

Abstract
R-spondins (Rspos) potentiate Wnt/β-catenin signaling, an important pathway in embryonic development that is constitutively active in many cancers. To analyze Rspo structure and function, we expressed full-length wild-type Rspo2 and Rspo2 point mutants corresponding to Rspo4 variants that have been linked to developmental defects. The Rspo2 mutants had markedly reduced potency relative to the wild-type protein, demonstrating for the first time specific amino acid residues in Rspos that are critical for β-catenin signaling. The diminished activity of Rspo2/C78Y and Rspo2/C113R was attributable to a defect in their secretion, while Rspo2/Q70R exhibited a decrease in its intrinsic activity. Cysteine assignments in a Rspo2 derivative containing only the two furin-like domains (Rspo2-2F) provided the first information about the disulfide-bonding pattern of this motif, which was characterized by multiple short loops and unpaired cysteine residues, and established that the loss-of-function cysteine mutants disrupted disulfide bond formation. Moreover, Rspo2-2F demonstrated potent activity and synergized strongly with Wnt-3a in a β-catenin reporter assay. In contrast, an Rspo2-2F derivative containing the Q70R substitution showed significantly reduced activity, although it still synergized with Wnt-3a in the reporter assay. Rspo2-2F derivatives elicited an unusually sustained phosphorylation (20 h) of the Wnt co-receptor, low density lipoprotein receptor-related protein 6 (LRP6), as well as an increase in cell surface LRP6. Co-immunoprecipitation experiments involving LRP6 and Kremens suggested that these associations contribute to Rspo2 activity, although the lack of major differences between wild-type and Q70R derivatives implied that additional interactions may be important.
Keywords: R-spondin2, Wnt signaling, β-catenin, LRP6, furin domain, disulfide mapping
1. Introduction
The R-spondins (Rspos)1 comprise a family of four highly conserved proteins that are widely expressed in vertebrate embryos and in the adult [1, 2]. They contain a thrombospondin type I repeat and initially were detected in the roof plate of the neural tube, hence the name “R-spondin” [3]. Gene targeting experiments have shown that they have critical functions during embryogenesis. Rspo2 is required for limb, laryngeal-tracheal and lung development [4–6], as well as myogenesis [7], while Rspo3 is essential for placental formation [8]. Human syndromes characterized by specific developmental abnormalities have been attributed to putative loss-of-function mutations in particular Rspo genes. Rspo1 mutations result in female to male gender reversal [9–12], whereas point mutations in Rspo4 cause defects in the formation of fingernails and toenails (anonychia) [13–16].
The relationship of Rspos to normal and malignant growth has not yet been firmly established. Administration of purified recombinant Rspo1 protein to mice elicited a dramatic increase in the size of the small intestines due to a massive stimulation of cell proliferation [17, 18]. Insertional activation of the Rspo2 and Rspo3 genes has been observed in the mouse mammary tumor virus model system, suggesting a potential positive contribution of Rspos to neoplasia [19, 20]. Alternatively, Rspo1 loss-of-function mutations were associated with an increased incidence of squamous cell skin carcinoma affecting the palmar and plantar surfaces [9]. This implied that Rspos might have a tumor suppressive effect in specific contexts. Current information suggests that changes in Rspo expression are relatively rare in human cancers [7].
Several observations have linked Rspos to the expression and activity of Wnt proteins. The Wnts form a large family of extracellular, lipid-modified glycoproteins that have a plethora of activities during embryonic development and in the adult, regulating cell proliferation, differentiation, polarity, motility and survival, organogenesis, body axis formation and tissue homeostasis [21]. Constitutive activation of Wnt signaling is common in many tumors [21]. An overlapping pattern of expression has been reported for Rspos and Wnts in many tissues, including the neural tube, developing muscle, and elsewhere [2, 3, 7, 22]. Importantly, they have been shown to cooperate with each other during development, specifically by promoting the transcriptional activity of β-catenin [7], a well-known mechanism of Wnt signaling that mediates both cell proliferation and differentiation, depending on the setting [23]. Several studies involving the use of transient expression or purified recombinant proteins in cell culture have verified that Rspos potentiate the Wnt/β-catenin signaling pathway [1, 7, 24–27]. Rspo deletion analysis demonstrated that its two furin-like domains, but not the thrombospondin type I repeat or positively charged carboxyl-terminal region, were required for synergistic activity [7, 24, 25].
Activation of the β-catenin pathway requires Wnt binding to both a seven-pass transmembrane receptor in the Frizzled (Fzd) family and either low density lipoprotein receptor-related proteins 5 or 6 (LRP5/6). Association of Wnt/Fzd/LRP results in disruption of a multi-protein complex that includes the scaffolding protein, Axin, Adenomatosis Polyposis Coli protein, β-catenin, as well as casein kinase Iα and glycogen synthase kinase 3, kinases that phorphorylate β-catenin to facilitate its proteasomal degradation [28]. Dissociation of this β-catenin degradation complex is preceded by Wnt-dependent phosphorylation of LRP6, which requires Fzd, Dishevelled, Axin, glycogen synthase kinase 3 and casein kinase Iγ [29–32]. Phosphorylated LRP6 provides high affinity binding sites for Axin, presumably enhancing its recruitment to the plasma membrane and, consequently, disruption of the β-catenin degradation complex.
While the evidence for Rspo/Wnt synergy in β-catenin signaling is extensive, its molecular mechanism is controversial. Contradictory reports support or refute the possibility that Rspos bind to Fzds, LRPs and/or Wnts [2, 25, 26]. One group has presented evidence that Rspos promote Wnt/β-catenin signaling by blocking the internalization and down-regulation of LRP6 that allegedly is induced by the binding of Dickkopf (Dkk) proteins to LRP5/6 and Kremens (Krm1/2) [24, 33]. According to this research, Rspos interact with Krm1/2, but not LRP6, to prevent LRP6 endocytosis [33]. However, others have questioned the relevance of LRP5/6 internalization as a mechanism of Dkk-dependent suppression of Wnt/β-catenin signaling [34], or the requirement of Krms for Rspo activity [35].
The present investigation was undertaken to identify structural features of Rspo2 that were critical for its activity in the β-catenin pathway. We chose to study Rspo2 because of its insertional activation in the mouse mammary tumor virus model of breast cancer [19]. Point mutations analogous to ones that occur naturally at highly conserved sites in the Rspo4 gene of humans with anonychia were introduced into the Rspo2 sequence to assess their impact on Wnt/β-catenin signaling. Rspo2 two-furin domain derivatives (Rspo2-2F) also were examined, as others noted that these domains were necessary and sufficient for activity [7, 24, 26]. Rspo2-2F was analyzed to determine the disulfide-bonding pattern of the furin-like domains, while wild type and mutant variants of Rspo2-2F and full-length Rspo2 were used to investigate mechanisms of Rspo activity.
