

Abstract
Transposable elements (TEs) are mobile DNA sequences that make up a large fraction of eukaryotic genomes. Recently it was discovered that PIWI-interacting RNAs (piRNAs), a class of small RNA molecules that are mainly generated from transposable elements, are crucial repressors of active TEs in the germline of fruit flies. By quantifying expression levels of 32 TE families in piRNA pathway mutants relative to wild-type fruit flies, we provide evidence that piRNAs can severely silence the activities of retrotransposons. We incorporate piRNAs into a population genetic framework for retrotransposons and perform forward simulations to model the population dynamics of piRNA loci and their targets. Using parameters optimized for Drosophila melanogaster, our simulation results indicate that (1) piRNAs can significantly reduce the fitness cost of retrotransposons; (2) retrotransposons that generate piRNAs (piRTs) are selectively more advantageous, and such retrotransposon insertions more easily attain high frequency or fixation; (3) retrotransposons that are repressed by piRNAs (targetRTs), however, also have an elevated probability of reaching high frequency or fixation in the population because their deleterious effects are attenuated. By surveying the polymorphisms of piRT and targetRT insertions across nine strains of D. melanogaster, we verified these theoretical predictions with population genomic data. Our theoretical and empirical analysis suggests that piRNAs can significantly increase the fitness of individuals that bear them; however, piRNAs may provide a shelter or Trojan horse for retrotransposons, allowing them to increase in frequency in a population by shielding the host from the deleterious consequences of retrotransposition.
Transposable elements (TEs) are mobile DNA sequences that make up a large fraction of eukaryotic genomes, contributing 45% of the human and 5.3% of the fruit fly genomes (Lander et al. 2001; Quesneville et al. 2005; Drosophila 12 Genomes Consortium et al. 2007). Due to their high copy number, TEs have had profound effects on the structure, content, and evolution of genomes (Biemont et al. 1999; Kidwell and Holyoake 2001; Kazazian 2004; Ashburner and Bergman 2005; Biemont and Vieira 2005; Bergman et al. 2006). TEs can mediate evolution of genome structures through their tendency to nucleate chromosomal rearrangements (Hoogland and Biemont 1996; Petrov et al. 2003; Biemont and Vieira 2006), their contribution to the creation of new genes (Britten 2006; Yang et al. 2008; Kaessmann et al. 2009), and their de novo generation of new regulatory motifs for neighboring genes (Lowe et al. 2007). TE insertions can be adaptive through their influence on gene expression levels (Daborn et al. 2002; Brookfield 2004; Schlenke and Begun 2004; Aminetzach et al. 2005; Gonzalez et al. 2008), and recently it was demonstrated that such adaptive TE insertion events have occurred often in the genome of Drosophila melanogaster (Gonzalez et al. 2008).
Like other kinds of mutations, however, most mutations created by TE insertions are deleterious to the host and are thus selected against. Approximately 50%–80% of mutations arising in D. melanogaster can be attributed to TEs (Finnegan 1992; Ashburner et al. 2004). Based on the copy number of TEs in D. melanogaster, it was estimated that TEs can decrease the fitness of hosts by 0.4%–5% (Eanes et al. 1987; Charlesworth and Langley 1989; Mackay et al. 1992; Pasyukova et al. 2004). The fitness costs of TEs are generally mediated through the following mechanisms: (1) TE insertions disrupt genes (Charlesworth and Charlesworth 1983; Finnegan 1992; McDonald et al. 1997); (2) transcription and translation of TE-encoded genes are costly (Brookfield 1991; Nuzhdin 1999); and (3) ectopic recombination among dispersed and heterozygous TEs creates deleterious chromosomal rearrangements (Montgomery et al. 1987; Langley et al. 1988; Charlesworth and Langley 1989; Petrov et al. 2003). It has been demonstrated that different TE families are regulated by different mechanisms, although these mechanisms are not mutually exclusive (Biemont et al. 1994; Carr et al. 2002; Petrov et al. 2003). Despite their being under strong selective pressure, TEs still persist in genomes because they replicate rapidly (Ohta 1983). Decades of theoretical and experimental research have established a framework for the population genetics of TEs, after making nearly universal assumptions of equilibrium between transposition/retrotransposition, excision, migration, recurrent horizontal transfers, genetic drift, and natural selection (Charlesworth and Charlesworth 1983; Charlesworth 1988; Charlesworth and Langley 1989; Biemont 1992; Brookfield and Badge 1997; Charlesworth et al. 1997; Nuzhdin 1999; Bartolome et al. 2002; Brookfield 2005; Le Rouzic and Deceliere 2005).
Various mechanisms of repression of TE activities have been incorporated into our understanding of the population genetics of TEs, including self-regulation of copy number by reducing transposition rates (Charlesworth and Charlesworth 1983; Langley et al. 1983), cis-acting regulation (transposition immunity) and trans-acting regulation (transposition repression) of TEs (Charlesworth and Langley 1986), regulation of transposition by host factors (Badge and Brookfield 1998), or more specifically, regulation of transposition by the interaction between TEs and the host genome such as the P-M hybrid dysgenesis system (Engels 1986; Boussy et al. 1988; Brookfield 1991; Coen et al. 1994; Quesneville and Anxolabehere 1997, 1998) or the I-R hybrid dysgenesis system (Proust et al. 1992; Chaboissier et al. 1995; Jensen et al. 1995, 2002). Recently, it was discovered that PIWI-interacting RNAs (piRNAs), a class of small (26–31 bp) RNA molecules, are crucial repressors of active TEs in the germlines of fruit flies and worms (Aravin et al. 2001, 2007; Nishida and Siomi 2006; Vagin et al. 2006; Brennecke et al. 2007, 2008; Nishida et al. 2007; Yin and Lin 2007; Das et al. 2008; Ghildiyal et al. 2008; Girard and Hannon 2008; Siomi and Siomi 2008; Li et al. 2009; Malone and Hannon 2009; Malone et al. 2009). In Drosophila, three paralogs of the PIWI family—PIWI, AUB, and AGO3—are crucial components of the RNA-induced silencing complex (RISC). RISC incorporating a piRNA can bind and cleave RNAs that have complementary sequences to the piRNA. Using an RNA-seq method, Brennecke et al. (2007) comprehensively sequenced small RNAs bound to the three PIWI proteins and identified 142 piRNA loci in the genome of D. melanogaster. They discovered that the piRNA loci were mainly comprised of disrupted transposable elements, and more interestingly, were located in genome regions biased toward heterochromatin. They proposed that piRNAs are produced from inactive TEs through a ping-pong mechanism and form a surveillance system against active TE invasion (Brennecke et al. 2007). Interestingly, only a small fraction of piRNAs discovered by Brennecke et al. (2007) were recovered in the study of deep sequencing PIWI-bound rasiRNAs by Yin and Lin (2007), which reflects the complexity of piRNA populations.
Genes encoding small RNAs, like other kinds of genes, should experience a history of birth, persistence, and death. It was recently demonstrated that microRNAs, a class of small RNAs that are generally conserved, stand as a dynamic class of genes in terms of birth and death at both the macro- and microevolutionary levels (Allen et al. 2004; Fahlgren et al. 2007; Grimson et al. 2008; Lu et al. 2008a,b). It was also found that the piRNAs are evolutionarily dynamic in rodents (Assis and Kondrashov 2009) as well as in Drosophila (Malone et al. 2009). In Drosophila, transposition/retrotransposition and excision of TEs occur frequently, and TE insertions are highly polymorphic among individuals (Montgomery and Langley 1983; Charlesworth and Langley 1989; Nuzhdin 1999; Petrov et al. 2003; Gonzalez et al. 2008). Since the majority of piRNAs were generated from TE loci, one would expect that piRNAs would similarly experience rapid population dynamics. Previous studies have surveyed population dynamics of individual piRNA loci such as Su(Ste) (Lyckegaard and Clark 1989; Kalmykova et al. 1998), flamenco (Bucheton 1995), and the I element (Chambeyron et al. 2008). Due to their recent discovery, piRNAs as a class of master regulators have yet to be considered in the framework of the theoretical population genetics of TEs.
In this study, we first demonstrate that activities of a large number of retrotransposons are severely silenced by piRNAs. Next, we carry out forward simulations and empirical data analysis to investigate the population dynamics of retrotransposons that generate piRNAs (piRTs) and retrotransposons that do not generate piRNAs but are repressed by piRNAs (targetRTs) in D. melanogaster. Our theoretical and empirical results indicate that piRNAs can significantly increase the fitness of organisms by reducing the fitness cost of retrotransposons. However, targetRTs also have a higher probability of reaching high frequency or fixation because their deleterious effects are (partially) attenuated. Once retrotransposons attain high frequency in the population, the host can only evade the deleterious consequences of frequent retrotransposition if the piRNA can successfully continue to repress the retrotransposons.
Results
Activities of retrotransposons are severely silenced by piRNAs
There are about 120 TE families that have been characterized in the reference genome of D. melanogaster (Kaminker et al. 2002; Quesneville et al. 2005). In the annotation of FlyBase (Release 5.13), there are 100 retrotransposon families, 21 transposon families, and one family of INE-1 whose biology remains largely unknown (Quesneville et al. 2005). We quantified expression levels of 32 TE families (26 retrotransposon and six transposon families) in the ovaries of one wild-type and three piRNA mutants with real-time PCR. The three mutants include one piwi allele (piwi06843/CyO), and two aub alleles (aubQC42/CyO and aubHN/CyO). Our PCR primers can potentially detect 486 TEs annotated in the reference genome, and the majority of them are putatively autonomous or virulent (Methods). For the 26 families of retrotransposons tested, expression levels were significantly elevated in the heterozygous piRNA mutants compared to the wild type. The magnitude of the expression increase was approximately twofold in both piwi06843/CyO and aubQC42/CyO and ∼10-fold in aubHN/CyO (Fig. 1) (P < 0.0001 in all the three cases; paired t-tests were used to compare the original Ct values). Mixed results were obtained for the six families of transposons tested in piwi06843/CyO and aubQC42/CyO. However, in aubHN/CyO, we still observed increased expression levels (ranging from approximately twofold to 16-fold) across all six transposon families (Fig. 1). (In Supplemental Figs. S1 and S2 and Supplemental Methods, we provide evidence that the elevated expression of TE mRNAs in the mutants was not due to the bias in the copy number of TEs.) The differences among transposons and retrotransposons in piRNA repression efficiency might be shaped by natural selection. The activities of retrotransposons are mainly mediated at the RNA level, while transposons move by a “cut-and-paste” mechanism, and therefore the expression levels of transposon-encoded genes might not be a crucial factor for their activity.
Figure 1.

Fold changes of TE mRNAs in piRNA pathway mutants relative to the wild type. The x-axis provides the names of the 32 TE families tested, and the y-axis is the log2 value of the fold changes. (A) piwi06843/CyO; (B) aubQC42/CyO; (C) aubHN/CyO. In each panel, the horizontal line at 0 represents the case when the expression level is the same in the mutants and wild-type flies. Three replicates were performed, and the average values ± standard errors are presented.
