

Abstract
Ventricular tachycardia and fibrillation (VT/VF) complicating Brugada syndrome, a genetic disorder linked to SCN5A mutations, and VF complicating acute myocardial infarction (AMI) have both been linked to phase 2 reentry. Because of these mechanistic similarities in arrhythmogenesis, we examined the contribution of SCN5A mutations to VT/VF complicating AMI. Nineteen consecutive patients developing VF during AMI were enrolled. Wild-type (WT) and mutant SCN5A genes were co-expressed with SCN1B in TSA201 cells and studied using whole-cell patch-clamp techniques. One missense mutation (G400A) in SCN5A was detected in a conserved region among the cohort of 19 patients. A H558R polymorphism was detected on the same allele. Unlike the other 18 patients who each developed 1-2 VF episodes during acute MI, the mutation carrier developed six episodes of VT/VF within the first 12 hours. All VT/VF episodes were associated with ST segment changes and were initiated by short-coupled extrasystoles. We describe the first sodium channel mutation to be associated with the development of an arrhythmic storm during acute ischemia. These findings suggest that a loss of function in SCN5A may predispose to ischemia induced arrhythmic storm. These results could be very useful for forensic implications regarding genetic screening in relatives.
Keywords: Ventricular tachycardia, Fibrillation, Arrhythmia, Ischemia, Sudden cardiac death
1. Introduction
The SCN5A gene encodes for the α-subunit of the human cardiac voltage-gated sodium channel Nav1.5. SCN5A mutations thus far described have been linked to sudden cardiac death associated with a number of inherited arrhythmic syndromes, including the LQT3 form of the long QT syndrome, conduction disease, atrial standstill and Brugada Syndrome (BS) [1,2]. However, their association with acquired forms of VF is not well defined. Since mechanistic similarities exist between the VT/VF caused by Brugada syndrome and that caused by VT/VF associated with myocardial ischemia (both are linked to phase 2 reentry), we hypothesized that SCN5A mutations similar to those linked to Brugada syndrome, may predispose to the development of VT/VF during AMI. The purpose of this study was to examine the contribution of SCN5A mutations to arrhythmogenesis in a group of patients who developed one or more episodes of VT/VF during acute myocardial infarction (AMI).
2. Methods
2.1. Clinical analysis
Nineteen consecutive patients admitted with AMI who developed ventricular fibrillation (VF) immediately prior to, or shortly after arrival to the intensive care unit, were studied. All patients had obvious ST segment elevation and elevation of cardiac enzymes diagnostic of AMI.
2.2. Mutation analysis of SCN5A
Genetic analysis was performed to examine the contribution of SCN5A mutations to arrhythmogenesis during AMI following written informed consent and approval from the regional Institutional Review Board. Genomic DNA was isolated from peripheral blood leukocytes using a commercial kit (Gentra System, Pure-gene). The exons of SCN5A gene were amplified and analyzed by direct sequencing. Polymerase chain reaction (PCR) products were purified with a commercial reagent (ExoSAP-IT, USB) and were directly sequenced from both directions using ABI PRISM 3100-Avant Automatic DNA sequencer. Genomic DNA from 182 ethnically-matched (Caucasian) healthy subjects, including 80 Ashkenazi Jews, was used as controls. We performed two independent PCRs to validate the nucleotide change. To assess whether G400A and H558R variations reside on the same allele, PCR experiments using the following primer sets: SCN5A exon 10 sense CTAGACTAGGTG ACTTGGAAATG and SCN5A exon 12a antisense GCTGTTCTTTTTG CCATGGAGG were performed, followed by cloning of the PCR products into a Topo TA vector (Invitrogen, Carlsbad, CA). Nineteen clones were directly sequenced in both directions using ABI PRISM 3100-Avant Automatic DNA sequencer (Applied Biosystems, Foster City, CA).