2. Materials and methods
2.1 Antibodies and chemicals
Anti-β-catenin antibody (catalog no. 610154) was from BD Biosciences (San Jose, CA). Anti-LRP6 (catalog no. 2560) and anti-phospho-LRP6 (Ser1490) antibodies (catalog no. 2568) were from Cell Signaling Technology (Danvers, MA). Anti-V5 (catalog no. R960-25), Alexa Fluor 488 phalloidin (catalog no. A12379), Alexa Fluor 568 phalloidin (catalog no. A12380) were from Invitrogen (Carlsbad, CA). Anti-FLAG (catalog no. F1804) and DAPI (catalog no. D9542) were from Sigma-Aldrich (St. Louis, MO). Anti-human Rspo2 antibody (catalog no. AF3266) was from R&D Systems (Minneapolis, MN). Anti-Myc antibody (catalog no. sc-40) was from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2 Plasmid constructs
The full-length coding sequence of mouse Rspo2, originally obtained from mouse mammary tumor tissue [19], was amplified by PCR and subcloned into pEF6-V5/His B vector. The Rspo2 mutants, Rspo2/Q70R, Rspo2/C78Y and Rspo2/C113R, were generated with the QuikChange Site-Directed Mutagenesis Kit (catalog no. 200519, Stratagene, La Jolla, CA) using pEF6-V5/His B-Rspo2 as template, and verified by DNA sequencing analysis. Full length human Dkk1 was cloned into pEF6-V5/His TOPO vector. The plasmid, pcDNA3.1-LRP6-Myc6 was a generous gift from Dr. Xi He, Harvard Medical School. The plasmid pCS2-flag-mouse Krm2 was kindly provided by Prof. Dr. C. Niehrs, German Cancer Research Center. Using the full-length mouse Rspo2 coding sequence as template, sequence encoding the two furin domains and a short stretch of upstream sequence (Rspo2-2F, corresponding to amino acid residues 17–140 of full-length Rspo2) and its mutant Rspo2-2F/Q70R were PCR-amplified with XhoI/XbaI sites and cloned into pPICZα A to create His6-tagged constructs. A KR sequence was introduced immediately upstream of the Rspo sequence to enable its cleavage from the yeast α-factor signal peptide by Kex2.
2.3 Cell culture
STF cells, a HEK293 cell line stably expressing a SuperTopFlash reporter gene [36], was kindly made available by Dr. Jeremy Nathans, Johns Hopkins University. STF and HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum in a 5% CO2 humidified 37°C incubator. CHO cells were cultured in F-12 medium supplemented with 10% fetal bovine serum in a 5% CO2 humidified 37°C incubator.
2.4 DNA transfection and detection of protein in conditioned media or cell lysates
STF cells (1.8 × 106) were seeded in a 10-cm cell culture dish, and the next day transfected with 6 μg of the indicated plasmids or empty vector using FuGENE 6 (catalog no. 11814443001, Roche, Indianapolis, IN) according to the manufacturer’s protocol. After 48 h, the conditioned medium (CM) was collected and concentrated ~20-fold (Amicon Ultra-15, Millipore, Billerica, MA). Anti-V5 antibody (1 μg) and 40 μl Protein A/G PLUS-Agarose slurry (catalog no. SC-2003, Santa Cruz Biotechnology) were added to the concentrated medium. After incubation at 4°C for 3 h with rotation, the beads were washed three times and proteins were resolved by SDS-PAGE in 10% polyacrylamide gels prior to western blot analysis. To obtain lysates, cells were washed twice with phosphate-buffered saline (PBS) and resuspended in 1 ml of the same solution. After removing 0.2 ml for the luciferase reporter assay, the remaining cells were pelleted, resuspended in 0.5 ml of PYLB buffer (10 mM Na4O7P2, 50 mM NaF, 50 mM NaCl, 1 mM EDTA, 50 mM HEPES, 1% Triton X-100, 1 mM Na3VO4, 10 μg/ml Leupeptin, 10 μg/ml Aprotinin, 1 mM PMSF) and incubated on ice for 10 min. Cell lysates were clarified by centrifugation and 50 μg samples were resolved by SDS-PAGE. Immunoblotting was performed with Immobilon-P PVDF membrane (catalog no. IPVH304F0, Millipore) and the indicated primary and appropriate secondary antibodies as previously described [37]. For detection of β-catenin and LRP6, samples were electrophoresed in 4–20% polyacrylamide gels.
2.5 GST-E-cadherin pull down assay
Soluble β-catenin was detected with a GST-E-cadherin pull-down assay performed as previously described [37].
2.6 Immunofluorescent analysis
CHO cells (2 × 104) were seeded on a 12-mm-diameter glass coverslip (catalog no. 12-545-80, Fisher Scientific, Pittsburgh, PA), placed in a 24-well plate and transfected with 0.375 μg of the indicated Rspo2 plasmids using FuGENE 6. After 24 h, cells were washed three times with PBS, and fixed in 3.65% formaldehyde for 20 min at room temperature. Following a wash with PBS, samples were either incubated with permeabilization buffer (0.1 % Triton X-100 in PBS) for 10 min, and then blocked with 5% BSA in PBS, or simply blocked with 5% BSA solution (non-permeabilized samples). After blocking for 1 h at room temperature, coverslips were incubated with mouse anti-V5 antibody (1:200) overnight at 4oC, washed three times with PBS, and incubated with Alexa 488-conjugated goat anti-mouse IgG secondary antibody (1:1000), Alexa 568 phalloidin (1:1000) and DAPI (1:1000) for 1 h at room temperature. Stained specimens were then mounted on a glass slide with Prolong Gold anti-fade reagent (catalog no. P36930, Invitrogen). Images were obtained and processed as previously described [37]. In parallel, CHO cells (5 × 105/well) were seeded in 6-well plates, transfected with 1.5 μg of corresponding plasmid, and processed for western blotting to evaluate the protein expression level.
2.7 Protein expression of Rspo2 derivatives in Pichia pastori
The pPICZα A-Rspo2-2F or pPICZα A-Rspo2-2F/Q70R plasmid was linearized with the restriction endonuclease SacI. The Pichia pastoris strain X-33 was transformed by using Pichia EasyComp™ transformation kit (catalog no. K1730-01, Invitrogen). Transformants were selected with YPDS plates containing 100 μg/ml Zeocin (catalog no. R250-01, Invitrogen). PCR analysis confirmed the presence of inserts in Zeocin-resistant colonies. Positive colonies were grown in 10 ml cultures and induced with 0.5% methanol for 7 days according to the protocol from Invitrogen, and CM was immunoblotted with anti-Rspo2 to assess the production of recombinant protein. The highest expresser was selected for larger scale expression.
2.8 Purification of Rspo2 derivatives
For preparation of sample by nickel chelating chromatography, a single transformant colony was inoculated into 500 ml BMGY medium and incubated at 30°C, 225 rpm. Cells were pelleted when OD600 was ~2 and resuspended in 2 L of BMMY medium supplemented with 0.5% methanol. 0.5% methanol was added to the medium daily to induce protein expression. After 7 days, the medium was concentrated 10-fold by ultrafiltration (Model 2000, Millipore) at 4°C using a YM3 membrane (Millipore). Rspo2-2F proteins were eluted from a HiTrap chelating affinity column (1.0 ml, GE Healthcare, Piscataway, NJ) with buffer (20 mM Tris-HCl pH 8.0, 0.5 M NaCl) containing 100 mM imidazole. Proteins were then dialyzed against PBS overnight and used for N-terminal Edman degradation sequence analysis and circular dichroism spectroscopy (Jasco J720 spectropolarimeter).
Alternatively, Rspo2-2F proteins were purified with cation-exchange chromatography. Yeast transformant was cultured as described in the previous paragraph, except that BMG and BMM media were used instead of BMGY and BMMY, respectively (see Invitrogen protocol for details) and BMM medium was supplemented with 1% casamino acids (catalog no. C-366, LabScientific, Livingston, NJ) to reduce proteolysis. Following ultrafiltration, the concentrated sample was diluted 10-fold with 20 mM sodium phosphate buffer (pH 6.0), and loaded on a HiTrap SP FF column (5 ml, GE Healthcare) that had been equilibrated in the same buffer at 4°C. Rspo2-2F protein was eluted with the sodium phosphate buffer containing 1 M NaCl. Peak fractions were collected and dialyzed overnight against 20 mM sodium phosphate buffer, pH 6.0. The dialyzed sample was applied to a Mono S column (1ml, GE Healthcare) equilibrated in this buffer, and protein eluted with a two-step linear NaCl gradient (0.1 M to 0.4 M in 20 ml, then 0.4 M to 0.7 M in 5 ml). Fractions containing Rspo2-2F proteins were identified by immunoblotting and Coomassie blue staining of gels after SDS-PAGE. Aliquots from peak fractions were directly used for disulfide bond assignment by liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS) analysis. The remainder was dialyzed against PBS overnight and stored at −80°C until use for functional studies.