In addition to our real-time PCR results, other studies also demonstrated that piRNAs can repress activities of retrotransposons such as gypsy, Idefix, ZAM, roo, mdg1, copia, I element, and Het-A (Sarot et al. 2004; Vagin et al. 2006; Klenov et al. 2007; Mevel-Ninio et al. 2007; Pelisson et al. 2007; Brennecke et al. 2008; Chambeyron et al. 2008; Desset et al. 2008). Using whole-genome tiling microarrays and quantitative RT-PCR, Li et al. (2009) found that expression levels of a large number of retrotransposon families are up-regulated in the absence of AGO3. Our study, together with others, suggests that activities of a large number of retrotransposons are severely silenced by piRNAs, while mixed results were obtained for transposons. In this study, we focus on the population dynamics of retrotransposons only.
Forward simulations of the population genetics of piRTs and targetRTs
To model the population dynamics of retrotransposons that can generate piRNAs (piRTs) as well as the retrotransposons that do not generate piRNAs but are repressed by piRNAs (targetRTs), we performed forward simulations. The simulation processes are similar to those proposed by Dolgin and Charlesworth (2008) except that we incorporate piRNAs into the population dynamics and we only focus on retrotransposons. We assume that
The population is diploid, panmictic, constant-sized, and has no overlapping generations.
We only consider one chromosome with a size (L) of 40 Mb, which is close to the actual size of chromosomes 2 and 3 of D. melanogaster.
The recombination rate is 2.5 × 10−8 per nucleotide per generation so that in each generation one chromosome will have roughly one crossover.
- Although different retrotransposon families impose detrimental effects on the host through different mechanisms (Charlesworth and Langley 1989; Carr et al. 2002; Petrov et al. 2003), the fitness (w) of a chromosome overall can be modeled by a linear function,
where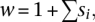
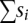 is the sum of the selective effects (si < 0) of all the retrotransposons carried on one chromosome (Le Rouzic and Capy 2006), or modeled by an exponential quadratic function,
is the sum of the selective effects (si < 0) of all the retrotransposons carried on one chromosome (Le Rouzic and Capy 2006), or modeled by an exponential quadratic function,
where a and b are constants and n is the number of retrotransposons (Charlesworth 1990). We use the latter function in our simulations.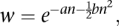
piRNAs are generated from long precursors, and any retrotransposons inserted into the piRNA-generating regions will be recruited by the piRNA biogenesis machinery and thus will result in suppression of activities of the retrotransposons.
4.2% of the chromosomal genes are potential piRNA-generating regions as estimated by the size of the 142 piRNA loci in the genome of D. melanogaster.
There is no sequence divergence between paralogous copies of retrotransposons so that one piRNA can potentially repress all the retrotransposons.
Inside a cell, piRNAs from one gamete can silence retrotransposons of both gametes.
Retrotransposons located inside piRNA loci lose the ability to retrotranspose.
For a retrotransposon located outside piRNA regions, its retrotransposition rate is u1 if the piRNA is not expressed in the cell; it is u2 if the piRNA is expressed in the cell.
The excision rate is v.
The insertion of retrotransposons does not change the chromosome size.
Ectopic recombination was assumed to be strongly deleterious, and products of ectopic recombination are rapidly eliminated from the population (for more details, see Methods).
The following parameter settings in our simulations are crucial for the biological outcomes: (1) the effective population size Ne, (2) the retrotransposition rate u1 and u2, (3) the excision rate v, and (4) the constants a and b in the exponential quadratic function of fitness. The effective population size of D. melanogaster has been estimated to be between 106 (Kreitman 1983) and 105 (Schug et al. 1998). The retrotransposition/transposition rates of TEs vary greatly across families in D. melanogaster, ranging from 0 to 3.94 × 10−3 per element per generation (Ising and Block 1981; Biemont and Aouar 1987; Biemont et al. 1990; Harada et al. 1990; Nuzhdin and Mackay 1994, 1995; Suh et al. 1995; Maside et al. 2000, 2001). (Rates estimated from previous studies are summarized in Supplemental Table S1.) The retrotransposition/transposition rate estimates for individual TE families often differ among studies, but the genomic average retrotransposition/transposition rate is usually within the range of 1.15 × 10−4 to 3.5 × 10−4 per element per generation (summarized in Supplemental Table S2), with the excision rate one or two orders of magnitude lower (Eggleston et al. 1988; Harada et al. 1990; Nuzhdin and Mackay 1995; Nuzhdin et al. 1997; Maside et al. 2000). In natural populations of D. melanogaster, the selective intensity against a segregating TE has been estimated to be on the order of 10−5–10−4 per element, thus in the quadratic function of fitness, a is usually set at 10−5 and b is in the range of 10−6–10−5 (Charlesworth et al. 1994; Dolgin and Charlesworth 2006, 2008). In our simulations we followed the parameter estimates for D. melanogaster used by Dolgin and Charlesworth (2008) and set Ne = 105 (or 5 × 104), u1 = 10−4, a = 10−5, and v = 0 (or 10−6), with b varying between 10−6 and 10−5. We also set Ne = 106, u1 = 3 × 10−4, a = 10−5, and v = 0 (or 3 × 10−6), and b varying between 10−6 and 10−5, based on the effective population size estimation by Kreitman (1983) and retrotransposition/excision rates estimated by previous studies (Supplemental Tables S1, S2). To expedite the simulation processes, we scaled up the retrotransposition/excision rates and selection parameters (u1, u2, v, a, and b) by 100-fold and scaled down the effective population size (Ne) and generation by 100-fold. (The recombination rate r is not scaled so that one chromosome will roughly have one crossover in each generation.) This reciprocal scaling technique does not affect the evolutionary consequences, as demonstrated and validated by previous studies (Dolgin and Charlesworth 2006, 2008; Le Rouzic et al. 2007; Soderberg and Berg 2007).
The simulations were performed under four different scenarios with regard to the repressing capabilities of piRNAs on their target retrotransposons. In scenario I, piRNAs have no repression effect on the activities of their targets (u1 = u2). Thus, scenario I serves as a reference and is a canonical population genetic framework based on the assumption of equilibrium between retrotransposition and natural selection. In scenarios II, III, and IV, we assume the existence of piRNAs that reduce retrotransposition rates to 10%, 1%, and 0.1% of their original rates, respectively (i.e., u2 is 0.1u1, 0.01u1, and 0.001u1 under scenarios II, III, and IV). The rates of recombination, retrotransposition, and excision are all modeled as Poisson processes.
piRNAs significantly reduce the fitness cost of retrotransposons
After 1000 generations, the number of retrotransposons carried by each chromosome reaches equilibrium under all parameter combinations used. In Figure 2A, we present the simulation results by varying u2 while keeping other parameters fixed. The parameters after scaling are set as follows: Ne = 1000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, and v = 0. The corresponding unscaled parameters are Ne = 105, a = 10−5, b = 5 × 10−6, r = 2.5 × 10−8, u1 = 10−4, and v = 0. To obtain the biologically relevant values, one should use the unscaled parameters and scale up the generation number in our simulations by 100-fold. Under scenario I, where piRNAs are assumed not to have any impact on the activities of the target retrotransposons (u2 = 0.01 after scaling), at equilibrium each chromosome on average bears 16.4 retrotransposons with a 90% confidence interval of (14.4–18.4). In scenario II, where piRNAs are assumed to repress the retrotransposons to 10% of their original retrotransposition rates (u2 = 0.001 after scaling), the number of retrotransposons on each chromosome at equilibrium is 6.4 with 90% CI (1.4–10). In other words, piRNAs can reduce the number of retrotransposons persisting in the genomes by 60%. Correspondingly, in our simulations (a = 0.001 and b = 0.0005 after scaling), the fitness of a chromosome imposed by the retrotransposons increases from 0.92 under scenario I to 0.98 under scenario II, an increase of 6.5% in each generation (Fig. 2B). However, since a and b are scaled up 100-fold in our simulations, the effect of piRNAs on host fitness improvement is also upscaled by 100-fold. With the unscaled settings (a = 10−5 and b = 5 × 10−6), the fitness of a chromosome imposed by the retrotransposons increases from 0.99916 under scenario I to 0.99983 under scenario II, which suggests that piRNAs can increase the host fitness by 0.067% in each generation in natural populations (Fig. 2C). Since only one family of retrotransposons on one chromosome was considered in our simulations, piRNAs can potentially improve the fitness of hosts by 0.9998330/0.9991630 − 1 = 2.03% in each generation if the genome hosts 30 similarly active retrotransposon families and there are no synergistic effects between families. The effect of piRNAs on host fitness improvement might be conservative here because (1) in our simulation we only consider one chromosome which accounts for 25%–30% of the genome size of D. melanogaster, (2) the number of active retrotransposon families might be greater than 30 in the genome (Kaminker et al. 2002; Quesneville et al. 2005), and (3) piRNAs might reduce the retrotransposition rates by more than 10-fold (Das et al. 2008). More pronounced effects of piRNAs were observed if we increase the effective population size by 10-fold and the retrotransposition rates by threefold. (The scaled parameters are Ne = 10,000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.03, and v = 0 in our simulation, and the corresponding unscaled parameters are Ne = 106, a = 10−5, b = 5 × 10−6, r = 2.5 × 10−8, u1 = 3 × 10−4, and v = 0.) After reaching equilibrium, the average number of retrotransposons carried by one chromosome decreases from 45 under scenario I to 10 under scenario II (Supplemental Fig. S3), and under these parameter settings, piRNAs can potentially improve the fitness of hosts by 19.3% if the genome hosts 30 similarly active retrotransposon families and there are no synergistic effects between families. Interestingly, the number of retrotransposons carried by each chromosome does not reduce in scale with increasing piRNA silencing efficiency. Under scenarios III and IV, where piRNAs inhibit activities of TEs to 1% (u2 = 0.0001 after scaling) and 0.1% (u2 = 0.00001 after scaling) of the original levels, at equilibrium the number of retrotransposons carried by each chromosome is around five in both cases given the parameter settings in Figure 2. The reduction of fitness cost by piRNAs can be consistently observed if we incorporate excision (Supplemental Figs. S4, S5) or use different settings of parameter b (Supplemental Fig. S6).
Figure 2.

The number of retrotransposons carried by one chromosome and the fitness costs to the host. (A) The number of retrotransposons carried by one chromosome. (B) The fitness of the host using the scaled a and b values in the simulations. (C) The fitness of the host using the unscaled a and b values. In scenario I, piRNAs have no repression effect on the activities of their targets. In scenarios II, III, and IV, piRNAs reduce a retrotransposon's retrotransposition rates to 10%, 1%, and 0.1% of its original rate, respectively. The solid line in each panel is the mean value, and the thin dashed lines represent the upper and lower 90% confidence bounds (Ne = 1000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, v = 0, after scaling for all the four scenarios, where Ne is the effective number of individuals, a and b are constants in the quadratic fitness function, u1 is the retrotransposition rate for each retrotransposon outside piRNA regions in the cells where piRNA is not expressed, v is the excision rate, and r is the recombination rate per nucleotide per generation; u2 is the retrotransposition rate for each retrotransposon outside piRNA regions in the cells where piRNA is expressed, u2 = 0.01, 0.001, 0.0001, and 0.00001, after scaling for scenarios I, II, III, and IV, respectively). In C, a = 10−5 and b = 5 × 10−6 were used in the quadratic fitness function.
piRTs are selectively more advantageous than targetRTs
In the above simulations (Fig. 2; Supplemental Figs. S3–S6), we assume that piRTs (retrotransposons that generate piRNAs) and targetRTs (retrotransposons that are repressed by piRNAs and outside piRNA loci) contribute equally to the fitness costs as defined by the exponential quadratic function. The reduction of fitness cost by piRNAs is more pronounced when we assume only targetRTs contribute to the fitness costs and piRTs are selectively neutral (Supplemental Fig. S7). The mechanism of piRNAs repressing the active targetRTs is essentially compatible with the “transposition repression” model proposed by Charlesworth and Langley (1986), a model that showed that the selective advantage of regulation of repressors can be promoted by a sufficiently high frequency of dominant lethal or sterile mutations associated with transpositions in diploid organisms. In our simulations, even when we assume a piTR insertion confers the same degree of fitness cost to its host as does a targetRT insertion, we still find that fixation probabilities differ in piRT and targetRT insertions because piRTs can repress targetRTs and are thus selectively more advantageous. In Figure 3, we plot the proportion of piRTs out of the total number of retrotransposons carried on each chromosome over generations (Ne = 500, a = 0.001, b = 0.0005, u1 = 0.01, v = 0 after scaling). Under scenario I, where piRTs have no impact on targetRTs (u2 = 0.01 after scaling), the proportion of piRTs does not change after reaching equilibrium, while under scenarios II–IV, where piRNAs inhibit the activities of targetRTs (u2 = 0.001, 0.0001, and 0.00001 after scaling, respectively), the proportions of piRTs increase steadily over time (the slope of regression of piRT proportion on generation number is greater than 0 for scenarios II, III, and IV, respectively; P < 10−15 for all three scenarios; the trend becomes weaker when larger population size was used) (Supplemental Figs. S8, S10).
