

Abstract
Perry syndrome consists of early-onset parkinsonism, depression, severe weight loss and hypoventilation, in which brain pathology is characterized by TDP-43 immunostaining. Through genome-wide linkage analysis we have identified five disease-segregating dynactin (DCTN1) CAP-Gly domain substitutions in 8 families that diminish microtubule binding and lead to intracytoplasmic inclusions. DCTN1 mutations were previously associated with motor neuron disease but can underlie the selective vulnerability of other neuronal populations in distinct neurodegenerative disorders.
Keywords: Dynactin, DCTN1, Perry syndrome, parkinsonism, neurodegeneration, TDP-43
Perry syndrome is a rapidly progressive, autosomal-dominant, neurodegenerative disorder with onset in the fifth decade (OMIM %168605). The cardinal symptoms consist of parkinsonism and weight loss, often accompanied by depression, apathy and social withdrawal with suicidal attempts. Hypoventilation is a late and predominantly nocturnal feature that leads to respiratory insufficiency, insomnia and ultimately death within 2–10 years (reviewed by 1). Pathologically, Perry syndrome has been recognized as a transactive-response DNA-binding protein 43 (TDP-43) proteinopathy although the physiologic relevance is unclear 2. Morphologically, TDP-43 and ubiquitin immunopositive inclusions are comparable to those found in amyotrophic lateral sclerosis and frontotemporal dementia. However, in Perry syndrome TDP-43 inclusions affect the extrapyramidal system and spare both motor neurons and cortical regions 2.
First described in two Canadian families, the condition has since been recognized in seven other families around the world 1. Despite different ethnicities, given the phenotype we hypothesized Perry syndrome may be caused by mutations within the same gene. An International Consortium was established in 2001 by ZKW with the help of local neurologists. Genealogies were constructed and clinical data and blood samples were obtained for families from Canada (605), England (396), France (395), Hawaii (originally of Japanese descent, 5), Japan (537 & 711), Turkey (513) and the United States (730) (pedigree identifiers are provided in parentheses). Autopsy material was reviewed and living affected and at-risk family members were prospectively enrolled using Mayo IRB-approved Brain Bank protocols. The study was also approved by local Ethical Committees from each participating Institution. Genes linked to parkinsonism were excluded as the cause of disease in the 8 families available to study 1.
A 4cM genome-wide scan was performed for DNA samples from four pedigrees likely to be most informative for linkage (395, 605, 711 & 730). Short tandem repeat (STR) genotyping for 1033 markers was carried out fee-for-service by DeCODE genetics (http://www.decode.com/). Linkage analysis was performed using MLINK 3 with a dominant model for two-point LOD scores, and SIMWALK2 for nonparametric statistics and haplotype analysis 4. There were only two loci with two-point LOD scores >2 (θ=0) with a haplotype shared by affected subjects, in each single family. These regions of suggestive linkage were a 14cM interval between D2S2368 and D2S1777 and an 18cM interval between D10S185 and D10S562. To maximize the information content, both loci were saturated with additional STRs at 0.5–1.0cM resolution in all family members and genotypes were re-analyzed. The potential locus on chromosome 10q23–25 was excluded whereas the locus on chromosome 2p12–14 became significantly linked (two-point LOD=3.52, θ=0 at D2S3046). Obligate recombinants in two families (395 and 730) narrowed the region to ~6cM between D2S1389 and D2S3049 and a genomic interval with 46 candidate genes (Supplementary table 1). Gene sequencing was performed on an ABI 3730 sequencer with SeqScape v2.5 analysis software (Applied Biosystems).
All 8 families with Perry syndrome were found to have one of five novel mutations located in exon 2 of the DCTN1 gene that segregated with disease: G71R (513 & 605), G71E (395), G71A (5, 396 & 711), T72P (730) and Q74P (537) (Fig. 1). The clinical and pathological phenotype caused by these five substitutions is remarkably similar with the exception of the English family (396) in which affected individuals did not exhibit hypoventilation or weight loss 1. Assuming each of these mutations caused dominantly inherited disease, it was possible to obtain a maximum two-point LOD = 11.5, θ=0 at D2S3072 and a multipoint LOD = 14.1, θ=0 at D2S3039. With the exception of G71A in two families of Japanese descent (5 and 711), haplotype analysis does not provide evidence of a common founder (Supplementary table 2). To confirm the rarity of these mutations and provide supporting evidence for pathogenicity, DCTN1 exon 2 was also sequenced in 949 unrelated US control subjects and a multiethnic panel of 475 probands with familial parkinsonism including subjects from continental America, Europe, Asia and N.Africa. None of the five mutations identified in Perry syndrome were found in unrelated subjects (Supplementary table 3).
Figure 1. Pedigrees with Perry syndrome and DCTN1 mutations.