2.9 Identification of free Cys residues and disulfide bonds by liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS) analysis
Rspo2-2F was alkylated with N-ethylmaleimide (20 mM) in the dark, at room temperature for 1 h and subsequently denatured with 8 M urea. The mixture was split equally and transferred into two Microcon YM-30 (Millipore) filters. Both filters were centrifuged at 8,000 × g to remove residual solvent. Proteolytic digestion was performed in the upper chamber of the filters using trypsin in one filter, and chymotrypsin in the other filter. The proteolytic enzyme to protein ratio was 1/40 (w/w). The proteolytic digestion was carried out overnight at 37oC, and the digested solution collected for LC/ESI-MS/MS analysis.
The resulting tryptic and chymotryptic peptides of Rspo2-2F were analyzed by LC/ESI-MS/MS using an LTQ ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). LC/ESI-MS/MS analyses were conducted using a dual pump Thermo Surveyor HPLC system. Peptide mixtures were chromatographically separated using a reverse phase nanoLC column (C18, 75μm × 130mm). The A and B mobile phases (mobile phase A is 0.1% HCOOH/water and mobile phase B is 0.1% HCOOH in acetonitrile) were used to create a three-step linear gradient of 5% to 30% B in the first 65 min, followed by 30% to 80% B in the next 10 min, and 80% B in the final 10 min. The LC/ESI-MS/MS data acquisition was set up to collect ion signals from the eluted peptides using an automatic, data-dependent scan procedure in which a cyclic series of three different scan modes (1 full scan, 4 zoom scans, and 4 MS/MS scans for top four abundant ions) were performed. The MS/MS analysis exclusion rule for the same precursor ion was set to a value of 2 during a 75 s period and the full scan mass range was set from m/z 400 to 1,800. The resulting MS/MS spectra were searched against the SwissProt human protein database using the Sequest program to identify the sequences of peptides.
2.10 Immunoblot analysis of cells treated with purified Rspo2-2F derivatives
STF cells were cultured in 6-well plates with serum-containing DMEM until monolayers were fully confluent. Subsequently, the cells were switched to serum-free DMEM and treated with the indicated proteins or PBS (negative control). Recombinant Wnt-3a was from R&D Systems (catalog no. 1324-WN, Minneapolis, MN). Cells were harvested after 1, 6 or 20 h, and lysates were prepared for LRP6 or phospho-LRP6 western blot analysis.
2.11 Luciferase reporter assay
For transfection experiments, STF cells were maintained in serum-containing DMEM prior to harvesting 48 h after transfection. In experiments with purified recombinant proteins, after STF cells were grown to confluence in serum-containing DMEM, the medium was changed to serum-free DMEM and cells were incubated with the indicated proteins for 20 h. STF cell lysates were prepared with reporter lysis buffer from the Luciferase assay system kit (catalog no. E1501, Promega, Madison, WI), and clarified by centrifugation. The supernatant was then analyzed in the reporter assay according to the manufacturer’s protocol. Luciferase activity was normalized to the protein concentration of cell lysates. In transfection experiments, three aliquots from each cell lysate were analyzed in parallel, while experiments involving recombinant proteins were performed with triplicate wells for each treatment group.
2.12 Biotinylation of LRP6 at the cell surface
STF cells were grown in 6-well plates with serum-containing DMEM until monolayers were 100% confluent. Following overnight incubation in serum-free DMEM, cultures were incubated for 20 h with the indicated proteins. Cell culture plates were then placed on ice and monolayers were washed three times with ice-cold PBS (pH 8.0), followed by treatment for 1 h on ice with 1 ml/well (0.5 mg/ml) of EZ-Link Sulfo-NHS-LC-Biotin (catalog no. 21217, Thermo Scientific Rockford, IL). After washing with 0.1 M glycine/PBS (pH 8.0) twice to stop the reaction, cells were lysed with PYLB buffer and biotinylated proteins (400 μg cell lysate) were precipitated using high capacity streptavidin agarose resin (catalog no. 20359, Thermo Scientific, Rockford, IL). Pellets were washed with PYLB buffer and then analyzed by western blotting using anti-LRP6 antibody.
2.13 Co-immunoprecipitation (co-IP) experiments
HEK293 cells (7 × 106) were seeded in a 10-cm dish and, after overnight incubation, co-transfected with 6 μg of pcDNA3-LRP6-Myc6 and 9 μg of pEF6-Rspo2-V5/His, pEF6-Rspo2/Q70R-V5/His or pEF6-Dkk1-V5/His TOPO using Lipofectamine 2000 (catalog no.11668-019, Invitrogen). CHO cells (1.5 × 106) were seeded in a 10-cm dish and, after overnight incubation, co-transfected with 6 μg of pCS2-flag-mouse Krm2 and 6 μg of pEF6-Rspo2-V5/His, pEF6-Rspo2/Q70R-V5/His or pEF6-Dkk1-V5/His TOPO using FuGENE 6. After 48 h, cells were washed with PBS and lysed with buffer (20 mM Tris-HCl, 150 mM NaCl, 10 mM EDTA, 0.2% Triton X-100, 0.2% NP-40, pH 8.0, 1 mM Na3VO4, 10 μg/ml Leupeptin, 10 μg/ml Aprotinin, 1 mM PMSF). Cleared lysates were subjected to immunoprecipitation with anti-V5, anti-Myc or anti-FLAG antibody. Immunoprecipitates were analyzed by western blotting using the indicated antibodies and corresponding secondary antibodies.
2.14 Statistical analysis
The significance of data obtained from densitometric analysis of immunoblots was determined with Student’s t test. The differences were considered to be significant when the P value was less than 0.05.
3. Results
3.1 Rspo2 point mutants lack activity in β-catenin pathway
Using mutations in the Rspo4 gene associated with anonychia as a guide [13–16], we generated a set of Rspo2 mutants each containing one of the following substitutions: Q70R, C78Y or C113R. To evaluate the activity of these Rspo2 derivatives in a β-catenin reporter assay, we transiently expressed their cDNA constructs in STF cells, a HEK293 line stably expressing a SuperTopFlash promoter. The activity of each mutant was markedly reduced relative to wild type Rspo2 (Fig. 1A). There was a similar decrease in the amount of free β-catenin detected in these cells by immunoblot analysis, as well as a decline in β-catenin measured in whole cell lysates (Fig. 1B). Consistent with this weak activity, cells transiently expressing the Rspo2 point mutations also showed little evidence of phosphorylated LRP6, an upstream marker of β-catenin pathway activation (Fig. 1B). Diminished activity was not attributable to low levels of protein expression or rapid protein turnover, as the quantity of V5 epitope-tagged Rspo2 protein detected in these cells was similar to or greater than the amount seen in lysate of cells expressing the wild type protein (Fig. 1B and Suppl. Fig. 1).
Figure 1. Rspo2/C78Y, Rspo2/C113R and Rspo2/Q70R mutants had markedly reduced activity in Wnt/β-catenin pathway.