Figure 3.

The dynamics over time (generations) of the mean proportion (%) of all retrotransposons that are piRTs (Ne = 500, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, v = 0, after scaling for all four scenarios; u2 = 0.01, 0.001, 0.0001, and 0.00001, after scaling for scenarios I–IV, respectively).
The frequency spectrum of TE insertions is a useful metric to understand the evolutionary forces underlying the insertions (Langley et al. 1983; Montgomery and Langley 1983; Capy et al. 1991; Petrov et al. 2003; Neafsey et al. 2004; Gonzalez et al. 2008, 2009). The relatively greater advantage of piRT over targetRT insertions is also manifested by the difference in the frequency spectra of new insertions. Given the same settings of parameters as in Figure 3, we observe that under scenario I, where piRNAs do not silence targetRTs at all, more than 90% of the piRT insertions persist in the population at a very low frequency (<10% of the population), and only rarely do they attain intermediate frequency or fixation (Fig. 4A). In contrast, under scenarios II–IV, ∼2%–4% of piRT insertions become fixed in the population. It is notable that insertions of piRTs with higher repression capacities tend to have a greater chance to reach high frequency or fixation—the proportion of piRT insertions that is fixed is 2%, 3.4%, and 4% of all new insertions when piRNAs reduce activities of targetRTs to 10%, 1%, and 0.1% of their original levels, respectively.
Figure 4.

(A) Retrotransposon insertions that generate piRNAs (piRT insertions) have a higher probability of fixation or of attaining high frequency in the population because of their relatively advantageous effects. (B) Retrotransposon insertions that are repressed by piRNAs (targetRT insertions) also have a higher probability to be fixed or attain high frequency, because their deleterious effects are (partially) alleviated by piRNAs. (Ne = 500, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, v = 0, after scaling for all four scenarios; u2 = 0.01, 0.001, 0.0001, and 0.00001, after scaling for scenarios I–IV, respectively, for both A and B.)
The suppressing effect of piRNAs can drive targetRT insertions to high frequency or fixation
It is intriguing that compared with scenario I, targetRT insertions under scenarios II–IV also have higher probabilities of achieving elevated frequency or fixation (Fig. 4B). Under scenario I, no targetRT insertions can expand to 50% or higher frequency in the population, while under scenarios II–IV, ∼0.2%–0.5% of the total targetRT insertions can be fixed in the population (Fig. 4B). In other words, insertions of targetRTs that are repressed by piRNAs have a higher probability of reaching high frequency or fixation. A plausible argument is that under scenario I, where targetRTs are not repressed by piRNAs, retrotransposition of targetRTs is strongly selected against so that the TEs cannot reach high frequency in the population. Conversely, under scenarios II–IV, where targetRTs are repressed by piRNAs, the strong deleterious effects of targetRTs are (partially) alleviated because their retrotransposition capacity is impaired, so they could segregate like (nearly) neutrally evolving elements to drift to higher frequency or even fixation (Ohta 1973). However, under scenarios II–IV, the frequency spectra of targetRT insertions are still significantly skewed to lower frequencies compared to those of piRTs (P < 10−15 in all three cases, χ2 tests) (Fig. 4), which reveals a potentially negative consequence of piRNA repression.
The shifts in the frequency spectra for both piRTs and targetRTs are still observed when we use Ne = 1000 (after scaling) while keeping the other parameter settings at the similar levels as in Figures 3 and 4 (Supplemental Figs. S8, S9) or when we use the scaled parameter settings of Ne = 10,000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.03, and v = 0 (Supplemental Figs. S10, S11). However, given the generation numbers used in our simulations (15,000 generations for both Ne = 500 and 1000 after scaling, and 9000 generations for Ne = 10,000 after scaling), the difference in population dynamics between piRTs and targetRTs are more pronounced when Ne = 500 (after scaling) is used. This is because increasing the effective population size by a factor of 2 (or 20) will increase the expected sojourn time for a neutral mutation by a factor of two (or 20), and the generation numbers used in our simulations (for Ne = 1000 or 10,000 after scaling) are not large enough for the difference of piRT and targetRT insertions to be reliably manifested.
In summary, our simulation results indicate that (1) piRNAs significantly increase the fitness of organisms by silencing retrotransposons, but (2) retrotransposons that are silenced by piRNAs also have higher probabilities to attain high frequency or fixation in the population.
Population genomics of piRTs and targetRTs in Drosophila
Our theoretical analysis demonstrates the double-edged effects of piRNAs on the accumulation of retrotransposons. In this section, we test these predictions using population genomics data.
High-frequency piRT insertions observed in D. melanogaster
First, let us examine the evolutionary dynamics of piRTs (retrotransposons that generate piRNAs). A large number of piRNA loci are located in heterochromatic regions (Brennecke et al. 2007). However, since annotations of TEs are only available for the euchromatic regions, we only focus on piRTs located there. It is challenging to determine the retrotransposons in the Drosophila genome that are not repressed by piRNAs, i.e., the piRTs strictly under scenario I in our simulations. Our simulations indicate that relative to scenario I, both piRT insertions and targetRT insertions are driven to higher frequencies in scenarios II–IV; however, under all three latter scenarios, the frequency spectra of piRT insertions are significantly skewed to higher frequencies than the targetRT insertions (Fig. 4). Thus, rather than directly testing our predictions, we compared the frequency spectra of retrotransposon insertions that are located inside (piRTs) and outside the piRNA loci (targetRTs). Genome sequences of nine D. melanogaster strains obtained with light whole-genome shotgun 454 Life Sciences (Roche) pyrosequencing runs were used to survey the frequency of TEs (Sackton et al. 2009).
For each of the 5408 TEs annotated in the euchromatic regions of the D. melanogaster reference genome (y1; cn1bw1, sp1), we determined its presence/absence status in other strains by mapping the boundary sequences of TEs with shotgun sequencing results at two levels of precision (Methods). We excluded 344 TEs because their boundary sites cannot be unambiguously mapped on the reference genome, and thus the strategy we used cannot accurately determine the frequencies of the insertions in the nine strains (Methods). TE insertions are generally rare in the populations of D. melanogaster; however, some of them can be fixed or even conserved across divergent Drosophila species (Petrov et al. 2003; Caspi and Pachter 2006; Begun et al. 2007; Gonzalez et al. 2008). To exclude the possibility that the frequency spectrum is biased by genealogy, in this analysis we further exclude 835 TE insertions that putatively have conserved boundary sequences in Drosophila simulans (Methods). These 835 TE insertions might be formed before the split of D. melanogaster and D. simulans, or are trans-species polymorphisms in the two species, or are recently horizontally transferred between the two species.
Many TE insertions are present in the reference genome but not in any of the strains we investigated (Table 1; Supplemental Fig. S12). Overall, ∼52% of all the TE insertions are restricted only to the reference genome and are found in none of the nine strains we surveyed. Among the 4239 (5408 − 344 − 835 = 4239) TE insertions, 1695 (40%) are from the INE-1 family, which are highly abundant across the Drosophila genus, while little is known about their biology (Quesneville et al. 2005; Yang and Barbash 2008). Of the remaining TE insertions, 652 (15%) are from transposons and 1892 (45%) are from retrotransposons. The three classes of TE insertions exhibit distinct frequency spectra: the proportion of insertions that are restricted to the reference genome is ∼43% for INE-1, ∼54% for transposons, and ∼60% for retrotransposons (Table 1; Supplemental Fig. S12). For all 4239 TE insertions, TEs with length <500 nt (or 1000 nt) are significantly skewed to higher frequency than TEs >500 nt (or 1000 nt) (P < 10−15 in both cases, χ2 tests) (Supplemental Fig. S13, A vs. B; Supplemental Fig. S14, A vs. B). However, the overall difference seems to be contributed solely by retrotransposons (P < 10−15 in both cases, χ2 tests) (Supplemental Fig. S13, G vs. H; Supplemental Fig. S14, G vs. H), but by neither INE-1 nor transposons (Supplemental Fig. S13, C vs. D, E vs. F; Supplemental Fig. S14, C vs. D, E vs. F). To understand the mechanisms regulating distinct retrotransposon families, we followed the analysis of Carr et al. (2002) and contrasted the observed proportion of X-linked retrotransposon insertions against the expected values under various selection mechanisms (Supplemental Tables S3, S4). Our results suggest that the regulatory mechanisms against the retrotransposon insertions are not mutually exclusive for the majority of the families examined. Detailed analysis is presented in Supplemental material.
Table 1.
Frequency spectrum of TE insertions across nine strains of D. melanogaster

Among the 5408 TEs annotated in the reference genome (FlyBase R5.13), 344 TE insertions mapped on multiple locations and 835 insertions that have putatively homologous sequences in D. simulans were excluded. Frequency 0 means the TE insertion was only detected in the reference genome but in none of the nine strains surveyed. Frequencies 1–5 mean that the TE insertions exist in one to five out of the nine strains surveyed. No TE insertions were detected in more than six out of the nine strains.
For the 1892 retrotransposons, the 309 that are located inside piRNA loci defined by Brennecke et al. (2007) are treated as piRTs, and the remaining 1583 that are outside those loci are treated as targetRTs. (Many of the targetRTs might be nonautonomous or inactive, which makes our analysis conservative; see below for details.) The frequency spectra of piRT insertions are significantly skewed to higher frequencies than targetRT insertions (P = 2.5 × 10−15, χ2 test) (Supplemental Fig. S15, A vs. B). This pattern holds true if we only consider the retrotransposons that are >500 nt (P = 2 × 10−8, χ2 test) (Fig. 5, A vs. B) or >1000 nt (P = 6.2 × 10−10, χ2 test) (Supplemental Fig. S15, C vs. D). It is well known that different families of retrotransposons might be heterogeneous in their insertion history (Bergman et al. 2006; Bergman and Bensasson 2007). In Supplemental Table S5, we present the frequency spectra for retrotransposon insertions of the 60 families that have at least one insertion located inside piRNA loci (only retrotransposons >500 nt were considered here). Due to the small number of retrotransposon insertions within each family, we do not have enough statistical power to detect the difference in frequency spectra between insertions located inside and outside piRNA loci. However, these 60 families are likely to be sufficiently old in the genome of D. melanogaster since at least one member of each family had the chance to be inserted into the piRNA regions. The pooled result of the 60 families strongly suggests that piRT insertions are significantly skewed to higher frequencies than their counterparts outside piRNA loci (P = 1.6 × 10−8, χ2 test) (Supplemental Table S6). The difference between the frequency spectra is congruent with what we observed in the simulation—piRTs are selectively more advantageous than targetRTs. In our simulations, the relatively advantageous nature of piRTs over targetRTs can be manifested even when we assume piRTs are strictly neutral or deleterious. The frequency spectrum of piRT insertions observed in D. melanogaster might be shaped by compound effects of purifying selection (because they mediate ectopic recombination) and positive selection (because they repress active retrotransposons) or hitchhiking. However, we are not sure about the overall consequences of the compound effects on a piRT insertion, because whether it is overall under negative, neutral, or positive selection, the relatively advantageous nature of piRT relative to targetRT insertions can always be detected by the difference in the frequency spectra.