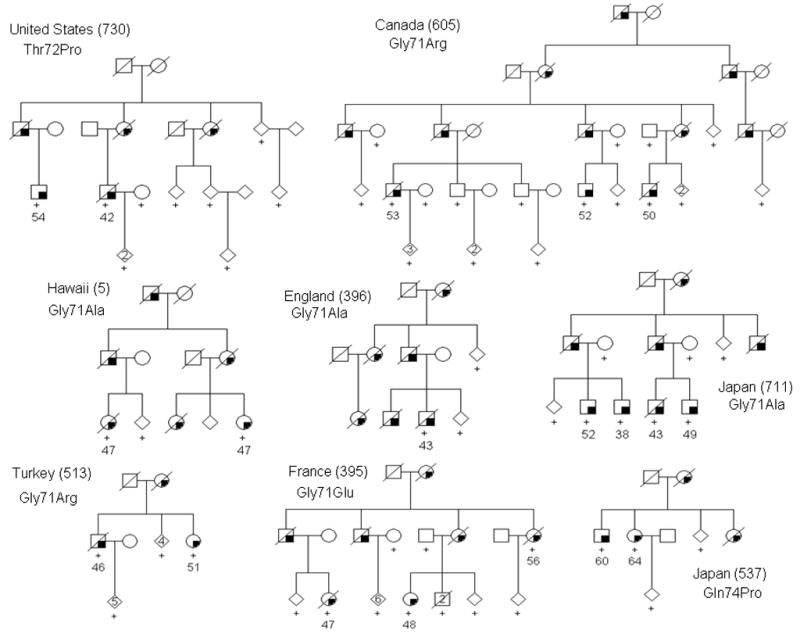
Filled symbols indicate affected individuals with age of symptom onset beneath. A + indicates DNA was available at the time of study. To protect confidentiality some individuals are not shown and gender is portrayed using a diamond.
DCTN1 encodes the large subunit of the dynactin complex, p150glued, essential in many intracellular transport functions and non-motile processes 5. p150glued protein interactions are partly mediated by the N-terminal cytoskeleton-associated protein-glycine-rich (CAP-Gly) domain and the ‘GKNDG’ binding motif 6,7. This includes an affinity for C-terminal acidic-aromatic sequences found in cytoplasmic linker protein CLIP170, α-tubulin and end-binding protein 16. Notably all five coding substitutions discovered in pedigrees with Perry syndrome affect evolutionarily conserved amino acids within or immediately adjacent to the p150glued CAP-Gly GKNDG motif (Supplementary fig. 1).
DCTN1 G59S was previously identified in a family with distal hereditary motor neuronopathy with a distinct vocal fold paresis (laryngeal dysfunction) 8,9. Clinically and pathologically this disorder is very different from Perry syndrome. The mean age at onset is typically a decade earlier, at 34 years, and has a far longer duration, on average ~17 years (range 7–31) 9. Electrophysiologic exams in affected family members revealed chronic motor neuron denervation, while post-mortem examination of the medulla in one case showed severe reduction of hypoglossal motor neurons. Dynactin p50 subunit immunostaining revealed cytoplasmic inclusions in enlarged motor neurons, patient fibroblasts and DCTN1 G59S transfected cells 8–10. In vitro there is a modest reduction in the affinity of mutant protein for microtubules, which has been suggested to impair axonal transport 8,10.
In contrast, in Perry syndrome there is no evidence of motor neuron pathology; on exam the hypoglossal nucleus (5/8 cases), motor cortex (2/8) and spinal cord (2/8) remain intact. The brunt of pathology is in surviving dopaminergic neurons of the substantia nigra, in the locus ceruleus, dorsal raphe nucleus and respiratory neurons in the ventrolateral medulla 2,11. Neuronal inclusions are observed with TDP-43 immuno-histochemistry and to a lesser extent with dynactin p62 and p50 antibodies (Fig. 2). Although no autopsy material was available for muscle or peripheral nerve, there was no clinical indication of motor neuron disease 1. One affected subject from US family 730, with DCTN1 T72P, developed mild polyneuropathy and muscle denervation but this could be incidental 12.
Figure 2. Dynactin and TDP-43 pathology in Perry syndrome.

Dynactin p150 antibodies are not useful for immunohistochemistry. Dynactin subunits p50 and p62 (that may also exist as unbound monomer 5) stain inclusions with the same morphology and distribution as TDP-43. However, double immunostaining for p62/TDP-43 and p50/TDP-43 shows p62 and p50 are in ≤5% of the TDP-43 positive inclusions. In contrast, only a very small number of inclusions are positive for only p62, or only p50, and not for TDP-43. Panels a–f illustrate a range of p62 immunoreactive inclusions in the globus pallidus in Perry syndrome neuronal cytoplasmic inclusions (NCI) (a–d), neurites (e) and juxta-vascular glia (f), which are morphologically similar to inclusions detected in Perry syndrome with TDP-43 immunohistochemistry 2. Double immunohistochemistry for p62 (blue chromogen in g–j) and TDP-43 (brown chromogen in g–j) demonstrate that some NCI are double labeled (i), while others show predominantly p62 (g) or TDP-43 (h). Granular axonal spheroids show a heterogenous mixture of p62 and TDP-43 (j). Immunofluorescence with p62 (green fluorochrome) and TDP-43 (red fluorochrome) (k and l) shows a heterogenous mixture of p62 and TDP-43 in granular axonal spheroid (k) and complete overlap of fluorochromes (yellow) in a subset of NCI (l). Sections of substantia nigra double stained for p50 (blue chromogen in m–r) and TDP-43 (brown chromogen in m-r) demonstrates that some NCI (m–o) are double labeled (o), while others show predominantly p62 (n) or TDP-43 (m). Similarly, some neurites (p–r) are double labeled (r), while others show predominantly p62 (q) or TDP-43 (p). [g–i, m–r are 100x, all others images are 40x].
