degenerative aortic valve disease. J Am Coll Cardiol. 2008;51(14):1415. doi: 10.1016/j.jacc.2007.11.068. [DOI] [PubMed] [Google Scholar]
- Merryman WD, Huang HY, et al. The effects of cellular contraction on aortic valve leaflet flexural stiffness. J Biomech. 2006;39(1):88–96. doi: 10.1016/j.jbiomech.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Merryman WD, Liao J, et al. Differences in tissue-remodeling potential of aortic and pulmonary heart valve interstitial cells. Tissue Eng. 2007;13(9):2281–9. doi: 10.1089/ten.2006.0324. [DOI] [PubMed] [Google Scholar]
- Merryman WD, Lukoff HD, et al. Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast. Cardiovasc Pathol. 2007;16(5):268–76. doi: 10.1016/j.carpath.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merryman WD, Youn I, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. Am J Physiol Heart Circ Physiol. 2006;290(1):H224–31. doi: 10.1152/ajpheart.00521.2005. [DOI] [PubMed] [Google Scholar]
- O'Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more) Arterioscler Thromb Vasc Biol. 2006;26(8):1721–8. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- Rabkin-Aikawa E, Farber M, et al. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13(5):841–7. [PubMed] [Google Scholar]
- Robicsek F, Thubrikar MJ, et al. Cause of degenerative disease of the trileaflet aortic valve: review of subject and presentation of a new theory. Ann Thorac Surg. 2002;73(4):1346–54. doi: 10.1016/s0003-4975(01)03001-6. [DOI] [PubMed] [Google Scholar]
- Sacks MS, Smith DB, et al. The aortic valve microstructure: effects of transvalvular pressure. Journal of Biomedical Materials Research. 1998;41(1):131–41. doi: 10.1002/(sici)1097-4636(199807)41:1<131::aid-jbm16>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Schoen F. Aortic valve structure-function correlations: Role of elastic fibers no longer a stretch of the imagination. J Heart Valve Dis. 1997;6:1–6. [PubMed] [Google Scholar]
- Simmons CA, Grant GR, et al. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96(7):792–9. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BF, Siscovick D, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29(3):630–4. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- Thubrikar M. The Aortic Valve. Boca Raton: CRC; 1990. [Google Scholar]
- Vesely I. The role of elastin in aortic valve mechanics. J Biomech. 1998;31(2):115–123. doi: 10.1016/s0021-9290(97)00122-x. [DOI] [PubMed] [Google Scholar]
- Weston MW, LaBorde DV, et al. Estimation of the shear stress on the surface of an aortic valve leaflet. Ann Biomed Eng. 1999;27(4):572–9. doi: 10.1114/1.199. [DOI] [PubMed] [Google Scholar]
- Weston MW, Yoganathan AP. Biosynthetic activity in heart valve leaflets in response to in vitro flow environments. Annals of Biomedical Engineering. 2001;29(9):752–63. doi: 10.1114/1.1397794. [DOI] [PubMed] [Google Scholar]
- Taking Care: Ethical Caregiving in Our Aging Society. The President's Council on Bioethics; 2005. p. 309. www.bioethics.gov. [Google Scholar]
- Yip CY, Chen JH, et al. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29(6):936–42. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]