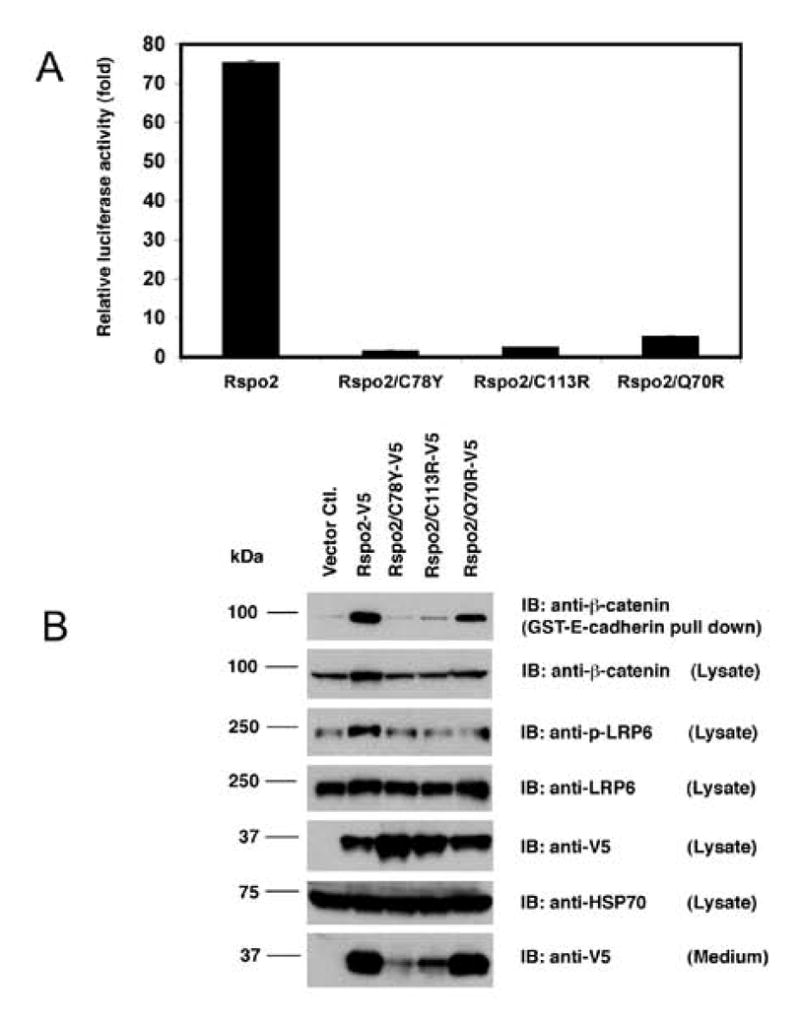
(A) Luciferase reporter assay. STF cells were transfected with 6 μg of empty vector or indicated Rspo2 construct and lysed 48 h later for luminescence measurements. Values were normalized according to total protein concentration of cell lysates, and luciferase activity was expressed as fold stimulation relative to results with the empty vector. Lysates were assayed in triplicate; error bars (often not visible in figure) represent S.D. (B) Immunoblot analysis of Rspo2 derivatives, β-catenin and LRP6 in transfected cells. After 48 h, lysates were prepared from transfected cells and blotted for β-catenin, phospho-LRP6, total LRP6, Rspo2 derivatives and HSP70 as a loading control. Free β-catenin was detected after pull-down with GST-E-cadherin. Rspo2 proteins also were visualized in concentrated CM. Note that the entire CM from transfected monolayer cultures were loaded on the gel, whereas only 1/15 of cell lysates were subjected to western blotting.
3.2 Cysteine mutants were defective in secretion
For all the Rspo2 proteins, including the wild type derivative, most of the protein was detected in the cell lysate rather than the CM. While comparable amounts of wild type and Q70R proteins accumulated in CM, much lower concentrations of the cysteine mutants were observed in this fraction (Fig. 1B), implying that Rspo2/C78Y and Rspo2/C113R proteins were not efficiently secreted. To test this hypothesis, we stained subconfluent monolayer cultures of non-permeabilized, transiently transfected CHO cells with antibody to V5 and determined that only small amounts of the Rspo2 cysteine mutants were detected outside of the cells (Fig. 2). In contrast, wild type Rspo2 and the Q70R mutant were readily seen along the outer edges of cells, and in the proximal extracellular space (Fig. 2). When cells were permeabilized prior to immunostaining, all the transfectants exhibited strong signal inside the cells (Suppl. Fig. 2A). Western blot analysis of CHO cell lysates showed similar expression of the cysteine mutants relative to the other Rspo2 derivatives (Suppl. Fig. 2B), comparable to the pattern in STF cells. Combined with the results presented in Fig. 1B, these data indicated that the Rspo2 cysteine mutants were specifically deficient in secretion.
Figure 2. Rspo2 cysteine mutants exhibited defective secretion in CHO cell culture.

CHO cells transiently transfected with empty vector or the indicated Rspo2 construct were fixed and stained under non-permeabilization conditions to visualize Rspo2 proteins on the cell surface or in the extracellular space. Rspo2 proteins were detected with V5 antibody, while cell boundaries were indicated by phalloidin staining of polymerized actin and DAPI stained nuclei.
3.3 Isolation and structural analysis of Rspo2-2F derivatives
Based on deletion experiments showing that the two furin-like domains were necessary and sufficient for stimulation of β-catenin reporter activity [7], we expressed and purified Rspo2-2F for structural and functional studies. Pichia pastoris was chosen for recombinant expression because of the likely fidelity of anticipated disulfide bond formation and the potential convenience of yeast culture for isotope labeling used in NMR solution structure analysis. A poly-histidine tag was included at the carboxyl-terminus to facilitate purification by chelating chromatography. Initial experiments indicated that a substantial fraction of the Rspo2-2F protein did not bind to nickel-charged resin, apparently due to partial proteolysis of the histidine tag (data not shown). We subsequently adopted sequential cation exchange chromatography to isolate both wild type Rspo2-2F and a derivative containing the Q70R substitution. Typical yields from shake flask cultures were 0.1–0.25 mg/liter, with purity estimated to be ≥ 90% (see Fig. 3A and 3B for analysis of purified protein by Coomassie blue staining after SDS-PAGE and Rspo2 immunoblotting, respectively).
Figure 3. Characterization of purified Rspo2-2F and Rspo2-2F/Q70R proteins.

(A) Coomassie blue staining of purified Rspo2-2F and Rspo2-2F/Q70R proteins (700 ng/lane). (B) Western blot of purified Rspo2-2F and Rspo2-2F/Q70R proteins (50 ng/lane) with anti-Rspo2 antibody. (C) Circular dichroism spectrum of nickel-purified Rspo2-2F. The circular dichroism spectrum was collected using a Jasco J720 spectropolarimeter, with a 30 μM protein solution in PBS. (D) Amino acid sequence and cysteine assignments of Rspo2-2F. The sequence of the purified protein is shown in standard font; the italicized segment was upstream sequence removed by Kex2 cleavage. Lines between cysteines indicate disulfide linkages and asterisks highlight free cysteines; the status of underlined cysteines was not been determined. Numbering corresponds to the sequence of full-length mouse Rspo2.
A circular dichroism spectrum of the purified protein indicated an absence of α-helix (Fig. 3C, lack of peak at 208 and 222 nm), consistent with software prediction that most of the protein is comprised of random coil (unpublished observations, SJL). Amino-terminal sequence analysis by Edman degradation revealed that the yeast protease, Kex2, expected to remove a yeast signal peptide from the recombinant protein, also cleaved the protein after an internal KR sequence. This resulted in a preparation consisting primarily of protein with an amino terminus beginning at Ala32 (Fig. 3D). This amino acid residue is immediately upstream of sequence encoded by exon 2, which corresponds to the first furin-like domain [13, 16]. Therefore, the two-furin domain structure of the recombinant proteins was intact.