Figure 5.

Frequency spectra of retrotransposons and other TEs. (A vs. B) The frequency spectra of piRT insertions are significantly skewed to higher frequencies than those of the targetRT insertions (only retrotransposons longer than 500 nt are considered). The difference in frequency spectra is statistically significant in the regions where recombination occurred (C vs. D), but vanishes in regions where recombination does not occur (E vs. F). (G vs. H) The difference in frequency spectra of piRT versus targetRT insertions is statistically significant if we use the piRNA loci defined by both Brennecke et al. (2007) and Yin and Lin (2007). (BY) piRNA locus defined by both Brennecke et al. (2007) and Yin and Lin (2007). The numbers in parentheses are the number of retrotransposons in that category. The x-axis is the frequency of insertion out of the nine strains of D. melanogaster. “0” means the insertion is only restricted in the reference genome of D. melanogaster. The y-axis is the proportion (%) of retrotransposon insertions with distinct frequency across the nine strains.
The difference in frequency spectra of retrotransposon insertions is more significant in the regions where recombination is frequent (retrotransposons >500 nt were examined here; P = 2 × 10−9, χ2 test) (Fig. 5, C vs. D). In genomic regions without recombination events (r = 0), however, the relative advantage of piRTs over targetRTs vanishes—the frequency spectra of piRT and targetRT insertions are not statistically different (P = 0.70, χ2 test) (Fig. 5, E vs. F). In other words, we only observed piRTs to be selectively more advantageous than targetRTs in genomic regions that undergo recombination. The observation might be attributed to the Hill-Robertson effect, which argues that tight linkage limits the power of natural selection (both positive and purifying selection) (Hill and Robertson 1966). It is well known that in no (or low) recombination regions, the efficacy of purifying selection is reduced so that both transposons and retrotransposons can accumulate (Charlesworth and Langley 1989; Bartolome et al. 2002; Dolgin and Charlesworth 2008). In regions where recombination occurs, however, ectopic recombination occurs sufficiently frequently that retrotransposons are more intensively selected against. It should be noted, however, that both piRTs and targetRTs participate in ectopic recombination so that neither has an advantage over the other. As a result, the fitness advantage of piRTs relative to targetRTs is manifested only in genomic regions where recombination occurs.
Only a small fraction of piRNAs discovered by Brennecke et al. (2007) was recovered in deep sequencing of PIWI-bound rasiRNAs by Yin and Lin (2007). A plausible explanation to reconcile this discrepancy is that piRNA populations are complex (Li et al. 2009), the sequencing platforms used by both studies might have recovered a subset of piRNAs from each locus, and thus individual piRNAs obtained in both studies are not necessarily the same ones. Out of the 12,903 unique piRNA reads obtained by Yin and Lin (2007), we find that 8604 (67%) are mapped on the piRNA loci defined by Brennecke et al. (2007). In Figure 5, G and H, we show that RT insertions located inside piRNA loci defined by both Brennecke et al. (2007) and Yin and Lin (2007) also have a frequency spectrum that is significantly skewed to higher frequency compared to insertions outside piRNA loci defined by both studies (retrotransposons >500 nt were used in comparison, P = 0.0007, χ2 test) (Fig. 5, G vs. H).
Identifying retrotransposons targeted by piRNAs
Before we provide evidence that piRNAs can increase the frequency of retrotransposons in the population of D. melanogaster, we shall describe the procedure for identifying TEs that are targeted by piRNAs. We retrieved extensive sequencing results of piRNAs from ovaries of four strains of D. melanogaster, mapped them on the piRNA loci, normalized the number of reads, and calculated the mean read number for each piRNA species (Methods). The average read numbers are assumed to be the expression levels of piRNA species across the population of D. melanogaster. We predicted the targets of piRNAs by requiring the piRNAs to be perfectly complementary to the target TEs (all the 5408 TEs annotated in FlyBase R5.13 were used here). We assigned a score S to each TE with the formula
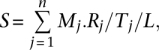 |
where n is the number of piRNA species that are perfectly complementary to this TE; Mj is the number of target sites of a piRNA species j on this TE; Rj is the read number of a piRNA species j (normalized to reads per million reads); Tj is the total number of target sites for piRNA species j; and L is the length of this TE (in kilobases). Thus, S is the density of piRNAs whose sequence is complementary to one TE, normalized by the length of the respective TE and corrected for multiple hits of the piRNAs. In our target prediction model, we not only took into account of the number of target sites, but also the expression levels of piRNAs. The S score is inversely related to the activities of TEs. A TE with a lower S score means that it has higher activity. In the extreme case of S = 0, that is, in the piRNA mutant background, the activities of autonomous TEs would be maximal.
Elevated frequencies of targetRT insertions due to piRNA silencing
For retrotransposons, the S scores are the densities of piRNAs targeting the targetRTs after correcting for differences in the lengths of the retrotransposons. However, we find that S is significantly positively correlated with the length of the 1583 putative targetRTs analyzed above in the section High-Frequency piRT Insertions Observed in D. melanogaster (the Pearson correlation coefficient is 0.23, P < 10−16 for all the targetRTs; the Pearson correlation coefficient is 0.11, P = 0.0007, for the 969 targetRTs that are >500 nt). One plausible explanation is that longer retrotransposons are generally more virulent and kept at low frequency in the population (Petrov et al. 2003), so that piRNAs that target such elements are selectively more advantageous to the host. In this case, such piRNAs are kept in greater abundance in the transcriptome. Nine-hundred-eighty-one (62%) of the 1583 targetRT insertions are restricted to just the reference genome of D. melanogaster, and the remaining 602 insertions can be found in at least one of the nine strains we surveyed. The 981 rare targetRTs are significantly longer than the 602 targetRTs that are more common (length is 3678 ± 107 nt for the former and 1319 ± 100 nt for the latter category, P < 10−16, Kolmogorov-Smirnov test). Thus, it is not surprising that the score S is significantly higher in the former than in the latter category (S is 75.2 ± 3.9 for the 981 targetRTs in the former and 33.9 ± 4.3 for the 602 targetRTs in the latter category; P < 10−16, Kolmogorov-Smirnov test).
One should keep in mind that the 1583 targetRTs (as well as other TEs in the genome) can be divided into three classes based on their retrotransposition capabilities: (1) autonomous, which have ORFs that encode the products required for retrotransposition and have full capabilities to be autonomously retrotransposed; (2) nonautonomous, which do not encode retrotransposition proteins but are able to retrotranspose with the aid of autonomous retrotransposons; and (3) inactive, which are inactive relics of the former two classes.
It is well documented that the three classes of retrotransposons have distinct evolutionary dynamics—the inactive retrotransposons might evolve like neutral elements, while autonomous retrotransposons, which are generally much longer and most virulent among the three classes, are strongly selected against (Petrov et al. 2003; Bergman and Bensasson 2007).
Since the targetRTs in our simulations are all autonomous and such elements are more intensively silenced by piRNAs (see below), we will only focus on the relationship between S scores and population frequency of the autonomous retrotransposons. Among the 5408 annotated TEs, 537 (520 retrotransposons and 17 transposons) have nearly intact open reading frames (ORFs), and therefore are considered putatively autonomous TEs (Methods). Four-hundred-sixty-two autonomous retrotransposon (34 piRT and 428 targetRT) insertions can be uniquely mapped to the reference genome of D. melanogaster and do not have conserved boundary sites in D. simulans (Methods). Four-hundred-thirty-two (93.5%) are found only in the reference genome and not in any of the nine strains we surveyed. Overall, the autonomous retrotransposon insertions have significantly lower frequencies than the nonautonomous or inactive retrotransposons (P < 10−16, χ2 test) (Supplemental Fig. S16, A vs. B). This pattern is observed even after we correct the possible bias caused by the length difference between the two categories (the length is 6994 ± 201 nt for the putative autonomous retrotransposons and only 1468 ± 61 nt for the nonautonomous and inactive retrotransposons; if we only consider nonautonomous retrotransposons with length >4500 nt so that the mean ± SE is 7038 ± 240 nt for this category, the same pattern is still observed: P < 10−16, χ2 test [Supplemental Fig. S16, B vs. C]). This comparison suggests that the putative autonomous retrotransposons are more virulent and are selected against because of their ability to retrotranspose. It is notable that the 38 putative autonomous piRT insertions have significantly higher frequencies than the 428 autonomous targetRT insertions (P < 10−10, χ2 test) (Supplemental Fig. S16, D vs. E), which is consistent with the frequency spectrum comparisons between the total piRT and targetRT insertions in the above section High-Frequency piRT Insertions Observed in D. melanogaster. Another salient observation is that autonomous targetRTs on average have higher S scores than the nonautonomous and inactive targetRTs (P < 10−16, Kolmogorov-Smirnov test) (Fig. 6, A vs. B). Since the average length is 6603 ± 125 nt for the autonomous targetRTs and 1365 ± 63 nt for the nonautonomous and inactive targetRTs (P < 10−16, Kolmogorov-Smirnov test), this observation is consistent with the positive correlation between S scores and the lengths of targetRTs as observed above.
Figure 6.

The boxplots of S scores for targetRTs (retrotransposons that are outside piRNA loci). (A) Nonautonomous/inactive retrotransposons. (B–H) Putatively autonomous targetRTs: (B) total putatively autonomous targetRTs; (C) rare autonomous targetRT insertions (the insertions only exist in the reference genome); (D) common autonomous targetRT insertions (the insertions exist in the reference genome and in at least one strain surveyed in this study or Gonzalez et al. 2008); (E) rare autonomous targetRT insertions in regions where recombination occurs; (F) common autonomous targetRT insertions in regions where recombination occurs; (G) rare autonomous targetRT insertions in nonrecombining regions; (H) common autonomous targetRT insertions in nonrecombining regions. The S scores of A–H are based on the small RNAs from the four piRNA libraries that are located inside the piRNA loci and perfectly antisense to TEs annotated in FlyBase R5.13 (see text). (I,J) S scores for all the putatively autonomous retrotransposons (inside and outside piRNA loci) with all the small RNAs in the four libraries (they can be inside piRNA loci or not). (I) The rare class; (J) the common class. The number of retrotransposon insertions in each category, the mean and standard error (SE) of S scores for each category appears below each boxplot. Kolmogorov-Smirnov tests were used to assess the statistical significance, and the P-values appear above the boxplots. In each boxplot, the minimum, 25%, 50%, and 75% quantiles, and the maximum S scores are represented by horizontal lines. The black dot is the mean S score. S scores greater than 500 are not plotted.
