

Summary
A comprehensive two phase ‘hot spot’ saturation mutagenesis strategy to rapidly evolve glycosyltransferase specificity for non-natural acceptors is described. Specifically, the application of a high throughput screen (based upon the fluorescent acceptor umbelliferone) was used to identify key amino acid ‘hot spots’ that contribute to GT proficiency and/or promiscuity. Saturation mutagenesis of the corresponding hot spots facilitated the utilization of a lower throughput screen to provide OleD prodigy capable of efficiently glycosylating the non-natural acceptor novobiocic acid with an array of unique sugars. Incredibly, even in the absence of a high-throughput screen for novobiocic acid glycosylation, this approach rapidly led to improvements in the desired catalytic activity of several hundred-fold.
Keywords: directed evolution, glycosyltransferase, enzyme engineering, sugar, carbohydrate
Introduction
Glycosylated secondary metabolites continue to serve as an important source for drug discovery. As a key element of these natural pharmacophores, the attached sugars are typically critical for biological activity and subtle alterations in natural product glycosylation can transform a natural agent’s pharmacological properties, molecular and cellular specificity, and even mechanism of action [1–3]. ‘Glycorandomization’ is an emerging method for natural product glycodiversification which employs the inherent or engineered substrate promiscuity of anomeric kinases and nucleotidyltransferases for the in vitro synthesis of sugar nucleotide libraries. As the culminating step, this diverse range of sugar donors is processed by natural product glycosyltransferases (GTs) to provide a library of variant natural product glycosides [4, 5]. The successful glycorandomization of glycopeptides [6], avermectins [7], and enediynes [8] highlights the notable promiscuity displayed by several natural product GTs. Yet, attempts to utilize other natural product GTs for glycorandomization have been severely restricted by the catalysts’ stringent substrate specificity [9, 10]. For example, the noviosyltransferase NovM, which catalyzes the glycosylation of novobiocic acid (1) (Figure 1A) en route to novobiocin (8) (Figure 1D) [11, 12], was found to accept only 4 of 54 alternative sugar nucleotides examined [11, 12]. Thus, while permissive secondary metabolite GTs open new opportunities for drug discovery, the stringent specificity of others limits enzymatic natural product diversification and highlights a need for general GT engineering and/or evolution platforms.
Figure 1. Relevant GT-catalyzed coumarin glycosylation reactions.

(A) The reaction catalyzed by WT NovM. (B) The reaction employed for the fluorescence-based screening assay used to evolve OleD. (C) Representation of the novobiocic acid glucosylation reaction catalyzed by WT and variant OleD. (D) The structures of representative naturally-occurring aminocoumarin antibiotics novobiocin (8), clorobiocin (9) and coumermycin A1 (10).
Despite a wealth of GT structural and biochemical information, few successful applications of rational structure-based engineering to alter natural product GT donor/acceptor specificities exist [13, 14]. Among the most recent, a combination of sequence-guided and structure-guided mutagenesis allowed the ability to modulate the N- versus O-glucosylation activities of xenobiotic GTs from Arabidopsis thaliana [15]. Alternatively, the general lack of high-throughput GT screens/selections has also limited the directed evolution of GTs [16, 17]. As the first example in the context of natural products, the directed evolution of the oleandomycin GT (OleD) from Streptomyces antibioticus using a simple fluorescence-based screen was recently described [16, 17]. Specifically, a small library of OleD variants, screened for the ability to glucosylate the fluorescent surrogate acceptor 4-methylumbelliferone (4), led to a number of variants with improved ability to produce the β-D-glucopyranoside 6 (Figure 1B). The functional amino mutations were subsequently recombined to afford a triple mutant enzyme which displayed a marked improvement in both acceptor and donor promiscuity. Interestingly, this variant also displayed a modest (5-fold) improvement in glucosylation toward the novobiocin aglycon 1 (Figure 1C) - a potential starting point for circumventing the stringency of the native noviosyltransferase NovM. Moreover, novobiocin presents an excellent model as the aminocoumarins have long been known as potent bacterial type II DNA topoisomerase DNA gyrase inhibitors [18], and can also augment the activity of various anticancer agents via their ability to inhibit Hsp90 [19, 20].