3.4 Identification of disulfide bonds and free cysteine residues in Rspo2 furin domains
The peptide sequence of Rspo2-2F was analyzed using an LC/ESI-MS/MS method described previously which involves the resolution of peptides by nano-LC, followed by detection and selection of peptide ions produced by the electrospray process [38]. Subsequently, each selected peptide ion was fragmented by collision induced dissociation and analyzed by MS/MS. Based on the mass of the peptide ion and its MS/MS fragmentation pattern, the sequence of the peptide was conclusively verified using the protein database searching program Sequest. With a combination of two proteolytic enzymes, trypsin and chymotrypsin, nearly the entire amino acid sequence of Rspo2-2F was confirmed, beginning with Ala32. Only ten amino acids out a total of 109 were not detected in the tryptic or chymotryptic fragments. No significant ion signals were detected for other contaminating proteins.
The sequence of purified Rspo2-2F contains fifteen Cys residues at Cys40, Cys43, Cys46 Cys52, Cys55, Cys74, Cys78, Cys93, Cys96, Cys101, Cys104, Cys110, Cys113, Cys124 and Cys128 (Fig. 3D). To determine which Cys residues are present as free cysteine, unreduced Rspo2-2F was treated with N-ethylmaleimide to react with sulfhydryl groups. Following N-ethylmaleimide treatment, Rspo2-2F was subjected to chymotryptic or tryptic digestion, the resulting digested peptides were resolved by HPLC, and subsequently analyzed by ESI-MS/MS. Two cysteine-containing peptides (Cys55 of the tryptic peptide 55–64 and Cys96 of tryptic peptide 96–107) were found to be alkylated with N-ethylmaleimide, demonstrating that Cys55 and Cys96 are free cysteine residues. As shown below, the peptide containing Cys96 also has an intramolecular disulfide bond formed between Cys101 and Cys104. The detected masses and the corresponding amino acid sequences of these two peptides are summarized in Table 1.
Table 1.
Peptides containing free cysteine residues
| Peptide Sequence6 | Detected m/z (Charged State of Ion) |
|---|---|
| C55QQKLFFFLR | 727.8 (2+) |
| C96RIENC101DSC104FSK | 764.2 (2+) |
Chymotryptic and tryptic digests of Rspo2-2F were analyzed to determine which of the remaining Cys residues are linked together in disulfide bonds. Five chymotryptic peptides (AA# 35–44, AA# 69–83, AA# 72–82, AA# 106–118 and AA# 110–118) were shown to contain intramolecular disulfide bonds formed between Cys40 and Cys43, Cys74 and Cys78, and Cys110 and Cys113 (Table 2). Tryptic digest of Rspo2-2F produced peptides (AA# 70–86, AA# 96–107, AA# 98–107 and AA# 122–139) that contain three intramolecular disulfide bonds formed between Cys74 and Cys78, Cys101 and Cys104, and Cys124 and Cys128 (Table 2). These data established that both cysteine mutants analyzed in this study involve residues that contribute to disulfide bonds, one in each furin domain. Furthermore, the other cysteine mutations in Rspo4 associated with anonychia also correspond to residues in Rspo2 that form disulfide linkages (Cys101 and Cys124) (13). Peptides containing the three remaining cysteine residues (Cys46, Cys52 and Cys93) were not recovered and therefore, we were unable to determine whether the sulfhydryl groups of these residues are free or involved in disulfide bonds (see Fig. 3D for a summary of cysteine assignments).
Table 2.
Peptides containing disulfide bonds
| Peptide Sequence | Disulfide Linkage | Detected m/z (Charged State of Ion) |
|---|---|---|
| VSNPIC40KGC43L | C40-C43 | 516.2 (2+) |
| RQYGEC74LHSC78PSGYY | C74- C78 | 880.8 (2+) |
| GEC74LHSC78PSGY | C74- C78 | 575.7 (2+) |
| SKDFC110TKC113KVGFY | C110- C113 | 762.3 (2+) |
| C110TKC113KVGFY | C110- C113 | 523.7 (2+) |
| QYGEC74LHSC78PSGYYGHR | C74- C78 | 977.8 (2+) |
| C96RIENC101DSC104FSK | C101- C104 | 764.2 (2+) |
| IENC101DSC104FSK | C101- C104 | 572.2 (2+) |
| GRC124FDEC128PDGFAPLDETM | C124- C128 | 1008.9 (2+) |
3.5 Wild type Rspo2-2F and the Q70R derivative have contrasting activity in β-catenin reporter assay
Wild type Rspo2-2F induced a dose-dependent increase in reporter activity using the STF cell line, achieving a maximal stimulation of ~150-fold at 100 ng/ml (Fig. 4A). This effect was similar to that observed after transient transfection of a construct encoding full-length Rspo2 (Fig. 1A), implying that the truncated protein expressed in Pichia was properly folded and sufficient for strong activity. An Rspo2-2F derivative containing the Q70R mutation was much less active than Rspo2-2F (Fig. 4A), further demonstrating the negative impact of this mutation on Rspo2 biological activity. In a separate experiment, we compared the ability of the Rspo2-2F derivatives to potentiate Wnt-3a signaling in the reporter assay. Wnt-3a at a concentration of 50 ng/ml induced a ~140-fold increase in reporter activity. Both Rspo2-2F proteins synergized with Wnt-3a, but the combination of wild type Rspo2-2F and Wnt-3a was far more potent than Rspo2-2F/Q70R plus Wnt-3a (~8,000-fold vs. ~1,500-fold induction) (Fig. 4B).
Figure 4. Comparison and contrast of Rspo2-2F and Rspo2-2F/Q70R activity in β-catenin pathway.

(A) Luciferase reporter activity of purified Rspo2-2F (◆) or Rspo2-2F/Q70R (■) at different concentration (10, 30, 100 and 300 ng/ml). (B) Luciferase reporter activity of purified Rspo2-2F (100 ng/ml), Rspo2-2F/Q70R (100 ng/ml) or Wnt-3a (50 ng/ml) alone or in combination. STF cells were treated with purified proteins for 20 h in serum-free DMEM and luminescence was normalized to total protein concentration of cell lysates. Relative luciferase activity was defined as the fold stimulation of data obtained from cells treated with PBS. (C) Time course analysis of LRP6 phosphorylation and total LRP6 content in cells treated with Rspo2-2F, Rspo2-2F/Q70R or Wnt-3a alone or in combination. Cells were incubated for 1, 6 or 20 h with the indicated factors prior to processing for immunoblotting. (D) Biotinylated and total LRP6 protein in cells treated with Rspo2-2F, Rspo2-2F/Q70R or Wnt-3a alone or in combination. Following 20 h incubation with the indicated factors, intact cells were subjected to biotinylation and biotinylated proteins were pelleted with streptavidin-conjugated beads. LRP6 recovered in these pellets and LRP6 present in whole cell lysates (50 μg/lane) were detected by immunoblotting. HSP70 detection served as a loading control for the cell lysates in (C) and (D). The numbers above lanes in (D) indicate band signal intensities, after normalization to the corresponding HSP70 signal, relative to the zero time point from one of three experiments with similar results.
3.6 Wild type Rspo2-2F and the Q70R mutant each induced a sustained phosphorylation and accumulation of LRP6 at cell surface
The Rspo2-2F proteins stimulated LRP6 phosphorylation, consistent with β-catenin pathway activation (Fig. 4C and Suppl. Fig. 3). Interestingly, the timing of LRP6 phosphorylation in response to Rspo-2F differed from that seen with Wnt-3a. While the effect of Wnt-3a was maximal at 1 h and much reduced at 20 h, the reverse was true for Rspo2-2F (Fig. 4C). Rspo2-2F/Q70R also had a delayed maximal effect on LRP6 phosphorylation (Fig. 4C). The combination of Wnt-3a and Rspo2-2F proteins elicited a strong, sustained increase in LRP6 phosphorylation, reflecting the synergy observed in the reporter assay (Fig. 4B and 4C, and Suppl. Fig. 3).