Only 18 (4.2%) of the 428 putative autonomous targetRT insertions are detected at least once in the nine strains we surveyed. To increase the statistical power of our analysis, we combined our data with the retrotransposon insertion frequencies determined by Gonzalez et al. (2008). Gonzalez et al. (2008) screened 902 TE insertions in the American and African populations of D. melanogaster based on PCR amplification. Among the 410 autonomous targetRT insertions that are not detected by the nine strains we surveyed, 124 are detected in at least one line of the American or African populations surveyed by Gonzalez et al. (2008). Thus, in the combined data set, 286 (410 − 124 = 286) autonomous targetRT insertions are found exclusively in the reference genome (referred to as the “rare” class), and 142 (18 + 124 = 142) are detected in at least one of the two studies (the “common” class).
It is compelling that the autonomous targetRTs in the common class have significantly higher S scores than those in the rare class (Fig. 6, C vs. D). The average S score is 97 for the 286 rare autonomous targetRTs and 136.4 for the 142 common autonomous targetRTs (P = 0.004, Kolmogorov-Smirnov test). The difference in the S score distribution is predominantly contributed by targetRTs in genomic regions where recombination occurs—the average S score is 100 for the 264 rare autonomous targetRTs and is 141 for the 132 common autonomous targetRTs (P = 0.0075, Kolmogorov-Smirnov test) (Fig. 6, G vs. H). In regions lacking recombination, the mean S score is 62 for the rare autonomous targetRTs and 73 for the common class (P = 0.95, Kolmogorov-Smirnov test) (Fig. 6, E vs. F). This observation is, once more, explained by the Hill-Robertson effect—only in recombining regions, where the efficacy of natural selection is not reduced, can the impact of piRNAs on the frequency spectrum of targetRT insertions be manifested. Thus, the positive correlation between the S scores and the population frequency of autonomous targetRTs validates our theoretical prediction that the repression effect of piRNAs on targetRTs will increase the chance that those targetRTs will reach higher frequencies. This observation is in strong contrast to the opposite pattern observed for the nonautonomous and inactive retrotransposons and makes our conclusions about the impact of piRNAs on the frequency spectrum of retrotransposons conservative.
In our piRNA target prediction, we only considered piRNAs that are perfectly antisense to targetRTs. However, imperfect piRNA:targetTE pairing can also lead to recognition and cleavage of target mRNAs. One well-documented example is Su(Ste), a piRNA locus capable of efficiently repressing the activity of Stellate, even though the sequence similarity between the two loci is ∼90% (Aravin et al. 2001). If we also allow up to three mismatches between piRNAs and the target sites, our observation still holds true (data not shown).
Discussion
In our theoretical modeling and empirical data analysis, we assume that piRNAs silence the activities of retrotransposons by antisense targeting their mRNAs. For a new retrotransposon insertion, it might be deleterious to the host, because (1) it disrupts genes, (2) its transcription and translation are costly, or (3) it mediates ectopic recombination. A piRNA cannot directly repress the ectopic recombination mediated by this newly inserted retrotransposon or the mutagenesis effects caused by this new insertion; however, a piRNA targeting this newly inserted retrotransposon can repress its activity and reduce its retrotransposition rate to other genomic regions, thereby potentially alleviating the fitness costs imposed by the retrotransposon. Recently it was demonstrated that piRNAs might direct the PIWI/SU(VAR)205 complex [SU(VAR)205 also known as HP1a] to piRNA-corresponding genomic regions to regulate the euchromatic histone modifications and hence the transcription activities of the target regions (Brower-Toland et al. 2007; Yin and Lin 2007; Lin and Yin 2008; Thomson and Lin 2009). It would be interesting to incorporate this new mechanism into our simulation model once we know more about the regulation of activities of retrotransposons and other classes of TEs by piRNAs through this mechanism.
The Ping-Pong model of piRNA biogenesis proposed by Brennecke et al. (2007) elegantly explains how piRNAs silence the target mRNAs and how piRNAs get amplified; however, the biogenesis mechanism of the original primary piRNAs is still not clear (Aravin et al. 2007). In our simulation model of piRNA biogenesis, we assume that once a retrotransposon has been inserted into a piRNA cluster, the retrotransposon will generate a piRNA. This assumption might be valid because the origin of piRNAs appears to have been rapid. For example, P-element-derived piRNAs were detected in D. melanogaster (Brennecke et al. 2008), although P-elements invaded the D. melanogaster genome only within the last 50 yr (Kidwell 1983).
In the above analysis, we only focused on piRNAs that are generated from the large piRNA loci defined by Brennecke et al. (2007) and updated by Malone et al. (2009). It is possible that some small individual loci would also generate piRNAs but are not covered by these defined piRNA loci. A second consideration is that siRNAs, which are ∼21 nt in length, can also silence transposable elements in somatic cells (Ghildiyal et al. 2008) and germline (Czech et al. 2008). Of the small RNAs retrieved from the four libraries (Table 2), a small fraction of them have a length of 21 nt (see Methods for details; also see Supplemental Fig. S17). Such possibilities are highlighted by 20 putatively autonomous retrotransposons (15 flea and five Transpac) with S scores of zero if we only consider small RNAs generated from the defined large piRNA loci. However, if we consider all the small RNAs obtained from the four aforementioned small RNA libraries, all 20 autonomous retrotransposons can be targeted by small RNAs with considerable S scores. Thus, we align all the small RNAs (the majority of them are piRNAs, and only a small fraction are siRNAs) that are perfectly antisense to all the TEs and calculate the S score for each TE using the same procedure as implemented above in the section Identifying Retrotransposons Targeted by piRNAs (Methods). The S score is significantly higher for the common class than the rare class (153 ± 13 vs. 113 ± 6, P = 0.0002, Kolmogorov-Smirnov test) (Fig. 6I,J) for all the autonomous retrotransposons. (Note that Figure 6, I and J, include insertions of both autonomous targetRTs and autonomous piRTs, because insertions of both targetRTs that have higher S scores and piRTs are predicted to attain higher frequencies in our simulations.)
Table 2.
Small RNA libraries from ovaries of four distinct strains of D. melanogaster

The impact of piRNAs on the population dynamics of piRTs and targetRTs modeled in our theoretical studies were confirmed by the frequency spectrum analysis of retrotransposon insertions in nine strains of D. melanogaster. One should keep in mind that population dynamics of retrotransposon insertions are influenced by compound regulatory mechanisms: including the three purifying selection mechanisms mentioned above, genetic drift, as well as positive selection associated with the retrotransposon insertions. Recently, Gonzalez et al. (2008) demonstrated that 13 TE insertions in D. melanogaster were positively selected. (Six of them are retrotransposon insertions: FBti0019170, FBti0019386, FBti0019430, FBti0019443, FBti0020046, and FBti0020091.) Excluding those elements does not influence any of our analysis. Thus, the patterns revealed by our empirical analysis are not likely to be biased by these positively selected retrotransposon insertions. In our frequency spectrum analysis, we did not consider the TEs specifically present in any of the nine strains but not in the reference genome, because the low coverage of shotgun sequencing we used does not admit such an analysis. Thus, the approach we used to detect TE insertion frequencies will be biased toward insertions at medium or high frequencies. Another consideration is that, due to the light coverage of genome sequences of the nine strains we surveyed, we might have failed to detect some insertions at medium/high frequencies. However, since each of the nine strains was sequenced randomly, we do not expect the frequency spectrum to be biased to any of the categories we analyzed. Currently there remain uncertainties about the impact of piRNAs, relative to other regulatory mechanisms, on the population dynamics of new insertions of retrotransposons. The ongoing population genomics projects of D. melanogaster, such as the Drosophila Genetics Reference Panel (DGRP) and Drosophila Population Genomics Project (DPGP), will provide a more comprehensive picture of the population dynamics of TEs.
It is remarkable that the 142 piRNA loci identified in D. melanogaster are significantly enriched in pericentromeric or telomeric heterochromatin (Supplemental Table S6; Brennecke et al. 2007). It is possible that chromatin structure plays an important role in defining piRNA clusters (Aravin et al. 2007). It is notable that recombination does not occur in heterochromatic regions. We found those TEs are significantly enriched in low recombination regions, even in euchromatin (Supplemental Fig. S18). When recombination occurs, piRTs are advantageous because they silence targetRTs, but also disadvantageous because they can mediate ectopic recombination (which has deleterious consequences). However, when recombination is absent, ectopic recombination does not occur so that the overall advantageous effects of piRNAs are maximally manifested. To quantify the advantageous effect, we performed forward simulations under scenarios III and V (Ne = 1000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, u2 = 0.0001, and v = 0 after scaling). Under scenario III, we assume that piRNAs can repress the activities of targetRTs to 1% of their original level and piRTs have the same recombination rates as targetRTs; while under scenario V, we used the same parameter settings as under scenario III except that we assume there is no recombination on the piRTs, so that piRTs do not contribute to the fitness cost of retrotransposons because they do not retrotranspose and they are not involved in ectopic recombination. At equilibrium, the mean number of retrotransposons carried on one chromosome is approximately six under scenario III, but less than two under scenario V, an ∼70% reduction (Fig. 7). Thus, we propose that the enrichment of piRTs in heterochromatic and low recombination regions might be shaped by natural selection. (Similar observations were made when we assume that recombination occurs in piRNA regions, but they have no fitness cost; see Supplemental Fig. S7 for details.)
Figure 7.

Simulation results indicate that reducing recombination rates of piRNA loci can greatly reduce the number of retrotransposons carried in the genome. Under scenario III, the piRNAs can repress the activities of targetRTs to 1% of their original level, and piRTs have the same recombination rates as targetRTs; under scenario V, the same parameter settings were used as under scenario III except that there was no recombination on the piRTs so that piRTs do not contribute to the fitness cost of retrotransposons because they do not retrotranspose or get involved into ectopic recombination. At equilibrium, the mean number of retrotransposons carried on one chromosome is approximately six under scenario III, while it is fewer than two under scenario V (Ne = 1000, a = 0.001, b = 0.0005, r = 2.5 × 10−8, u1 = 0.01, u2 = 0.0001, and v = 0, after scaling for both scenarios III and V).
Conclusions
In this study, we provide experimental evidence that piRNAs can silence a large number of retrotransposon families in Drosophila. Our theoretical and empirical analysis suggests that piRNAs can significantly increase the fitness of individuals that bear them; however, piRNAs may provide a shelter or Trojan horse for retrotransposons to increase in frequency in a population by shielding the host from the deleterious consequences of retrotransposition. Once the piRNAs attain high frequency, the host fitness then depends on the piRNAs to successfully continue to repress the elements, making retention of the piRNAs vital to the host.
Methods
Fly stocks
piRNA mutant stocks are one piwi mutant (P{PZ}piwi06843cn1/CyO;ry506) and two aub mutants (w1118;aubQC42cn1bw1/CyO,P{sevRas1.V12}FK1 and aubHNcn1bw1/CyO). The heterozygotes of the three mutant strains were used for real-time PCR analysis. The three mutant strains were ordered from the Bloomington Drosophila Stock Center with the accession numbers of 12225, 4968, and 8517, respectively. The wild type is an out-crossing strain of D. melanogaster, outbred5.