2.3. Mutagenesis and patch clamp method
Mutant sodium channels were expressed in modified human embryonic kidney cell line TSA201 as previously described [3]. Briefly, TSA201 cells were cotransfected with 12 mg of SCN5A (WT, G400A or G400A/H558R) and 4 mg of SCN1B (β1 subunit) using the calcium phosphate precipitation method. Membrane currents were measured using whole-cell patch-clamp techniques in transfected TSA201 cells.
2.4. Statistical analysis
Data are expressed as mean ± SEM. Two-tailed Student's t-test and ANOVA coupled with a Student–Newman–Keuls test were used for statistical analysis, as appropriate (SigmaStat, Jandel Scientific Software). Differences were considered to be statistically significant at a value of P < 0.05.
3. Results
3.1. Clinical observations
Nineteen patients (18 males), aged 57 ± 10 years, admitted with anterior (9 patients) and inferior (10 patients) myocardial infarction (MI) were studied (Table 1). All patients had preserved left ventricular ejection fraction, reflecting the fact that (for all except 1 patient), this was the first myocardial infarction. One patient (MMRL23) presented with an arrhythmic storm. This 70-year old male with a history of coronary disease was hospitalized because of angina exacerbation. That hospitalization course was uneventful and the patient was discharged after 5 days. However, he returned 2 days later with an evolving anterior myocardial infarction. Shortly thereafter, he developed ventricular fibrillation (VF). This patient with the arrhythmic storm was the only one in which a SCN5A mutation was uncovered. The mutation carrier had no history of previous syncope or a history of familial sudden death. Moreover, he did not display ST segment elevation suggestive of Brugada syndrome at any time prior to the AMI.
Table 1.
Clinical observations and molecular genetic analysis of SCN5A in 19 AMI patients.
| Patient ID | Gender | Age | VT/VF episodes | MI location | Culprit artery | Number of diseased coronary arteries | Site where VF occured | SCN5A non-synonymous polymorphism | SCN5A mutation |
|---|---|---|---|---|---|---|---|---|---|
| MMRL23 | M | 70 | 6 | Ant | LAD | 1 | ICU | H558R | G400A |
| MMRL48 | M | 50 | 1 | Inf | RCA | 2 | E.R. | – | – |
| MMRL49 | M | 42 | 1 | Inf | RCA | 1 | Cath Lab | – | – |
| MMRL51* | M | 48 | 1 | Inf | LCX | 2 | E.R. | – | – |
| MMRL52 | M | 43 | 1 | Ant | LAD | 1 | COH | – | – |
| MMRL53** | M | 59 | 1 | Inf | RCA | 1 | E.R. | – | – |
| MMRL54 | M | 53 | 2 | Ant | LAD | 1 | Cath Lab | H558R | – |
| MMRL55* | M | 52 | 1 | Inf | RCA | 1 | COH | H558R | – |
| MMRL56 | M | 58 | 2 | Inf | RCA | 1 | OOH; Cath Lab | – | – |
| MMRL57 | F | 54 | 1 | Ant | LAD | 0a | OOH | – | – |
| MMRL58 | M | 63 | 1 | Inf | LCX | 1 | E.R. | – | – |
| MMRL59 | M | 71 | 1 | Inf | RCA | 1b | ICCU | – | – |
| MMRL60 | M | 59 | 1 | Inf | No | 0b | H | H558R | – |
| MMRL61 | M | 66 | 1 | Ant | LAD | 1 | E.R. | R34C | – |
| MMRL62 | M | 43 | 1 | Inf | RCA | 1 | E.R. | – | – |
| MMRL63 | M | 46 | 1 | Ant | LAD | 2 | COH | – | – |
| MMRL64 | M | 54 | 1 | Ant | LAD | 1 | E.R. | – | – |
| MMRL73 | M | 73 | 1 | Ant | LAD | 1 | E.R. | H558R | – |
| MMRL74 | M | 77 | 1 | Ant | LAD | 3 | E.R. | – | – |
Patient MMRL57 had irregular vessel with suspicious Myocardial infarction (MI) location.
Patients MMRL59 and 60 had Prinzmetal Angina.