Functional assessment of Perry syndrome mutants, DCTN1 G71R and Q74P, reveals a modest decrease in microtubule binding comparable to G59S (Supplementary fig. 2). In addition, cells transfected with DCTN1 G59S, G71R and Q74P demonstrate a dramatic redistribution of dynactin compared to over-expression of wild-type protein (Supplementary fig. 3). Together, these findings provide supporting evidence of pathogenicity. However, p150glued is known to have at least two microtubule-binding domains with quite different physiological roles 7. The CAP-Gly domain normally facilitates stable binding as a ‘parking brake’ to inhibit dynein motility, and it has yet to be assessed whether G59S or Perry mutations enhance dynein processivity or ‘skating’ along microtubules. Mutation in the p150glued CAP-Gly domain may also destabilize the entire dynactin complex promoting its degradation, of which a general consequence may be impaired mis-folded protein clearance and/or aggregation. Whether dynactin in TDP-43 immunopositive inclusions is due their functional interaction or to a secondary response accompanying protein aggregation and inclusion formation should be examined in other TDP-43 proteinopathies.
In summary, p150glued CAP-Gly mutations are likely to disrupt many physiologically-relevant moderate-affinity interactions of p150glued with its binding partners 5,6. Based on crystal structure, G59 is centrally located in the p150glued CAP-Gly domain 13. In contrast, Perry syndrome mutations at G71 (with T72 & Q74, adjacent) directly affect the GKNDG binding motif. The subtle differences of these mutations on binding interactions are now most important to resolve, as they may underlie the selective vulnerability of distinct neuronal populations which consequently manifest as quite different neurodegenerative diseases.
Supplementary Material
Acknowledgments
We are grateful to all family members who participated in the study. We thank Dr. Leon Thal posthumously for his clinical contribution. We must also thank Drs. Jan Aasly, Faycel Hentati, Tim Lynch, Yoshikuni Mizuno, Ryan Uitti and Ruey-Mei Wu for their contribution and continued collaboration. Caroline Kent, Sarah Lincoln and Stephanie Cobb provided technical support, whereas Fred Pishotta, Curt Younkin and Susan Hawley helped with computing and bioinformatics. MJF, ZKW and AJS are funded by the Pacific Alzheimer Research Foundation (PARF) grant C06-01. Mayo Clinic Jacksonville is a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 #NS40256).
MJF designed and directed the genetic study, wrote and edited the manuscript. MMH and JMK performed the STR and linkage analysis. JD performed the protein biochemistry and cell biology. AJS, DBC, BL, FC, YT, TY, LG, BE, KPB and ZKW performed clinical and neurologic exams, assisted by LLG, SC, CWW and LAB. CV-G and OAR performed sequence and nucleotide analysis. MC-C and DWD performed the neuropathology and immunohistochemistry. All authors contributed to manuscript revision.
References
- 1.Wider C, Wszolek ZK. Parkinsonism Relat Disord. 2008;14:1–7. doi: 10.1016/j.parkreldis.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Wider C, et al. Parkinsonism Relat Disord. 2008 doi: 10.1016/j.parkreldis.2007.06.012. Epub August 23rd . [DOI] [PubMed] [Google Scholar]
- 3.Ott J. Proc Natl Acad Sci U S A. 1989;86:4175–8. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sobel E, Sengul H, Weeks DE. Hum Hered. 2001;52:121–31. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 5.Schroer TA. Annu Rev Cell Dev Biol. 2004;20:759–79. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 6.Weisbrich A, et al. Nat Struct Mol Biol. 2007;14:959–67. doi: 10.1038/nsmb1291. [DOI] [PubMed] [Google Scholar]
- 7.Culver-Hanlon TL, et al. Nat Cell Biol. 2006;8:264–70. doi: 10.1038/ncb1370. [DOI] [PubMed] [Google Scholar]
- 8.Puls I, et al. Nat Genet. 2003;33:455–6. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 9.Puls I, et al. Ann Neurol. 2005;57:687–94. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy JR, et al. J Cell Biol. 2006;172:733–45. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuboi Y, et al. Acta Neuropathol. 2008;115:263–8. doi: 10.1007/s00401-007-0246-1. [DOI] [PubMed] [Google Scholar]
- 12.Roy EP, et al. Neurology. 1988;38:637–9. doi: 10.1212/wnl.38.4.637. [DOI] [PubMed] [Google Scholar]
- 13.Honnappa S, et al. Mol Cell. 2006;23:663–71. doi: 10.1016/j.molcel.2006.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.