

Abstract
The clinical course of patients with two relatively common LQT3 mutations has not been well described. In this study, we investigated the mutational-specific risk in patients with deletional (ΔKPQ) and missense (D1790G) mutations involving the SCN5A gene. The study population involved 50 patients with the ΔKPQ mutation and 35 patients with the D1790G mutation. The cumulative probability of a first cardiac event (syncope, aborted cardiac arrest, or LQTS-related sudden death) was evaluated using the Kaplan-Meier method. The Cox proportional-hazards survivorship model was used to determine the independent contribution of clinical and genetic factors to the first occurrence of cardiac events from birth through age 40 years. The Andersen-Gill proportional-intensity regression model was used to analyze the factors associated with recurrent syncope. Patients with a ΔKPQ mutation had a significantly higher probability of a first cardiac event from birth through age 40 years (34%) than those with D1790G mutation (20%) with p<0.001. Multivariate analysis demonstrated an increased risk of cardiac events among ΔKPQ carriers as compared to D1790G carriers (hazard ratio = 2.42, p<0.0001) after adjustment for sex and QTc duration. Patients with ΔKPQ mutations also had an increased risk for recurrent syncope (hazard ratio = 5.20, p<0.001). The clinical course of LQT3 patients with ΔKPQ mutations is more virulent than those with D1790G mutations, and this effect is independent of QTc duration. The findings highlight the importance of knowing the specific mutation in risk stratification of LQT3 patients.
Keywords: Long QT Syndrome, Long QT Syndrome Type-3, SCN5A, Genetics
The congenital long QT syndrome (LQTS) is an inherited disorder affecting the cardiac ion channels, with prolonged ventricular repolarization contributing to syncope, tachyarrhythmias, and sudden death. The LQT3 genotype, which includes a spectrum of different mutations, accounts for only 7-10% of patients with LQTS, and most studies have been restricted to case reports or small numbers of patients with limited follow-up.1-3 Patients with LQT3 have an increased risk of lethal cardiac events compared to patients with LQT1 or LQT2 forms of this disorder,4,5 but the reports to date are limited. The majority of these events typically occur when the patients are resting or asleep, presumably at a slow heart rate. 6,7 β-blocker therapy, the mainstay treatment for LQT1 and LQT2 patients, offers limited protection for LQT3 patients. 6,8 The lack of long-term, clinical follow-up involving patients with LQT3 and the concern about the high risk of sudden cardiac death have led many clinicians to consider an implantable defibrillator, even in the absence of symptoms.3 Within the International LQTS Registry, we identified 2 LQT3 mutations, D1790G and ΔKPQ, that have had detailed electrophysiologic study. The D1790G mutation exhibits a prolonged cardiac ventricular action potential due to calcium-sensitive exchange modulation.9 The ΔKPQ mutation causes transitions from the inactivated state back into the open state with abnormal late entry of sodium into the myocardial cells.10,11 Sodium-channel blockers such as mexiletine, flecainide and ranolazine have been shown to shorten the QTc interval in patients with D1790G and ΔKPQ mutations; however, the clinical efficacy of these drugs remains unknown in patients with these 2 mutations.12-14 In this study, we investigated the clinical course, ECG phenotype, and treatment efficacy in patients with these two distinct LQT3 mutations.
METHODS
The study population of 85 patients was drawn from the International Long QT Syndrome Registry, and consisted of 50 affected subjects from 3 unrelated families carrying the ΔKPQ mutation, and 35 subjects from 2 unrelated families from Israel carrying the D1790G mutation. The patients carrying the D1790G mutation were either from a large multigenerational kindred originating from Tunisia or a family from Israel. The SCN5A mutations were identified using standard genetic tests performed in academic molecular-genetic laboratories. The LQTS Registry study was approved by the University of Rochester Institutional Review Board, and informed consent was obtained from all study participants or their guardians. The first recorded ECG obtained at the time of patient enrollment in the Registry was used in the current analysis.