Patients MMRL51 and 55 reported a history of familial sudden death.
For patient MMRL53, this was his second inferior MI.
3.2. Molecular genetics
Genetic analysis showed that all but one patient tested negative for a SCN5A mutation. MMRL23, the only patient with the arrhythmic storm, presented with a novel missense mutation combined with H558R polymorphism in the same allele of the SCN5A gene (Fig. 1B). PCR-based sequencing analysis revealed a double peak in the sequence of exon 10 of the SCN5A gene (Fig. 1A) showing a G-to-C transversion at nucleotide 1199 predicting an amino acid substitution of Gly for Ala at codon 400 (designated G400A). This nucleotide change was not observed in 364 reference alleles, suggesting that this variation represents a disease-related mutation. Gly-400 is located in the S6 transmembrane segment of domain I of SCN5A (Fig. 1B) and is highly conserved among the members of the voltage-gated sodium channel α-subunit family and through evolution (Fig. 1C). No mutations were detected in any of long QT genes.
Fig. 1.
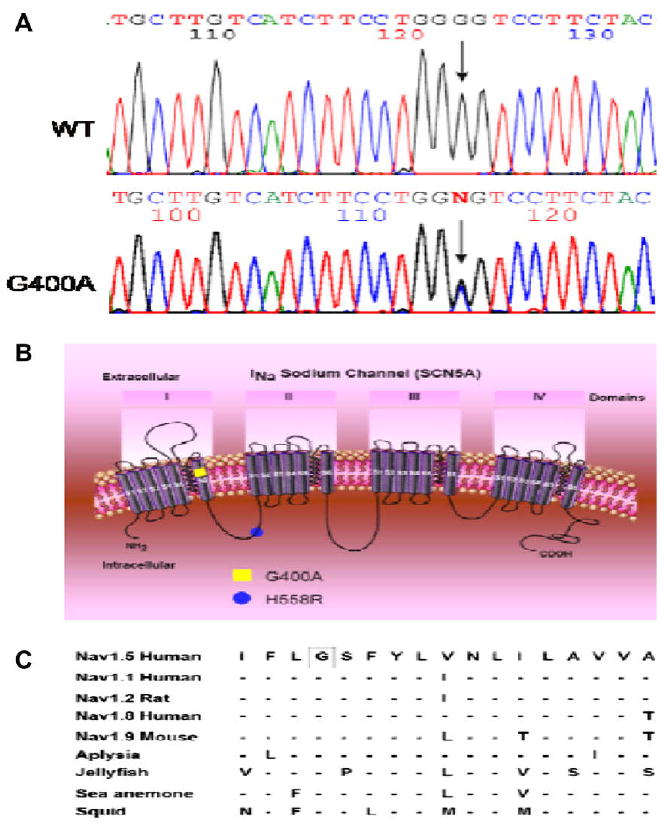
Genetic analysis of patient MMRL23. (A) PCR-based sequence of SCN5A exon 10 showing wild-type (WT) and G-to-C transversion at nucleotide 1199 (arrow) in patient MMRL23. The mutation predicts a substitution of Ala (GCG) for Gly (GGG) at position 400 (G400A). (B) Location of the G400A mutation and H558R polymorphism are indicated using the conventional transmembrane topology model. (C) Alignment of the voltage-gated sodium channel α-subunit family amino acid sequence, with related sequence shows that G400 is highly conserved among different sodium channels and different species. Dashes indicate identical residues to human SCN5A channel.