The delayed peak in LRP6 phosphorylation following addition of Rspo2-2F proteins was accompanied by a small or variable accumulation of total LRP6 in whole cell lysates at 20 h (Fig. 4C and Suppl. Fig. 3). To address the possibility that the increase in LRP6 phosphorylation coincided with an elevation in its concentration at the cell surface, we labeled intact cells with biotin, precipitated biotinylated proteins with streptavidin-beads and immunoblotted for LRP6. The results showed that there was a substantial increase in the amount of LRP6 at the cell surface after a 20 h treatment with both Rspo2-2F proteins (Fig. 4D).
3.7 Both wild type Rspo2 and Rspo2/Q70R interact with LRP6 and Krm2
We explored the potential association of Rspo2 with LRP6 or Krm2 in co-IP experiments using either V5-tagged full-length Rspo2 or the Rspo2/Q70R mutant and Myc6-tagged LRP6 or FLAG-tagged Krm2. As a positive control, we also examined the co-IP of LRP6 and Krm2 with the Wnt antagonist, Dkk1, which previously had been shown to bind directly to LRP6 [39–41] and Krms [42]. Immunoblotting of cell lysates confirmed that similar amounts of LRP6 were expressed in transiently transfected HEK293 cells, as well as comparable amounts of the potential ligands (Fig. 5A). All three ligands co-precipitated with LRP6, using either Myc (Fig. 5B) or V5 antibody (Fig. 5C). The interaction of LRP6 with Dkk1 appeared to be stronger than its interaction with the Rspo2 derivatives, and the association with wild type Rspo2 was somewhat stronger than with the Q70R mutant. However, some of these differences might be attributable to variation in the efficiency of immunoprecipitation. The analysis of Rspo2 and Dkk1 association with Krm2 yielded similar results, except the interactions with all the putative ligands were roughly equivalent (Fig. 6). Qualitatively similar data were obtained with Krm1 (unpublished observations, SJL and JSR). In all the co-IP experiments, the specificity of co-precipitation was supported by an absence of LRP6, Krm2, Dkk1 and Rspo2 proteins in pellets obtained with control IgG (Fig. 5 and 6). Moreover, co-IP of Rspo2 derivatives with Fzd5 and Fzd8 yielded negative results (unpublished observations, SJL and JSR), implying that the positive data obtained in Figs. 5 and 6 was not simply due to co-expression of recombinant proteins. Thus, our experiments were consistent with reports that Rspo proteins can associate with LRP6 and Krm2.
Figure 5. Rspo2 and Rspo2/Q70R association with LRP6.

(A) Co-expression of V5-tagged Rspo2, Rspo2/Q70R and Dkk1 with LRP6-Myc6 in lysates from transiently transfected HEK293 cells. (B) Co-IP of V5-tagged proteins with LRP6-Myc6, using Myc antibody or control mouse IgG. Precipitates were immunoblotted in parallel with V5 and Myc antibodies. (C) Co-IP of V5-tagged proteins with LRP6-Myc6, using V5 antibody or control mouse IgG. Precipitates were immunoblotted in parallel with Myc and V5 antibodies.
Figure 6. Rspo2 and Rspo2/Q70R association with Krm2.

(A) Co-expression of V5-tagged Rspo2, Rspo2/Q70R and Dkk1 with Krm2-FLAG in lysates from transiently transfected CHO cells. (B) Co-IP of V5-tagged proteins with Krm2-FLAG using FLAG antibody or control mouse IgG. Precipitates were immunoblotted in parallel with V5 and FLAG antibodies. (C) Co-IP of V5-tagged proteins with Krm2-FLAG, using V5 antibody or control mouse IgG. Precipitates were immunoblotted in parallel with FLAG and V5 antibodies.
4. Discussion
This study established that specific point mutations in the furin domains of Rspo2 markedly inhibit its ability to stimulate β-catenin transcriptional activity. While the loss of activity associated with C78Y and C113R substitutions was attributable to a defect in secretion, the Q70R mutation did not impair Rspo2 processing. Rather, this mutation dramatically reduced the intrinsic activity of Rspo2 in the β-catenin reporter assay. The data suggest that the Q70R substitution disrupts one or more molecular interactions important for Rspo signaling. Consistent with this view, software analysis of Rspo structure predicted that Q70 was present on the protein surface [[16] and unpublished data, SJL], and a surface location of R70 was inferred from our observation that Rspo2-2F/Q70R bound more tightly to a cationic exchange resin than Rspo2-2F (data not shown). A comparison of wild type and Rspo2 derivatives containing the Q70R substitution should provide useful insights into the mechanisms of Rspo activity, as illustrated in this report and discussed below.
The Rspo2 point mutants analyzed in this project were created to mimic mutations that occur at highly conserved sites in the Rspo4 gene of individuals with anonychia. Until now there was no evidence that such mutations decreased signaling in the β-catenin pathway. A recent report showed that Rspo4 potentiates Wnt-3a activity in a SuperTopFlash reporter assay [24]. Thus, it is likely that the mutations in Rspo4 also diminish signaling through the β-catenin pathway, thereby contributing to impaired fingernail and toenail formation.
The analysis of Cys linkages in Rspo2 was noteworthy in a number of respects. To our knowledge this is the first report of the disulfide-bonding pattern for a furin-like domain, and it indicated that short loops typify these motifs. Taken together, there were five disulfide bridges identified in the two domains, and all of them joined residues that were only separated by two or three intervening amino acids. Such short loops presumably would impose relatively limited constraints on protein folding and structure. In addition, there were two free Cys residues, one in each domain, suggesting that free Cys residues also may be characteristic of furin-like domains. While it is possible that the appearance of a free Cys in our two-furin domain derivative resulted from the loss of an interaction with a Cys elsewhere in full-length Rspo2, this seems unlikely because there probably is only one remaining unpaired Cys in the mature protein, located in the region linking the second furin-like domain to the thrombospondin type-1 repeat (Cys141). The six Cys residues in the thrombospondin domain presumably form three disulfide bonds within this motif [43], while Cys21 has been predicted to be the C-terminal residue of the signal peptide sequence [1, 25]. Moreover, the potent biological activity of Rspo2-2F demonstrated that the presence of the free Cys residues had little or no deleterious effect.
The strong activity of Rspo2-2F in the reporter assay reinforced the point mutant data in establishing the importance of the furin domains for β-catenin signaling. Moreover, our results provided new information about the mechanism whereby Rspos potentiate Wnt stimulation of the β-catenin pathway. As summarized above, LRP6 phosphorylation is an early event in activation of the pathway, and others have demonstrated that Rspos can elicit LRP6 phosphorylation and/or promote the response to Wnt treatment (24, 26, 33). Here we showed that the time course of LRP6 phosphorylation induced by Rspo2-2F is significantly different than that of Wnt-3a. While Wnt-3a triggered an increase in LRP6 phosphorylation that peaked within 1 h, the maximal effect of Rspo2-2F was seen at the latest time point, 20 h. The marked synergy of Rspo2-2F and Wnt-3a in stimulating β-catenin signaling presumably was mediated, at least in part, by the sustained, high level of LRP6 phosphorylation that was evident throughout the 20 h period. Consistent with this pattern, we also documented a substantial increase in the amount of LRP6 at the cell surface after 20 h (Fig. 4D). Somewhat surprisingly, similar effects were observed with Rspo2-2F/Q70R, despite the fact that this variant exhibited much less activity than Rspo2-2F in the reporter assay (Fig. 4A). However, the Q70R mutant still showed synergy with Wnt-3a in the reporter assay, suggesting that the shared effects of wild type and mutant Rspo2-2F on LRP6 phosphorylation and accumulation are important mechanisms contributing to their potentiation of Wnt activity in the β-catenin pathway. It is worth noting that Rspo4 also had little activity when tested alone in a SuperTopFlash assay, but dramatically enhanced the activity of Wnt-3a [24]. Thus, an ability to markedly enhance Wnt activity while having minimal impact when added alone is not unique to Rspo2-2F/Q70R. Therefore, the effects of Rspo2-2F proteins on LRP6 phosphorylation and accumulation may signify a common Rspo mechanism for the stimulation of Wnt/β-catenin signaling.