Subjects were included if they had a genetically confirmed D1790G or ΔKPQ mutation or if they were obligate carriers of the mutation. Family members who were untested for the D1790G or ΔKPQ mutation were included in the study sample if they had a QTc≥480 ms. Lead II of the baseline ECG was used for the analysis of T-wave measurements, which included QT onset, QT peak, T-wave duration, and T-wave amplitude.
Statistical analysis was performed using SAS 9.1.3 for Windows (SAS Institute, Cary, NC, USA). The cumulative probability of a first cardiac event (syncope, aborted cardiac arrest, or LQTS-related death) was assessed by the Kaplan-Meier method with significance testing by the log-rank statistic. The Cox proportional-hazards survivorship model was used to evaluate the independent contribution of clinical and genetic factors to the first occurrence of cardiac events from birth through the age of 40 years. In the analyses of the factors and outcomes associated with recurrent syncope, the end-point intensity function was adjusted for covariate effects by the Andersen Gill proportional-intensity regression model15. This method is analogous to the proportional-hazards method, but examines the risk of repeated events and not just a first event.
RESULTS
The clinical characteristics of the patients with ΔKPQ and D1790G mutations are shown in Table 1. Patients with ΔKPQ mutations were more likely to be treated with beta-blockers, implanted defibrillators, left cervicothoracic sympathetic ganglionectomy, and pacemakers than patients with D1790G mutations. Patients with D1790G mutations were primarily treated with flecainide, and this medication was discontinued in 3 patients after new onset of Brugada-type pattern on the ECG. Only 2 patients with the ΔKPQ mutation were treated with mexiletine. Among patients with the ΔKPQ mutation, 3 experienced atrial fibrillation before the age of 25. Six patients with the ΔKPQ mutation developed various degrees of atrio-ventricular block during follow-up, including 3 patients with first-degree block, 1 patient with second-degree block, and 2 patients with third-degree heart block. None of the patients with D1790G mutations suffered from atrial fibrillation or heart block.
Table 1.
Patient Characteristics
| ΔKPQ (n=50) |
D1790G (n=35) |
P value |
|
|---|---|---|---|
| Female | 21 (42%) | 16 (46%) | 0.73 |
| Any Cardiac Event, (%) | |||
| Total | 15 (30%) | 6 (17%) | 0.08 |
| Syncope | 12 (24%) | 2 (7%) | 0.12 |
| Aborted cardiac arrest | 2 (4%) | 1 (3%) | 0.77 |
| LQTS-related death | 3 (6%) | 3 (9%) | 0.65 |
| Syncope on Beta Blockers | 5 (10%) | 0 | 0.15 |
| 1st Cardiac Event (% of 1st events) | |||
| Syncope | 13 (26%) | 3 (9%) | 0.04 |
| Aborted cardiac arrest | 0 | 1 (3%) | 0.23 |
| LQTS-related death | 2 (4%) | 2 (6%) | 0.71 |
| Conduction Disorders | |||
| Atrioventricular block | 6 (12%) | 0 | 0.06 |
| Torsade de pointes | 1 (2%) | 2 (6%) | 0.36 |
| Atrial fibrillation | 3 (6%) | 0 | 0.55 |
| Therapy | |||
| Flecainide | 8 (16%) | 11 (31%) | 0.09 |
| Beta-Blockers | 31 (62%) | 4 (11%) | <0.0001 |
| Implanted defibrillator | 11 (22%) | 2 (6%) | 0.04 |
| Pacemaker | 9 (18%) | 0 | 0.001 |
| Left cardiac ganglionectomy | 4 (8%) | 0 | 0.09 |
The baseline ECG characteristics were analyzed by age groups consisting of patients <10 years of age, and patients ≥ 10 years of age, to account for the higher heart rate in pediatric patients. In the younger age group, the QRS and PR intervals were longer in D1790G carriers than in ΔKPQ mutation carriers; however measurements in both mutations were within normal limits. In adults, the QTc interval was longer and the frequency of QTc > 500 ms was greater in ΔKPQ mutation carriers than in D1790G carriers (Table 2).
Table 2.