Unlike the umbelliferone 4, novobiocic acid 1 is not fluorescent which severely limits the throughput of any engineering strategy dependent upon 1 as substrate. We postulated that the amino acid positions identified (‘hot spots’) as contributing to GT proficiency and/or promiscuity via the high throughput fluorescence-based screen (using 4), are also likely to contribute to utilization of other variant substrates (e.g. 1). To test this hypothesis, herein we describe the optimization of OleD toward the non-natural acceptor 1 via a comprehensive program of ‘hot spot’ saturation mutagenesis of functional positions previously identified via the high throughput fluorescence-based screen. Incredibly, even in the absence of a high-throughput screen for glucosylation of 1, this approach led to activity improvements toward 1 of several hundred-fold. In addition, this study reveals a correlation between catalyst proficiency and increased donor promiscuity in the context of 1, ultimately affording a new catalyst for the glycorandomization of novobiocin.
Results
Specificity of OleD mutants toward novobiocic acid 1
As previously described, the OleD triple mutant P67T/S132F/A242V displayed improvements in glucosylation activity with a panel of non-natural acceptors, including novobiocic acid 1 (5-fold improved) [16]. This variant was constructed following the identification of three clones from a library of OleD mutants, which had improved activity toward umbelliferone 4. One of these clones, 2C3, possessed a single amino acid mutation (A242V), while the other two, 8B3 and 7B9, possessed the mutations P67T/I112T and S132F/G340W, respectively. In order to identify functional mutations in 8B3 and 7B9, each single mutant was constructed by site-directed mutagenesis, and the specific activity of each purified enzyme determined with 4 [16]. These results clearly demonstrated that P67T, S132F, and A242V were responsible for improved activity toward 4 (~7, 2.6, and 2.3-fold improved compared to WT OleD), while G340W and I112T appeared non-functional (~0.5-fold reduced activity, compared to WT OleD) [16]. To further evaluate the impact of these mutations upon substrate specificity, the activity of each single mutant OleD was determined with the alternative acceptor 1 (Figure 2A). These results revealed that P67T, S132F, and A242V increased production of the putative glucoside 7 2–3 fold (Figure 2A), while G339W was non-beneficial. Surprisingly, I112T displayed the largest improvement (7.4-fold) in glucosylation activity toward 1. Thus, in the previously identified double mutant P67T/I112T (clone 8B3) [16], both P67T and I112T influence 1 specificity, while only P67T appears functional with respect to 4.
Figure 2. Creation of OleD variants improved toward novobiocic acid (1).

(A) Specific activities of WT and mutant OleDs with novobiocic acid (1) and UPD-Glc (5) as acceptor/donor, respectively. (B) The crude cell extract glucosylation activities of randomly selected colonies from saturation mutagenesis libraries P67X, I112X, and A242X, with 1 as acceptor. Activities are illustrated in descending order and arrows designate clones that were selected for in-depth characterization.
Given I112T, P67T, S132F, and A242V individually improve activity toward 1, combinations were next assessed for synergistic enhancements of the desired activity. For this secondary set of combinations, I112T was retained as an invariant substitution, given its significant impact upon 1-activity. Thus, three double mutants were generated by combining each of P67T, S132F, and A242V with I112T and each double mutant was over-expressed, purified, and the specific activity toward the substrate pair 1/5 determined by RP-HPLC (Figure 2A). This analysis revealed the activity of I112T variant could be further enhanced via the incorporation of either P67T or A242V (P67T/I112T and A242V/I112T, respectively) while the S132F/I112T variant was slightly less active than the I112T parent (Figure 2A). As predicted, further amalgamation of the three remaining mutations, to give the triple mutant P67T/I112T/A242V, provided a superior catalyst for 1 glucosylation. Among the two additional triple mutants possible, I112T/S132F/A242V was less active than P67T/I112T/A242V, consistent with the detrimental effect the S132F mutation in combination with other functional substitutions. Therefore, the remaining triple mutant (P67T/I112T/S132F) was not pursued. For unknown reasons, the quadruple mutant P67T/I112T/S132F/A242V failed to express under several different conditions tested (data not shown).