RNA extraction, cDNA synthesis, and real-time PCR
About 50 pairs of ovaries were dissected from the mutant and wild-type flies. RNA was isolated using TRIzol (Invitrogen) and reverse-transcribed with Oligo-dT (IDT DNA) by M-MLV Reverse Transcriptase (Promega). The resulting cDNAs were used as templates for real-time PCR. The experimental procedures followed the manufacturers' protocols. For each RNA sample, reverse transcription and real-time PCR were independently repeated three times. Primers of the 32 TE families were designed using Primer3 software (Rozen and Skaletsky 2000). The primer sequences were given in Supplemental Table S7. Our primer set can potentially amplify 486 unique TEs annotated in FlyBase R5.13. (375 of them are putative autonomous TEs that have alignable length >99% of the canonical proteins). The TEs that can be amplified with our primers are given in Supplemental Table S8. One housekeeping gene, RpL32 (also known as RP49), was used to normalize the cDNA concentrations. SYBR GreenER qPCR SuperMixes for ABI PRISM (Invitrogen) were used in real-time PCR reactions. All PCRs were run in an ABI 7000 Sequence Detection System (Applied Biosystems) with the following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 sec at 95°C and 1 min at 60°C. The computer program SDS 2.0 (Applied Biosystems) was used to analyze the real-time PCR data.
Simulation of population dynamics of piRTs and targetRTs
The forward simulation processes are similar to Dolgin and Charlesworth (2008) except we consider piRNAs in the model. The simulation procedures are fully described in Dolgin and Charlesworth (2008). To expedite the simulation process, we scaled the parameters by 100 times, as described in Results. The following are settings of parameters in the simulations:
The effective number of individuals (Ne) is 500, 1000, or 10,000 after scaling.
The recombination rate is r = 2.5 × 10−8 per nucleotide per generation so that one chromosome will have roughly one crossover in each generation.
u1 is the retrotransposition rate for a retrotransposon located outside piRNA regions when a piRNA is not expressed in the cell; it is 0.01 or 0.03 per element per generation after scaling.
u2 is the retrotransposition rate for a retrotransposon located outside piRNA regions when a piRNA is expressed in the cell; it is equal to u1, 0.1u1, 0.01u1, and 0.001u1 in scenarios I, II, III, and IV, respectively.
The excision rate (v) is 0, 0.001, 0.0001, or 0.0003 after scaling.
- The fitness (w) of a chromosome is
(Charlesworth 1990), where n is the number of TEs carried by the chromosome, a = 0.001 after scaling, and b is 0.005, 0.001, 0.0008, 0.0006, 0.0005, or 0.0003 after scaling in our simulations.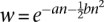
Simulations were initiated by randomly inserting a single transposable element on the non-piRNA-generating regions of each chromosome. Each generation, the two gametes of each offspring were sampled from the previous generation based on the fitness of the two gametes. The two gametes then recombine with the rates proportional to the physical distances of neighboring retrotransposons. Next, each retrotransposon retrotransposes at the rate u1 (or u2, depending on whether piRNA is expressed or not) per element per generation. Rates of recombination, retrotransposition, and excision are all modeled by Poisson distributions. The simulation was performed for 15,000 generations when scaled Ne is 500, or 1000, and 9000 generations when scaled Ne is 10,000. For every 10 generations, the number and frequencies of retrotransposons inside and outside piRNA loci were recorded. Each set of initial parameters was run in the simulation with 20–400 independent repetitions. For each run when Ne is 500 or 1000 after scaling, the frequency spectra of retrotransposon insertions from generations 13,000 to 15,000 were averaged and the mean value of the independent runs was obtained. Frequency spectra of retrotransposon insertions from generations 8000 to 9000 were summarized when Ne = 10,000 (after scaling) was used in the simulations.
Recombination rates of TEs were taken from Comeron and Kreitman (2002). All the simulations and statistical analyses were performed using the R environment (http://www.r-project.org). In testing the difference in frequency spectra of TE insertions, the frequency is binned into 0, 1, and >1 out of the nine strains we surveyed. A χ2 test (degrees of freedom [df] = 2) is used for a comparison between any two categories.
Polymorphisms of TE insertions
Sequences and annotations of TEs (FlyBase R5.13) were downloaded from the FlyBase website (http://www.flybase.net). Nine strains of D. melanogaster were sequenced by the Washington University Genome Center as part of an NHGRI pilot study (directed by D.L. Hartl and A.G. Clark) using the 454 Life Sciences (Roche) technique. Strains were North Carolina 301, 303, 306, 358, 375, 732, and Malawi 28-5, 56-4, and 63-5. The detailed sequencing procedure is described in Sackton et al. (in press). To determine TE insertion polymorphisms in the nine strains, we map all the 454 shotgun reads on the reference genome of D. melanogaster (FlyBase R5.13) using BLAT (Kent 2002). For each read, the highest score of BLAT alignment with the reference genome is recorded. If a read is mapped on more than one position with the highest BLAT score, it is removed in further analysis. The remaining shotgun reads that can be mapped on unique locations of the reference genome and covering at least one boundary site of a TE are used to infer the presence/absence of TE insertions. A TE insertion is determined to be present in the strain to which a shotgun read is assigned with the following strategies: (1) a shotgun read has ≥90 nt that can be perfectly mapped on the reference genome and at least 25 nt are mapped on both sides of a TE boundary site, or (2) a shotgun read has ≥100 nt mapped on the reference genome with the sequence identity ≥98% and at least 40 nt are mapped on both sides of a TE boundary site. Among the 5408 TEs annotated in FlyBase R5.13, 334 TEs are excluded from our analysis because they have flanking sequences (50 nt at each side) that can be perfectly mapped on multiple locations of the reference genome. The aligned length between the 454 shotgun reads and the reference genome is 111.7 ± 0.2 nt and the sequence identity is 99.66% ± 0.01% (Supplemental Fig. S19). A summary of the mapping results for the nine strains by Sackton et al. (2009) is presented in Table 1.
Mapping TE insertions in D. simulans
The shotgun reads of D. simulans were downloaded from ftp://ftp.ensembl.org/pub/traces/drosophila_simulans/. The shotgun reads were mapped on the reference genome of D. melanogaster using BLAST with an E cutoff value of 10−5. Only the reads that mapped to unique positions of D. melanogaster were used in this analysis. For a TE insertion annotated in D. melanogaster (R5.13), it is determined to be present in D. simulans if a shotgun read of D. simulans has ≥150 nt that can be best mapped on the genome of D. melanogaster with sequence ≥80% and at least 50 nt of the read is mapped on both sides of a TE boundary site. The aligned length between the shotgun reads and the reference genome is 415.1 ± 2.4 nt. The sequence identity is 91.19% ± 0.06% (Supplemental Fig. S20), which is close to the genomic average divergence between D. melanogaster and D. simulans (Begun et al. 2007; Drosophila 12 Genomes Consortium 2007). Eight-hundred-seventy-one TE insertions are conserved between D. melanogaster and D. simulans based on this criterion (36 TE insertions cannot be unambiguously mapped on D. melanogaster).
Identifying putative autonomous TEs
The 5408 TEs annotated consist of three categories in regard to their transposition/retrotransposition capabilities: (1) autonomous TEs, which have ORFs that encode the products required for transposition and have full capabilities to be transposed by themselves; (2) nonautonomous TEs, which do not encode transposition proteins but are able to transpose with the aid of autonomous TEs; and (3) inactive TEs, which are inactive relics of the former two classes. We identify putatively autonomous TEs based on whether they carry intact ORFs. Using the 140 protein sequences encoded by canonical transposable elements (Kaminker et al. 2002), we identified the ORFs from the 5408 TEs with the GeneWise2 program (Birney et al. 2004). The E-value cutoff was set at 10−6. TEs that have putatively functional ORFs with aligned protein length >99% of the canonical proteins were arbitrarily chosen as putative autonomous TEs. Five-hundred-thirty-seven putative autonomous TEs were identified by this set of criteria.
piRNA loci
Brennecke et al. (2007) sequenced small RNAs bound by PIWI, AUB, and AGO3 proteins from ovaries of D. melanogaster and defined 142 piRNA loci by using the piRNAs that can be uniquely mapped on the reference genome. Boundary regions of a subset of loci were updated in a recent study (Malone et al. 2009). Among the 5408 TEs annotated in FlyBase (R5.13), 680 are completely located inside (or overlapping at least 50 nt with) these piRNA loci. Yin and Lin (2007) sequenced 12,903 unique piRNAs bound by PIWI and defined 369 piRNA clusters. Among the 12,903 unique piRNA reads from Yin and Lin (2007), we find that 8604 (67%) can be mapped on the piRNA loci defined by Brennecke et al. (2007). Two-hundred-forty-one TEs in FlyBase (R5.13) are located in this set of piRNA clusters defined by Yin and Lin (2007). One-hundred-thirty-two TEs are located in the piRNA clusters defined by both studies.
Small RNA libraries
To obtain an unbiased expression level of distinct piRNAs in vivo, small RNAs from ovary libraries of four distinct strains of D. melanogaster were used (Czech et al. 2008; Li et al. 2009). The first strain is a wild-type (GEO accession no. GSM280082); the second strain is a Dcr-2L811Fsx homozygote (GSM280083); the third strain is a loqsf00791 homozygote (GSM280084); and the last strain is Oregon-R (SRA accession no. SRR010960). Since neither dcr-2 nor loqs is involved in the piRNA biogenesis and functionality pathways, all four libraries were treated as wild type for piRNAs. These four small RNA libraries provide 277,808 to 1,018,295 small RNAs that can be perfectly mapped as antisense to the 5408 TEs annotated in FlyBase R5.13 (Table 2). The reads of piRNAs from these four libraries were normalized by dividing by the total read count, and the average read number for each unique piRNA sequence was calculated. The small RNA libraries were summarized in Table 2. The length distributions of the small RNAs antisense to any of the 5408 TEs were presented in Supplemental Figure S17. Supplemental Figure S17A is the antisense small RNAs that can be mapped on the 142 piRNA loci, and the length distribution is bimodal: the major peak is 25 nt, which represents piRNAs, and a minor peak is 21 nt, which represents siRNAs. The minor peak is conspicuous in libraries GSM280082 and SRR010960 but vanishes in GSM280083 and GSM280084, because the latter two libraries are mutants for the siRNA pathways. Supplemental Figure S17B is the length distribution of all the small RNAs perfectly antisense to any of the 5408 TEs. The pattern is the same as in Supplemental Figure S17A. Overall, most of the small RNAs examined in this study are contributed by piRNAs.
Target prediction
A few cases have been reported where piRNAs can cleave the TE mRNAs to which they are perfectly complementary (Brennecke et al. 2007; Nishida et al. 2007). For the first step of target prediction, we focused on piRNAs:TEs with perfect antisense matches. For each TE, a score (S) is defined with the following formula:
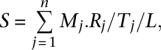 |
where n is the number of piRNA species that are perfectly complementary to this TE; Mj is the number of target sites of a piRNA species j on this TE; Rj is the read number of a piRNA species j (normalized to reads per million reads); Tj is the total number of target sites for piRNA species j; and L is the length of this TE (in kilobases). In other words, for each TE, S is the density of piRNAs complementary to this TE, normalized by the length of the TE and corrected for multiple hits of the piRNAs. Thus, in our target prediction model, we not only took into account the possible target sites, but also the expression levels of piRNAs. We also allowed up to three mismatches between each piRNA and the target sites (data not shown).