How Rspo proteins regulate LRP6 phosphorylation and stability is currently a matter of debate and speculation. Binnerts et al. presented evidence that Rspo1 prevented the Dkk-dependent internalization of LRP6, via an association with Krms [33]. However, others have argued that Krm1/2 are not required for Rspo activity, based on experiments with embryos that lack expression of Krms [35]. The idea that Dkk blocks Wnt/β-catenin signaling by down-regulating LRP6 also has been questioned [34]. Even if Rspos disrupt Dkk-dependent internalization of LRP6, additional mechanisms may contribute to Rspo activity. The delayed onset and sustained phosphorylation of LRP6, as well as its accumulation in response to Rspo2 derivatives might result from enhanced recycling of LRP6 to the plasma membrane, as recycling of LRP6 has been described (44). Our co-immunoprecipation experiments involving Rspo2 derivatives and LRP6 or Krms suggest that interactions between these molecules contribute to Rspo2 activity. Nonetheless, the large quantitative differences in reporter activity of wild type and Q70R derivatives, despite their similar behavior in the co-IP experiments, implied that additional interactions may be important.
In summary the present findings emphasize the importance of the furin domains for Rspo activity in the β-catenin pathway. Point mutation analysis revealed a critical role of individual amino acid residues for this biological activity, both intrinsic activity and activity dependent on the secretion of Rspo protein. The mapping of disulfide bonds and free cysteine residues in the furin domains provided new structural information about these functionally important motifs. The delayed, but sustained LRP6 phosphorylation and accumulation of LRP6 protein induced by Rspo2 offer new insights about Rspo activity in the β-catenin pathway.
Supplementary Material
Supplementary Figure 1. Relative expression of V5-tagged Rspo2 proteins in STF cells. Cells were transiently transfected with vectors encoding wild-type Rspo2, Rspo2/C78Y, Rspo2/C113R or Rspo2/Q70R and cell lysates were immunoblotted with anti-V5 or anti-HSP70, as illustrated in Fig. 1B. Anti-V5 (Rspo2) band intensity was analyzed by densitometry and normalized to the corresponding HSP70 band. Relative band intensity was defined as the ratio of this normalized value to the normalized value of wild-type Rspo2. Results are the means and SEM (error bars) from three separate experiments. Statistical analysis revealed that there was no significant difference in the relative expression of the wild-type and mutant Rspo2 proteins.
Supplementary Figure 2. Expression of V5-tagged Rspo2 proteins in CHO cells. (A) Immunofluorescent staining of permeabilized CHO cells transiently transfected with empty vector or constructs encoding wild-type Rspo2, Rspo2/C78Y, Rspo2/C113R or Rspo2/Q70R. Rspo2 proteins were detected with V5 antibody (green), while phalloidin stained polymerized actin (red) and DAPI stained nuclei (blue). (B) Immunoblot analysis of CHO cells transiently transfected with empty vector or constructs encoding Rspo2 derivatives. Rspo2 proteins in whole cell lysates were detected with antibody to V5, while blotting with antibody to HSP70 ensured that similar amounts of total protein were loaded in each of the lanes.
Supplementary Figure 3. Quantitative analysis of phosphorylated and total LRP6 in STF cells treated with Wnt-3a, Rspo2-2F or Rspo2-2F/Q70R protein alone or in combination. Densitometric analysis of (A) LRP6 phosphorylation and (B) total LRP6 content in cells treated with the indicated concentrations of Rspo2-2F, Rspo2-2F/Q70R and/or Wnt-3a for 1, 6 or 20 h prior to processing for immunoblotting. Cell lysates also were immunoblotted for HSP70 as a loading control. The intensity of the LRP6 bands from each of three separate experiments (illustrated in Fig. 4C) were normalized to the corresponding HSP70 band, and relative band intensity was defined as the ratio of this normalized value to the normalized value of the time zero control. Means and SEM (error bars) are presented in the histogram. *significant difference relative to the zero time point, p<0.05; #significant difference relative to the Wnt-3a 20 h time point, p<0.05; §trend to significant difference relative to the Wnt-3a 20 h time point, 0.05< p<0.06.
Acknowledgments
The authors thank Drs. Xi He and Christof Niehrs for providing plasmid constructs, Dr. Jeremy Nathans for providing STF cells, Dr. Eric Anderson for preliminary analysis of the disulfide bonding pattern of Rspo2-2F, and the NCI-Frederick Protein Chemistry Lab for performing the Edman degradation amino-terminal sequence analysis of Rspo2-2F. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. MS analysis was supported by grants from the National Institutes of Health, Grant P20 MD000262 and the National Science Foundation, Grant CHEM-0619163.
Footnotes
Abbreviations: Rspo, R-spondin; LRP5/6, low density lipoprotein receptor-related proteins 5 or 6; Fzd, Frizzled; Dkk, Dickkopf; Krm, Kremen; Rspo2-2F, R-spondin2 two furin-domain derivative; STF cells, a HEK293 cell line stably expressing a SuperTopFlash reporter gene; CM, conditioned medium; PBS, phosphate-buffered saline; LC/ESI-MS/MS, liquid chromatography/electrospray ionization-tandem mass spectrometry; co-IP, co-immunoprecipitation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim KA, Zhao J, Andarmani S, Kakitani M, Oshima T, Binnerts ME, Abo A, Tomizuka K, Funk WD. Cell Cycle. 2006;5:23. doi: 10.4161/cc.5.1.2305. [DOI] [PubMed] [Google Scholar]
- 2.Nam JS, Turcotte TJ, Yoon JK. Gene Expr Patterns. 2007;7:306. doi: 10.1016/j.modgep.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Chen JZ, Wang S, Tang R, Yang QS, Zhao E, Chao Y, Ying K, Xie Y, Mao YM. Mol Biol Rep. 2002;29:287. doi: 10.1023/a:1020479301379. [DOI] [PubMed] [Google Scholar]
- 4.Aoki M, Kiyonari H, Nakamura H, Okamoto H. Dev Growth Differ. 2008;50:85. doi: 10.1111/j.1440-169X.2007.00978.x. [DOI] [PubMed] [Google Scholar]
- 5.Nam JS, Park E, Turcotte TJ, Palencia S, Zhan X, Lee J, Yun K, Funk WD, Yoon JK. Dev Biol. 2007;311:124. doi: 10.1016/j.ydbio.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell SM, Schreiner CM, Wert SE, Mucenski ML, Scott WJ, Whitsett JA. Development. 2008;135:1049. doi: 10.1242/dev.013359. [DOI] [PubMed] [Google Scholar]
- 7.Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. Dev Cell. 2004;7:525. doi: 10.1016/j.devcel.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 8.Aoki M, Mieda M, Ikeda T, Hamada Y, Nakamura H, Okamoto H. Dev Biol. 2007;301:218. doi: 10.1016/j.ydbio.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, Guerra L, Schedl A, Camerino G. Nat Genet. 2006;38:1304. doi: 10.1038/ng1907. [DOI] [PubMed] [Google Scholar]
- 10.Capel B. Nat Genet. 2006;38:1233. doi: 10.1038/ng1106-1233. [DOI] [PubMed] [Google Scholar]
- 11.Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, Grammatico B, Remotti D, Saccucci P, Valentini F, Mazzilli MC, Majore S, Grammatico P. Hum Mutat. 2008;29:220. doi: 10.1002/humu.20665. [DOI] [PubMed] [Google Scholar]
- 12.Wilhelm D. Bioessays. 2007;29:314. doi: 10.1002/bies.20553. [DOI] [PubMed] [Google Scholar]
- 13.Blaydon DC, Ishii Y, O’Toole EA, Unsworth HC, Teh MT, Ruschendorf F, Sinclair C, Hopsu-Havu VK, Tidman N, Moss C, Watson R, de Berker D, Wajid M, Christiano AM, Kelsell DP. Nat Genet. 2006;38:1245. doi: 10.1038/ng1883. [DOI] [PubMed] [Google Scholar]