Electrocardiogram Characteristics in Lead II
| ΔKPQ | 1790G | P-value | |
|---|---|---|---|
| Pediatric ECG’s: Age <10 years | |||
| Number of ECG’s | 18 | 11 | |
| ECG on Beta Blockers | 4 | 1 | |
| ECG on Mexilitine | 0 | 0 | |
| ECG on Flecainide | 0 | 1 | |
| Mean age, (years) | 2.1±2.6 | 3.5±2.6 | 0.17 |
| QTc, (ms) | 489±48 | 496±55 | 0.72 |
| QTc> 500 ms | 8 (36%) | 6 (55%) | 0.32 |
| PR, (ms) | 129±39 | 156±47 | 0.01 |
| QRS, (ms) | 68±13 | 87±25 | 0.03 |
| RR, (ms) | 581±159 | 558±86 | 0.66 |
| Adult ECG’s: Age ≥10 years | |||
| Number of ECG’s | 28 | 24 | |
| ECG on Beta Blockers | 1 | 0 | |
| ECG on Mexilitine | 0 | 0 | |
| ECG on Flecainide | 0 | 1 | |
| Mean age, (years) | 31±16 | 30.9±7 | 0.96 |
| QTc, (ms) | 534±38 | 510±8 | 0.05 |
| QTc> 500 ms | 21 (78%) | 8 (42%) | 0.01 |
| PR, (ms) | 174±2 | 164±5 | 0.14 |
| QRS, (ms) | 90±21 | 80±18 | 0.06 |
| RR, (ms) | 991±316 | 930±184 | 0.39 |
Patients with ΔKPQ mutations had a higher cardiac event rate than those with D1790G mutations (34% vs. 20%, respectively, p<0.001, Fig. 1). In addition, most of the cardiac events occurred after the age of 10 (Fig. 1). Cox proportional hazards regression analysis demonstrated that patients with ΔKPQ mutations had an increased risk of cardiac events (HR= 2.42, p<0.001 Table 3) and recurrent syncope (HR=5.20, p=0.02, Table 4) compared to those with D1790G mutations.
Figure 1.
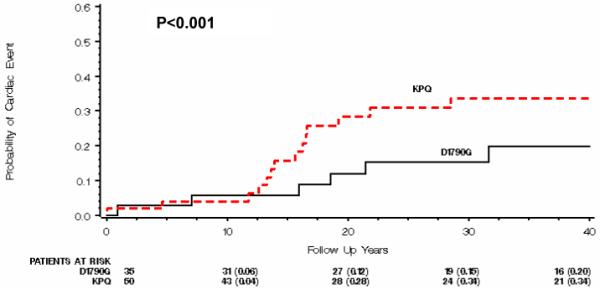
Kaplan-Meier estimates of the probability of a first cardiac event from birth to 40 years of age.
Table 3.
Multivariate Analysis: Predictors of First Cardiac Event
| Variable | HR, 95% CI | P-value |
|---|---|---|
| ΔKPQ | 2.42 (1.55-3.77) | <0.0001 |
| QTc≥500 | 1.63 (0.59-4.56) | 0.35 |
| Male | 1.80 (0.68-4.76) | 0.24 |
Analyses consisted of a total of 85 patients, 23 of whom had events.
Table 4.
Multivariate Analysis: Factors Associated with Recurrent Syncope
| Variable | HR (95% CI) | P-value |
|---|---|---|
| ΔKPQ | 5.20 (1.31, 20.70) | 0.02 |
| QTc≥ 500 ms | 0.95 (0.19, 4.62) | 0.95 |
| Male | 0.99 (0.25, 3.89) | 0.98 |
Analysis consisted of a total of 27events.
We explored the LQTS-related therapy at the time of cardiac events. Among the 15 ΔKPQ patients who experienced a first cardiac event through age 40 years, 12 (80%) were on beta-blockers during the event (average years prior to first cardiac event: 2.2 ± 4.7), and 1 patient was on flecainide during a cardiac event (average follow-up time to the event: 5.4±3.5 years). Among the 6 D1790G carriers who experienced a cardiac event, 1 (17%) was on flecainide during the event (average follow-up: 4.1±4.1 years). In addition, among ΔKPQ patients who experienced events, 1 had received a pacemaker and 2 had an ICD; none of the 4 ΔKPQ patients with a prior left cervicothoracic sympathetic ganglionectomy experienced a subsequent cardiac event.