Acknowledgments
We thank Anthony Greenberg, Roman Arguello, Zhenglong Gu, Xu Wang, Casey Bergman, and three anonymous reviewers for helpful comments and suggestions. We thank Daniel Hartl's lab for permission to use the shotgun reads of D. melanogaster, and Gregory Hannon's lab and Phillip D. Zamore's lab for making the large-scale sequences of piRNAs publicly available. Part of this work was carried out by using the resources of the Computational Biology Service Unit from Cornell University, which is partially funded by Microsoft Corporation. This work is supported by NIH grant R01 AI64950 to Andrew G. Clark and Brian P. Lazzaro.
Footnotes
[Supplemental material is available online at http://www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.095406.109.
References
- Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004;36:1282–1290. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- Aminetzach YT, Macpherson JM, Petrov DA. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science. 2005;309:764–767. doi: 10.1126/science.1112699. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol. 2001;11:1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Hannon GJ, Brennecke J. The Piwi–piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Bergman CM. Drosophila melanogaster: A case study of a model genomic sequence and its consequences. Genome Res. 2005;15:1661–1667. doi: 10.1101/gr.3726705. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Golic KG, Hawley RS. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2004. Drosophila: A Laboratory handbook. [Google Scholar]
- Assis R, Kondrashov AS. Rapid repetitive element-mediated expansion of piRNA clusters in mammalian evolution. Proc Natl Acad Sci. 2009;106:7079–7082. doi: 10.1073/pnas.0900523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badge RM, Brookfield JF. A novel repressor of P element transposition in Drosophila melanogaster. Genet Res. 1998;71:21–30. doi: 10.1017/s0016672397003066. [DOI] [PubMed] [Google Scholar]
- Bartolome C, Maside X, Charlesworth B. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002;19:926–937. doi: 10.1093/oxfordjournals.molbev.a004150. [DOI] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Bensasson D. Recent LTR retrotransposon insertion contrasts with waves of non-LTR insertion since speciation in Drosophila melanogaster. Proc Natl Acad Sci. 2007;104:11340–11345. doi: 10.1073/pnas.0702552104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman C, Quesneville H, Anxolabehere D, Ashburner M. Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 2006;7:R112. doi: 10.1186/gb-2006-7-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemont C. Population genetics of transposable DNA elements. A Drosophila point of view. Genetica. 1992;86:67–84. doi: 10.1007/BF00133712. [DOI] [PubMed] [Google Scholar]
- Biemont C, Aouar A. Copy-number dependent transpositions and excisions of the mdg-1 mobile element in inbred lines of Drosophila melanogaster. Heredity. 1987;58:39–47. [Google Scholar]
- Biemont C, Vieira C. What transposable elements tell us about genome organization and evolution: The case of Drosophila. Cytogenet Genome Res. 2005;110:25–34. doi: 10.1159/000084935. [DOI] [PubMed] [Google Scholar]
- Biemont C, Vieira C. Genetics: Junk DNA as an evolutionary force. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- Biemont C, Ronsseray S, Anxolabehere D, Izaabel H, Gautier C. Localization of P elements, copy number regulation, and cytotype determination in Drosophila melanogaster. Genet Res. 1990;56:3–14. doi: 10.1017/s0016672300028822. [DOI] [PubMed] [Google Scholar]
- Biemont C, Lemeunier F, Garcia Guerreiro MP, Brookfield JF, Gautier C, Aulard S, Pasyukova EG. Population dynamics of the copia, mdg1, mdg3, gypsy, and P transposable elements in a natural population of Drosophila melanogaster. Genet Res. 1994;63:197–212. doi: 10.1017/s0016672300032353. [DOI] [PubMed] [Google Scholar]
- Biemont C, Vieira C, Borie N, Lepetit D. Transposable elements and genome evolution: The case of Drosophila simulans. Genetica. 1999;107:113–120. [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussy IA, Healy MJ, Oakeshott JG, Kidwell MG. Molecular analysis of the P-M gonadal dysgenesis cline in eastern Australian Drosophila melanogaster. Genetics. 1988;119:889–902. doi: 10.1093/genetics/119.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science. 2008;322:1387–1392. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten R. Transposable elements have contributed to thousands of human proteins. Proc Natl Acad Sci. 2006;103:1798–1803. doi: 10.1073/pnas.0510007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookfield JFY. Models of repression of transposition in P-M hybrid dysgenesis by P cytotype and by zygotically encoded repressor proteins. Genetics. 1991;128:471–486. doi: 10.1093/genetics/128.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookfield JF. Evolutionary genetics: Mobile DNAs as sources of adaptive change? Curr Biol. 2004;14:R344–R345. doi: 10.1016/j.cub.2004.04.021. [DOI] [PubMed] [Google Scholar]
- Brookfield JFY. The ecology of the genome—mobile DNA elements and their hosts. Nat Rev Genet. 2005;6:128–136. doi: 10.1038/nrg1524. [DOI] [PubMed] [Google Scholar]
- Brookfield JF, Badge RM. Population genetics models of transposable elements. Genetica. 1997;100:281–294. [PubMed] [Google Scholar]
- Brower-Toland B, Findley SD, Jiang L, Liu L, Yin H, Dus M, Zhou P, Elgin SC, Lin H. Drosophila PIWI associates with chromatin and interacts directly with HP1a. Genes & Dev. 2007;21:2300–2311. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheton A. The relationship between the flamenco gene and gypsy in Drosophila: How to tame a retrovirus. Trends Genet. 1995;11:349–353. doi: 10.1016/s0168-9525(00)89105-2. [DOI] [PubMed] [Google Scholar]
- Capy P, Maruyama K, David JR, Hartl DL. Insertion sites of the transposable element mariner are fixed in the genome of Drosophila sechellia. J Mol Evol. 1991;33:450–456. doi: 10.1007/BF02103137. [DOI] [PubMed] [Google Scholar]
- Carr M, Soloway JR, Robinson TE, Brookfield JF. Mechanisms regulating the copy numbers of six LTR retrotransposons in the genome of Drosophila melanogaster. Chromosoma. 2002;110:511–518. doi: 10.1007/s00412-001-0174-0. [DOI] [PubMed] [Google Scholar]
- Caspi A, Pachter L. Identification of transposable elements using multiple alignments of related genomes. Genome Res. 2006;16:260–270. doi: 10.1101/gr.4361206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaboissier MC, Bornecque C, Busseau I, Bucheton A. A genetically tagged, defective I element can be complemented by actively transposing I factors in the germline of I-R dysgenic females in Drosophila melanogaster. Mol Gen Genet. 1995;248:434–438. doi: 10.1007/BF02191643. [DOI] [PubMed] [Google Scholar]
- Chambeyron S, Popkova A, Payen-Groschene G, Brun C, Laouini D, Pelisson A, Bucheton A. piRNA-mediated nuclear accumulation of retrotransposon transcripts in the Drosophila female germline. Proc Natl Acad Sci. 2008;105:14964–14969. doi: 10.1073/pnas.0805943105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The maintenance of transposable elements in natural populations. Basic Life Sci. 1988;47:189–212. doi: 10.1007/978-1-4684-5550-2_14. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Mutation–selection balance and the evolutionary advantage of sex and recombination. Genet Res. 1990;55:199–221. doi: 10.1017/s0016672300025532. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The population dynamics of transposable elements. Genet Res. 1983;42:1–27. [Google Scholar]
- Charlesworth B, Langley CH. The evolution of self-regulated transposition of transposable elements. Genetics. 1986;112:359–383. doi: 10.1093/genetics/112.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH. The population genetics of Drosophila transposable elements. Annu Rev Genet. 1989;23:251–287. doi: 10.1146/annurev.ge.23.120189.001343. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–220. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH, Sniegowski PD. Transposable element distributions in Drosophila. Genetics. 1997;147:1993–1995. doi: 10.1093/genetics/147.4.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen D, Lemaitre B, Delattre M, Quesneville H, Ronsseray S, Simonelig M, Higuet D, Lehmann M, Montchamp C, Nouaud D, et al. Drosophila P element: Transposition, regulation and evolution. Genetica. 1994;93:61–78. doi: 10.1007/BF01435240. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. Population, evolutionary and genomic consequences of interference selection. Genetics. 2002;161:389–410. doi: 10.1093/genetics/161.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P, et al. A single p450 allele associated with insecticide resistance in Drosophila. Science. 2002;297:2253–2256. doi: 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desset S, Buchon N, Meignin C, Coiffet M, Vaury C. In Drosophila melanogaster the COM locus directs the somatic silencing of two retrotransposons through both Piwi-dependent and -independent pathways. PLoS One. 2008;3:e1526. doi: 10.1371/journal.pone.0001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin ES, Charlesworth B. The fate of transposable elements in asexual populations. Genetics. 2006;174:817–827. doi: 10.1534/genetics.106.060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin ES, Charlesworth B. The effects of recombination rate on the distribution and abundance of transposable elements. Genetics. 2008;178:2169–2177. doi: 10.1534/genetics.107.082743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Eanes WF, Wesley D, Hey J, Houle D, Ajioka JW. The fitness consequences of P-element insertion in Drosophila melanogaster. Genet Res. 1987;52:17–26. [Google Scholar]
- Eggleston WB, Johnson-Schlitz DM, Engels WR. P-M hybrid dysgenesis does not mobilize other transposable element families in D. melanogaster. Nature. 1988;331:368–370. doi: 10.1038/331368a0. [DOI] [PubMed] [Google Scholar]
- Engels WR. On the evolution and population genetics of hybrid-dysgenesis-causing transposable elements in Drosophila. Philos Trans R Soc Lond B Biol Sci. 1986;312:205–215. doi: 10.1098/rstb.1986.0002. [DOI] [PubMed] [Google Scholar]
- Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan DJ. Transposable elements. Curr Opin Genet Dev. 1992;2:861–867. doi: 10.1016/s0959-437x(05)80108-x. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M, Seitz H, Horwich MD, Li C, Du T, Lee S, Xu J, Kittler EL, Zapp ML, Weng Z, et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Hannon GJ. Conserved themes in small-RNA-mediated transposon control. Trends Cell Biol. 2008;18:136–148. doi: 10.1016/j.tcb.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J, Lenkov K, Lipatov M, Macpherson JM, Petrov DA. High rate of recent transposable element-induced adaptation in Drosophila melanogaster. PLoS Biol. 2008;6:e251. doi: 10.1371/journal.pbio.0060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J, Macpherson JM, Messer PW, Petrov DA. Inferring the strength of selection in Drosophila under complex demographic models. Mol Biol Evol. 2009;26:513–526. doi: 10.1093/molbev/msn270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature. 2008;455:1193–1197. doi: 10.1038/nature07415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Yukuhiro K, Mukai T. Transposition rates of movable genetic elements in Drosophila melanogaster. Proc Natl Acad Sci. 1990;87:3248–3252. doi: 10.1073/pnas.87.8.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8:269–294. [PubMed] [Google Scholar]
- Hoogland C, Biemont C. Chromosomal distribution of transposable elements in Drosophila melanogaster: Test of the ectopic recombination model for maintenance of insertion site number. Genetics. 1996;144:197–204. doi: 10.1093/genetics/144.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising G, Block K. Derivation-dependent distribution of insertion sites for a Drosophila transposon. Cold Spring Harb Symp Quant Biol. 1981;45:527–544. doi: 10.1101/sqb.1981.045.01.069. [DOI] [PubMed] [Google Scholar]