4. Discussion
Recent clinical studies have provided support for this hypothesis demonstrating a synergism between ST segment elevation and arrhythmogenesis in patients with the Brugada syndrome when an ischemic insult is superimposed [4]. In the present study, we provide evidence in support of the corollary hypothesis that SCN5A mutations can exacerbate arrhythmogenesis in the setting of AMI. Our findings suggest a genetic predisposition for acquired (i.e., ischemia related) VF in 1 out of 19 patients with AMI complicated by VF. A similar percentage of genetic anomalies predisposing to ventricular arrhythmias have been reported for other forms of “acquired arrhythmic syndromes”, such as drug-induced long QT syndrome [5]. The patient carrying the SCN5A mutation was the only one who developed an arrhythmic storm during AMI in our small series. The fact that this patient developed his first VF only at 70 years of age and only in the setting of AMI supports the notion that the SCN5A mutation in this patient was a predisposing factor for the acquired arrhythmic syndrome. The G400A missense mutation in SCN5A was found to create several defects in the function of the sodium channel, including a markedly reduced current density, impaired recovery from inactivation, and shift in the voltage dependence of inactivation to hyperpolarized potentials. The functional consequence of these changes is likely to be a reduced Na+ current during the upstroke of the action potential. Our G400A carrier had a common polymorphism (H558R) on the same allele, which in the case of other mutations. Our study shows an effect of H558R to further accentuate the effect of a loss of function mutation Previous studies demonstrated that heterogeneous loss of the action potential dome during ischemia give rise to transmural dispersion of repolarization as well as phase 2 reentry, thus precipitating reentry in the form of VT/VF [6–8]. Because SCN5A mutations are known to contribute to arrhythmogenesis in a variety of inherited diseases, including long QT and Brugada syndromes, we made an effort to ascertain whether our G400A carrier has a subclinical form of these syndromes. The mutation carrier tested negative to a sodium block challenge involving flecainide designed to unmask the Brugada syndrome, as well as to adenosine, a provocative test designed to unmask the long QT syndrome. The common long QT genes were screened and found to contain no mutations.
Study limitations
Our data support the hypothesis that the loss of function mutation in SCN5A facilitates arrhythmogenesis attending acute MI, but because of their limited nature are no more than hypothesis-forming. A more extensive study is clearly needed to test this hypothesis.
Abbreviations
- AMI
acute myocardial infarction
- BS
Brugada syndrome
- INa
sodium channel current
- Ito
transient outward current
- LAD
left anterior descending
- LIMA
left internal mammary artery
- LQT3
form of the long QT syndrome
- MI
myocardial infarction
- PCR
polymerase chain reaction
- RV
right ventricular
- VF
ventricular fibrillation
- VT/VF
ventricular tachycardia and fibrillation
- WT
wild-type
Footnotes
Supported by Fondi di Ateneo Linea D1, Università Cattolica del Sacro Cuore, Roma, Italy and Grants HL47678 (C.A.) and HL66169 (R.B.) from NHLBI, and grants from the American Heart Association (R.B.), National Heart Foundation, a program of the American Health Assistance Foundation (J.M.C.), and NYS and Florida Grand Lodges, F. & A.M.
Conflict of interest: There are no conflicts of interest or financial disclosures.
References
- 1.Roden DM. Human genomics and its impact on arrhythmias. Trends Cardiovasc Med. 2004;14:112–6. doi: 10.1016/j.tcm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. Heart Rhythm. 2004;1:42C–56C. doi: 10.1016/j.hrthm.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–9. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 4.Noda T, Shimizu W, Taguchi A, Satomi K, Suyama K, Kurita T, et al. ST-segment elevation and ventricular fibrillation without coronary spasm by intracoronary injection of acetylcholine and/or ergonovine maleate in patients with Brugada syndrome. J Am Coll Cardiol. 2002;40:1841–7. doi: 10.1016/s0735-1097(02)02494-4. [DOI] [PubMed] [Google Scholar]
- 5.Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025–32. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 7.Yan GX, Joshi A, Guo D, Hlaing T, Martin J, Xu X, et al. Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation. 2004;110:1036–41. doi: 10.1161/01.CIR.0000140258.09964.19. [DOI] [PubMed] [Google Scholar]
- 8.Di Diego JM, Antzelevitch C. Cellular basis for ST-segment changes observed during ischemia. J Electrocardiol. 2003;36(Suppl):1–5. doi: 10.1016/j.jelectrocard.2003.09.001. [DOI] [PubMed] [Google Scholar]