- 14.Bergmann C, Senderek J, Anhuf D, Thiel CT, Ekici AB, Poblete-Gutierrez P, van Steensel M, Seelow D, Nurnberg G, Schild HH, Nurnberg P, Reis A, Frank J, Zerres K. Am J Hum Genet. 2006;79:1105. doi: 10.1086/509789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishii Y, Wajid M, Bazzi H, Fantauzzo KA, Barber AG, Blaydon DC, Nam JS, Yoon JK, Kelsell DP, Christiano AM. J Invest Dermatol. 2008;128:867. doi: 10.1038/sj.jid.5701078. [DOI] [PubMed] [Google Scholar]
- 16.Bruchle NO, Frank J, Frank V, Senderek J, Akar A, Koc E, Rigopoulos D, van Steensel M, Zerres K, Bergmann C. J Invest Dermatol. 2008;128:791. doi: 10.1038/sj.jid.5701088. [DOI] [PubMed] [Google Scholar]
- 17.Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, Binnerts M, Liu Y, Boyle B, Park E, Emtage P, Funk WD, Tomizuka K. Science. 2005;309:1256. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J, de Vera J, Narushima S, Beck EX, Palencia S, Shinkawa P, Kim KA, Liu Y, Levy MD, Berg DJ, Abo A, Funk WD. Gastroenterology. 2007;132:1331. doi: 10.1053/j.gastro.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Lowther W, Wiley K, Smith GH, Callahan R. J Virol. 2005;79:10093. doi: 10.1128/JVI.79.15.10093-10096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J. J Hilkens Nat Genet. 2007;39:759. doi: 10.1038/ng2034. [DOI] [PubMed] [Google Scholar]
- 21.Logan CY, Nusse R. Annu Rev Cell Dev Biol. 2004;20:781. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 22.Kamata T, Katsube K, Michikawa M, Yamada M, Takada S, Mizusawa H. Biochim Biophys Acta. 2004;1676:51. doi: 10.1016/j.bbaexp.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 23.Clevers H. Cell. 2006;127:469. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Kim KA, Wagle M, Tran K, Zhan X, Dixon MA, Liu S, Gros D, Korver W, Yonkovich S, Tomasevic N, Binnerts M, Abo A. Mol Biol Cell. 2008;19:2588. doi: 10.1091/mbc.E08-02-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK. J Biol Chem. 2006;281:13247. doi: 10.1074/jbc.M508324200. [DOI] [PubMed] [Google Scholar]
- 26.Wei Q, Yokota C, Semenov MV, Doble B, Woodgett J, He X. J Biol Chem. 2007;282:15903. doi: 10.1074/jbc.M701927200. [DOI] [PubMed] [Google Scholar]
- 27.Lu W, Kim KA, Liu J, Abo A, Feng X, Cao X, Li Y. FEBS Lett. 2008;582:643. doi: 10.1016/j.febslet.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 28.Huang H, He X. Curr Opin Cell Biol. 2008;20:119. doi: 10.1016/j.ceb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C. Nature. 2005;438:867. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- 30.Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X. Nature. 2005;438:873. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Science. 2007;316:1619. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- 32.Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, Almeida K, Wang J, Doble B, Woodgett J, Wynshaw-Boris A, Hsieh JC, He X. Development. 2008;135:367. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binnerts ME, Kim KA, Bright JM, Patel SM, Tran K, Zhou M, Leung JM, Liu Y, Lomas WE, 3rd, Dixon M, Hazell SA, Wagle M, Nie WS, Tomasevic N, Williams J, Zhan X, Levy MD, Funk WD, Abo A. Proc Natl Acad Sci U S A. 2007;104:14700. doi: 10.1073/pnas.0702305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semenov MV, Zhang X, He X. J Biol Chem. 2008;283:21427. doi: 10.1074/jbc.M800014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellwanger K, Saito H, Clement-Lacroix P, Maltry N, Niedermeyer J, Lee WK, Baron R, Rawadi G, Westphal H, Niehrs C. Mol Cell Biol. 2008;28:4875. doi: 10.1128/MCB.00222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Cell. 2004;116:883. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 37.Endo Y, Beauchamp E, Woods D, Taylor WG, Toretsky JA, Uren A, Rubin JS. Mol Cell Biol. 2008;28:2368. doi: 10.1128/MCB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yen TY, Macher BA. Methods Enzymol. 2006;415:103. doi: 10.1016/S0076-6879(06)15007-7. [DOI] [PubMed] [Google Scholar]
- 39.Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Curr Biol. 2001;11:951. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 40.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. Nature. 2001;411:321. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 41.Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Nat Cell Biol. 2001;3:683. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- 42.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Nature. 2002;417:664. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 43.Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang JH. J Cell Biol. 2002;159:373. doi: 10.1083/jcb.200206062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan Z, Vijayakumar S, de la Torre TV, Rotolo S, Bafico A. Mol Cell Biol. 2007;27:7291. doi: 10.1128/MCB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Relative expression of V5-tagged Rspo2 proteins in STF cells. Cells were transiently transfected with vectors encoding wild-type Rspo2, Rspo2/C78Y, Rspo2/C113R or Rspo2/Q70R and cell lysates were immunoblotted with anti-V5 or anti-HSP70, as illustrated in Fig. 1B. Anti-V5 (Rspo2) band intensity was analyzed by densitometry and normalized to the corresponding HSP70 band. Relative band intensity was defined as the ratio of this normalized value to the normalized value of wild-type Rspo2. Results are the means and SEM (error bars) from three separate experiments. Statistical analysis revealed that there was no significant difference in the relative expression of the wild-type and mutant Rspo2 proteins.
Supplementary Figure 2. Expression of V5-tagged Rspo2 proteins in CHO cells. (A) Immunofluorescent staining of permeabilized CHO cells transiently transfected with empty vector or constructs encoding wild-type Rspo2, Rspo2/C78Y, Rspo2/C113R or Rspo2/Q70R. Rspo2 proteins were detected with V5 antibody (green), while phalloidin stained polymerized actin (red) and DAPI stained nuclei (blue). (B) Immunoblot analysis of CHO cells transiently transfected with empty vector or constructs encoding Rspo2 derivatives. Rspo2 proteins in whole cell lysates were detected with antibody to V5, while blotting with antibody to HSP70 ensured that similar amounts of total protein were loaded in each of the lanes.
Supplementary Figure 3. Quantitative analysis of phosphorylated and total LRP6 in STF cells treated with Wnt-3a, Rspo2-2F or Rspo2-2F/Q70R protein alone or in combination. Densitometric analysis of (A) LRP6 phosphorylation and (B) total LRP6 content in cells treated with the indicated concentrations of Rspo2-2F, Rspo2-2F/Q70R and/or Wnt-3a for 1, 6 or 20 h prior to processing for immunoblotting. Cell lysates also were immunoblotted for HSP70 as a loading control. The intensity of the LRP6 bands from each of three separate experiments (illustrated in Fig. 4C) were normalized to the corresponding HSP70 band, and relative band intensity was defined as the ratio of this normalized value to the normalized value of the time zero control. Means and SEM (error bars) are presented in the histogram. *significant difference relative to the zero time point, p<0.05; #significant difference relative to the Wnt-3a 20 h time point, p<0.05; §trend to significant difference relative to the Wnt-3a 20 h time point, 0.05< p<0.06.