- Jensen S, Cavarec L, Gassama MP, Heidmann T. Defective I elements introduced into Drosophila as transgenes can regulate reactivity and prevent I-R hybrid dysgenesis. Mol Gen Genet. 1995;248:381–390. doi: 10.1007/BF02191637. [DOI] [PubMed] [Google Scholar]
- Jensen S, Gassama MP, Dramard X, Heidmann T. Regulation of I-transposon activity in Drosophila: Evidence for cosuppression of nonhomologous transgenes and possible role of ancestral I-related pericentromeric elements. Genetics. 2002;162:1197–1209. doi: 10.1093/genetics/162.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Vinckenbosch N, Long M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat Rev Genet. 2009;10:19–31. doi: 10.1038/nrg2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmykova AI, Dobritsa AA, Gvozdev VA. Su(Ste) diverged tandem repeats in a Y chromosome of Drosophila melanogaster are transcribed and variously processed. Genetics. 1998;148:243–249. doi: 10.1093/genetics/148.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002;3:RESEARCH0084. doi: 10.1186/gb-2002-3-12-research0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr Mobile elements: Drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG. Hybrid dysgenesis in Drosophila melanogaster: Factors affecting chromosomal contamination in the P-M system. Genetics. 1983;104:317–341. doi: 10.1093/genetics/104.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Holyoake AJ. Transposon-induced hotspots for genomic instability. Genome Res. 2001;11:1321–1322. doi: 10.1101/gr.201201. [DOI] [PubMed] [Google Scholar]
- Klenov MS, Lavrov SA, Stolyarenko AD, Ryazansky SS, Aravin AA, Tuschl T, Gvozdev VA. Repeat-associated siRNAs cause chromatin silencing of retrotransposons in the Drosophila melanogaster germline. Nucleic Acids Res. 2007;35:5430–5438. doi: 10.1093/nar/gkm576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman M. Nucleotide polymorphism at the alcohol dehydrogenase locus of Drosophila melanogaster. Nature. 1983;304:412–417. doi: 10.1038/304412a0. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langley CH, Brookfield JFY, Kaplan NL. Transposable elements in Mendelian populations. I. A theory. Genetics. 1983;104:457–471. doi: 10.1093/genetics/104.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet Res. 1988;52:223–235. doi: 10.1017/s0016672300027695. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Capy P. Population genetics models of competition between transposable element subfamilies. Genetics. 2006;174:785–793. doi: 10.1534/genetics.105.052241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic A, Deceliere G. Models of the population genetics of transposable elements. Genet Res. 2005;85:171–181. doi: 10.1017/S0016672305007585. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Boutin TS, Capy P. Long-term evolution of transposable elements. Proc Natl Acad Sci. 2007;104:19375–19380. doi: 10.1073/pnas.0705238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell. 2009;137:509–521. doi: 10.1016/j.cell.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Yin H. A novel epigenetic mechanism in Drosophila somatic cells mediated by Piwi and piRNAs. Cold Spring Harb Symp Quant Biol. 2008;73:273–281. doi: 10.1101/sqb.2008.73.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe CB, Bejerano G, Haussler D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc Natl Acad Sci. 2007;104:8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Fu Y, Kumar S, Shen Y, Zeng K, Xu A, Carthew R, Wu CI. Adaptive evolution of newly emerged micro-RNA genes in Drosophila. Mol Biol Evol. 2008a;25:929–938. doi: 10.1093/molbev/msn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI. The birth and death of microRNA genes in Drosophila. Nat Genet. 2008b;40:351–355. doi: 10.1038/ng.73. [DOI] [PubMed] [Google Scholar]
- Lyckegaard EM, Clark AG. Ribosomal DNA and Stellate gene copy number variation on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci. 1989;86:1944–1948. doi: 10.1073/pnas.86.6.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, Lyman RF, Jackson MS. Effects of P element insertions on quantitative traits in Drosophila melanogaster. Genetics. 1992;130:315–332. doi: 10.1093/genetics/130.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell. 2009;136:656–668. doi: 10.1016/j.cell.2009.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. doi: 10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maside X, Assimacopoulos S, Charlesworth B. Rates of movement of transposable elements on the second chromosome of Drosophila melanogaster. Genet Res. 2000;75:275–284. doi: 10.1017/s0016672399004474. [DOI] [PubMed] [Google Scholar]
- Maside X, Bartolome C, Assimacopoulos S, Charlesworth B. Rates of movement and distribution of transposable elements in Drosophila melanogaster: In situ hybridization vs Southern blotting data. Genet Res. 2001;78:121–136. doi: 10.1017/s0016672301005201. [DOI] [PubMed] [Google Scholar]
- McDonald JF, Matyunina LV, Wilson S, Jordan IK, Bowen NJ, Miller WJ. LTR retrotransposons and the evolution of eukaryotic enhancers. Genetica. 1997;100:3–13. [PubMed] [Google Scholar]
- Mevel-Ninio M, Pelisson A, Kinder J, Campos AR, Bucheton A. The flamenco locus controls the gypsy and ZAM retroviruses and is required for Drosophila oogenesis. Genetics. 2007;175:1615–1624. doi: 10.1534/genetics.106.068106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery EA, Langley CH. Transposable elements in Mendelian populations. II. Distribution of three COPIA-like elements in a natural population of Drosophila melanogaster. Genetics. 1983;104:473–483. doi: 10.1093/genetics/104.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery E, Charlesworth B, Langley CH. A test for the role of natural selection in the stabilization of transposable element copy number in a population of Drosophila melanogaster. Genet Res. 1987;49:31–41. doi: 10.1017/s0016672300026707. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Blumenstiel JP, Hartl DL. Different regulatory mechanisms underlie similar transposable element profiles in pufferfish and fruitflies. Mol Biol Evol. 2004;21:2310–2318. doi: 10.1093/molbev/msh243. [DOI] [PubMed] [Google Scholar]
- Nishida KM, Siomi MC. [Molecular mechanisms of RNA silencing by siRNA, miRNA and piRNA] Tanpakushitsu Kakusan Koso. 2006;51:2450–2455. [PubMed] [Google Scholar]
- Nishida KM, Saito K, Mori T, Kawamura Y, Nagami-Okada T, Inagaki S, Siomi H, Siomi MC. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA. 2007;13:1911–1922. doi: 10.1261/rna.744307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV. Sure facts, speculations, and open questions about the evolution of transposable element copy number. Genetica. 1999;107:129–137. [PubMed] [Google Scholar]
- Nuzhdin SV, Mackay TF. Direct determination of retrotransposon transposition rates in Drosophila melanogaster. Genet Res. 1994;63:139–144. doi: 10.1017/s0016672300032249. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV, Mackay TF. The genomic rate of transposable element movement in Drosophila melanogaster. Mol Biol Evol. 1995;12:180–181. doi: 10.1093/oxfordjournals.molbev.a040188. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV, Pasyukova EG, Mackay TF. Accumulation of transposable elements in laboratory lines of Drosophila melanogaster. Genetica. 1997;100:167–175. [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Ohta T. Theoretical study on the accumulation of selfish DNA. Genet Res. 1983;41:1–15. doi: 10.1017/s0016672300021029. [DOI] [PubMed] [Google Scholar]
- Pasyukova EG, Nuzhdin SV, Morozova TV, Mackay TF. Accumulation of transposable elements in the genome of Drosophila melanogaster is associated with a decrease in fitness. J Hered. 2004;95:284–290. doi: 10.1093/jhered/esh050. [DOI] [PubMed] [Google Scholar]
- Pelisson A, Sarot E, Payen-Groschene G, Bucheton A. A novel repeat-associated small interfering RNA-mediated silencing pathway downregulates complementary sense gypsy transcripts in somatic cells of the Drosophila ovary. J Virol. 2007;81:1951–1960. doi: 10.1128/JVI.01980-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA, Aminetzach YT, Davis JC, Bensasson D, Hirsh AE. Size matters: Non-LTR retrotransposable elements and ectopic recombination in Drosophila. Mol Biol Evol. 2003;20:880–892. doi: 10.1093/molbev/msg102. [DOI] [PubMed] [Google Scholar]
- Proust J, Prudhommeau C, Ladeveze V, Gotteland M, Fontyne-Branchard MC. I-R hybrid dysgenesis in Drosophila melanogaster. Use of in situ hybridization to show the association of I factor DNA with induced sex-linked recessive lethals. Mutat Res. 1992;268:265–285. doi: 10.1016/0027-5107(92)90233-r. [DOI] [PubMed] [Google Scholar]
- Quesneville H, Anxolabehere D. A simulation of P element horizontal transfer in Drosophila. Genetica. 1997;100:295–307. [PubMed] [Google Scholar]
- Quesneville H, Anxolabehere D. Dynamics of transposable elements in metapopulations: A model of P element invasion in Drosophila. Theor Popul Biol. 1998;54:175–193. doi: 10.1006/tpbi.1997.1353. [DOI] [PubMed] [Google Scholar]
- Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol. 2005;1:166–175. doi: 10.1371/journal.pcbi.0010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sackton TB, Kulathinal RJ, Bergman CM, Quinlan AR, Dopman ER, Carneiro M, Marth GT, Hartl DL, Clark AG. Population genomic inferences from sparse high-throughput sequencing of two populations of Drosophila melanogaster. Genome Biol Evol. 2009;1:439–455. doi: 10.1093/gbe/evp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarot E, Payen-Groschene G, Bucheton A, Pelisson A. Evidence for a piwi-dependent RNA silencing of the gypsy endogenous retrovirus by the Drosophila melanogaster flamenco gene. Genetics. 2004;166:1313–1321. doi: 10.1534/genetics.166.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ. Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc Natl Acad Sci. 2004;101:1626–1631. doi: 10.1073/pnas.0303793101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug MD, Hutter CM, Wetterstrand KA, Gaudette MS, Mackay TF, Aquadro CF. The mutation rates of di-, tri- and tetranucleotide repeats in Drosophila melanogaster. Mol Biol Evol. 1998;15:1751–1760. doi: 10.1093/oxfordjournals.molbev.a025901. [DOI] [PubMed] [Google Scholar]
- Siomi H, Siomi MC. Interactions between transposable elements and Argonautes have (probably) been shaping the Drosophila genome throughout evolution. Curr Opin Genet Dev. 2008;18:181–187. doi: 10.1016/j.gde.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Soderberg RJ, Berg OG. Mutational interference and the progression of Muller's ratchet when mutations have a broad range of deleterious effects. Genetics. 2007;177:971–986. doi: 10.1534/genetics.107.073791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh DS, Choi EH, Yamazaki T, Harada K. Studies on the transposition rates of mobile genetic elements in a natural population of Drosophila melanogaster. Mol Biol Evol. 1995;12:748–758. doi: 10.1093/oxfordjournals.molbev.a040253. [DOI] [PubMed] [Google Scholar]
- Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: Progress and prospect. Annu Rev Cell Dev Biol. 2009;25:355–376. doi: 10.1146/annurev.cellbio.24.110707.175327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- Yang HP, Barbash DA. Abundant and species-specific DINE-1 transposable elements in 12 Drosophila genomes. Genome Biol. 2008;9:R39. doi: 10.1186/gb-2008-9-2-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Arguello JR, Li X, Ding Y, Zhou Q, Chen Y, Zhang Y, Zhao R, Brunet F, Peng L, et al. Repetitive element-mediated recombination as a mechanism for new gene origination in Drosophila. PLoS Genet. 2008;4:e3. doi: 10.1371/journal.pgen.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Lin H. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature. 2007;450:304–308. doi: 10.1038/nature06263. [DOI] [PubMed] [Google Scholar]