DISCUSSION
This is the first study to investigate the clinical course of LQTS patients with 2 different LQT3 mutations. The findings show meaningful differences in the clinical severity between patients affected with ΔKPQ and D1790G mutations, with ΔKPQ patients at a considerably higher risk for first cardiac events and for recurrent syncope than those with the D1790 mutation.
LQT3 results from mutations of the SCN5A gene on Chromosome 3p21 that affect the structure of the cardiac α-subunit of the voltage-gated sodium ion channel. Mutations in SCN5A have also been associated with Brugada syndrome and cardiac conduction disorders 16,17,18 in what is referred to as the cardiac sodium channel overlap syndromes.18 The ΔKPQ mutation (in-frame deletion of residues Lys 1505, Pro 1506, and Gln 1507) is located within the intracellular linker between domains III and IV of the voltage-gated channel (Figure 2). This region is critical for fast inactivation of the channel and contains a 3-residue hydrophobic motif, which acts as a latch in closing the pore1. In vitro studies have shown that mutant ΔKPQ channels result in a sustained inward current during membrane depolarization, with multiple reopenings of the channels causing delayed repolarization 10. Animal studies indicate that ΔKPQ mutations are associated with rate-dependent early after depolarization-driven dysfunction as well as delayed after depolarizations. Arrhythmias may be triggered by sudden accelerations in heart rate or premature beats leading to lengthening of the action potential.19,20
Figure 2.

SCN5A Sodium Ion Channel and location of D1790G and DeltaKPQ mutations.
The D1790G mutation is located in the C-terminus region of the SCN5A protein channel (Figure 2). In vitro experiments show alterations in the steady-state inactivation of the channels, but also show conflicting data on the presence of persistent inward current 21,22. Based on computational models, the D1790G mutation may induce changes in sodium channel activity, with prolonged action potentials that lengthen with slower heart rates9. Additional research has shown that the D1790G mutation is modulated by protein kinase A (PKA), and the bursts of current from the non-inactivating channels are enhanced by PKA-dependent phosphorylation. Comparison of the D1790G mutation with the ΔKPQ mutation has shown that D1790G alters steady-state inactivation of the channels while ΔKPQ does not.22
Pharmacological studies have shown that mexiletine, flecainide, and ranolazine are effective in shortening the QTc interval and normalizing the repolarization T-wave pattern in patients with ΔKPQ or D1780G mutations.13,14,23 Carriers of ΔKPQ mutation have exhibited “Brugada-like” ECG changes during exposure to intravenous flecainide.24 At lower doses of flecainide, no side effects have been noted.23 In the current study, 3 patients discontinued flecainide due to Brugada-type ECG changes.
Patients with LQT3 mutations are considered to be at high risk for life-threatening cardiac events.4 The paucity of aborted cardiac arrest and death events together with the relatively high utilization of beta-blockers, flecainide, and implanted defibrillators in the current study limits data-based recommendations regarding therapy for patients with ΔKPQ or D1790G mutations. Beta-blockers do not seem to be helpful in these patients, and the experience with long-term mexiletine, flecainide, and ranolazine therapy is limited. The more than 4-fold increased risk for first cardiac events and for recurrent syncope in those with the ΔKPQ mutation compared to those with the D1790G mutation leads us to recommend more aggressive therapy in those with the ΔKPQ mutation. One needs to individualize therapy. In ΔKPQ patients who have experienced a syncopal episode, we recommend left cervico-thoracic sympathetic ganglionectomy in small-sized younger patients and an implantable defibrillator in older patients, especially in those with QTc intervals >500ms. Patients with an aborted cardiac arrest regardless of genotype or QTc interval should have an implanted defibrillator.
The number of families with the ΔKPQ and D1790G mutations was limited, and the affected family members were from different countries. Our findings did not account for any family-specific polymorphisms or non-hereditary factors that may have affected the mutation phenotype. The inclusion of non-genotyped patients with QTc≥480 ms may have biased the study sample towards more severe phenotypes.
The clinical course of LQT3 patients with ΔKPQ mutations is more severe than that of patients with D1790G mutations, and this effect is independent of QTc duration. The findings highlight the importance of knowing the specific mutation when risk stratifying LQT3 patients for preventive therapy.
Acknowledgments
Grant Support: Supported in part by research grant HL-33843 (Moss) and HL-51618 (Moss) from the National Institutes of Health, Bethesda, Maryland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm. 2009;6:113–120. doi: 10.1016/j.hrthm.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, Lehmann MH, Hall WJ, International Long-QT Syndrome Registry Research Group Influence of genotype on the clinical course of the long-QT syndrome. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 5.Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long-QT syndrome after age 40. Circulation. 2008;117:2192–2201. doi: 10.1161/CIRCULATIONAHA.107.729368. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47. doi: 10.1111/j.1365-2796.2005.01583.x. [DOI] [PubMed] [Google Scholar]
- 8.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, Moncalvo C, Tulipani C, Veia A, Bottelli G, Nastoli J. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 9.Wehrens XH, Abriel H, Cabo C, Benhorin J, Kass RS. Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na(+) channel alpha-subunit: A computational analysis. Circulation. 2000;102:584–590. doi: 10.1161/01.cir.102.5.584. [DOI] [PubMed] [Google Scholar]
- 10.Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 11.Chandra R, Starmer CF, Grant AO. Multiple effects of KPQ deletion mutation on gating of human cardiac Na+ channels expressed in mammalian cells. Am J Physiol. 1998;274:H1643–1654. doi: 10.1152/ajpheart.1998.274.5.H1643. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 13.Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol. 2005;10:59–66. doi: 10.1111/j.1542-474X.2005.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benhorin J, Taub R, Goldmit M, Kerem B, Kass RS, Windman I, Medina A. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation. 2000;101:1698–1706. doi: 10.1161/01.cir.101.14.1698. [DOI] [PubMed] [Google Scholar]
- 15.Andersen PK, Gill RD. Cox’s regression for counting processes: A large sample study. The Annals of Statistics. 1982;10:1100–1120. [Google Scholar]
- 16.Balser J. Biophysics of Normal and Abnormal Cardiac Sodium Channel Function. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology From Cell to Bedside. Fourth Edition Saunders; Philadelphia: PA: 2004. pp. 77–87. [Google Scholar]
- 17.Wang DW, Viswanathan PC, Balser JR, George AL, Jr., Benson DW. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation. 2002;105:341–346. doi: 10.1161/hc0302.102592. [DOI] [PubMed] [Google Scholar]
- 18.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 20.Fredj S, Lindegger N, Sampson KJ, Carmeliet P, Kass RS. Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ Res. 2006;99:1225–1232. doi: 10.1161/01.RES.0000251305.25604.b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baroudi G, Chahine M. Biophysical phenotypes of SCN5A mutations causing long QT and Brugada syndromes. FEBS Lett. 2000;487:224–228. doi: 10.1016/s0014-5793(00)02360-7. [DOI] [PubMed] [Google Scholar]
- 22.An RH, Wang XL, Kerem B, Benhorin J, Medina A, Goldmit M, Kass RS. Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ Res. 1998;83:141–146. doi: 10.1161/01.res.83.2.141. [DOI] [PubMed] [Google Scholar]
- 23.Windle JR, Geletka RC, Moss AJ, Zareba W, Atkins DL. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A:DeltaKPQ mutation. Ann Noninvasive Electrocardiol. 2001;6:153–158. doi: 10.1111/j.1542-474X.2001.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome: the role of flecainide challenge. Circulation. 2000;102:945–947. doi: 10.1161/01.cir.102.9.945. [DOI] [PubMed] [Google Scholar]