Saturation mutagenesis of Pro-67, Ile-112, and Ala-242
To further optimize the selected lead catalyst, single-site saturation mutagenesis at each of Pro-67, Ile-112, and Ala-242 in the scaffold P67T/I112T/A242V was performed, generating the libraries “P67X”, “I112X”, and “A242X”, respectively. A key constraint of this approach is that, unlike the screening target in our original directed evolution program (4), the acceptor 1 is non-fluorescent. Accordingly, each library was screened for activity toward 1 via RP-HPLC and, for convenience, we limited screening to ~100 colonies from each library, which represents around 95% coverage [21]. From this approach, library P67X failed to identify improved variants while several colonies from the I112X and A242X libraries displayed 2–3 fold enhancements of 1 glucosylation (Figure 2B). DNA sequencing revealed that substitution of Ala-242 with leucine was responsible for the hits identified from the A242X library while two hits from the I112X library were found to possess the mutation I112K. Subsequent enzyme assay, using purified enzymes, confirmed that these clones were more active than the parent P67T/I112T/A242V. Specifically, P67T/I112K/A242V and P67T/I112T/A242L were ~7- and ~4-fold improved over the parent P67T/I112T/A242V, respectively (Figure 2A). While a recombination of the three ‘best’ mutations (P67T/I112K/A242L) lead to a slightly less active variant, notably, this approach rapidly identified a variant (P67T/I112K/A242V) 150-fold improved compared to WT OleD, and 28-fold improved over the previously described P67T/S132F/A242V [16] in terms of specific activity with 1 and 5.
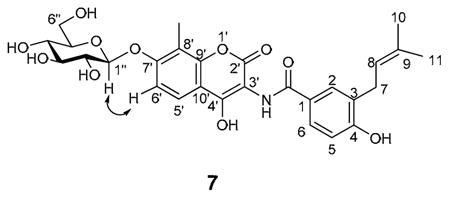
Product characterization
All OleDs described, including WT OleD, catalyzed the formation of a single, identical product based upon RP-HPLC and LC-MS (data not shown), consistent with the formation of a single mono-glucoside with conserved regio- and stereochemistry. The architectural similarities between 1 and umbelliferone 4 implicate the 1 C7′-OH as the likely position for glucosylation, to provide 7 (Figure 1C). Scale-up of the P67T/I112K/A242L-catalyzed 1-glucosylation reaction followed by structural elucidation confirmed glucoside 7 as the product (see Experimental Procedures and Supplementary Figure 1). Interestingly, LC-MS analysis of this large scale reaction also revealed trace production of a putative diglucosyl-substituted product (data not shown).
Kinetic characterization of WT and mutant OleDs
The WT and the mutant OleD P67T/I112K/A242L were compared by determining steady-state kinetic parameters with 1 or 5 as variable substrates, as described in the Experimental Procedures. The WT enzyme displayed hyperbolic saturation with both 1 and 5 as variable substrates, providing a Km for 1 and 5 of 2 and 2.81 mM, respectively (Figure 3A–B). The kcat determined with either 1 or 5 (0.041 and 0.073 min−1, respectively) were not in complete agreement, likely because at the concentration of 1 used (5mM, the solubility limit) with 5 as variable substrate, WT OleD was not completely saturated with acceptor. Nevertheless, these results demonstrated 1 to be a poor substrate for WT OleD.
Figure 3. Steady-state kinetic analysis of WT and P67T/I112K/A242V OleD.

(A) WT OleD with novobiocic acid (1) as variable substrate and [UDP-Glc (5)] fixed at 5 mM. (B) WT OleD with UDP-Glc (5) as variable substrate and [novobiocic acid (1)] fixed at 5 mM. (C) P67T/I112K/A242V OleD with novobiocic acid (1) as variable substrate and [UDP-Glc (5)] fixed at 5 mM. (D) P67T/I112K/A242V OleD with UDP-Glc (5) as variable substrate and [novobiocic acid (1)] fixed at 5 mM.
The steady state kinetics of the triple mutant P67T/I112K/A242V were very different from that of the WT enzyme (Figure 3C–D). Saturation with both 1 and 5 was easily achieved, with apparent Kms of 0.8 mM and 0.41 mM respectively − 2.5-fold and 6.9-fold improved over WT OleD. Moreover, the kcat determined with either acceptor 1 or donor 5 as the variable substrate (5.13 and 3.67 min−1) were in closer agreement, reflecting the improved Km for the acceptor. Thus, in terms of catalytic efficiency (kcat/Km) with 1 as acceptor, the triple mutant P67T/I112K/A242V is ~300-fold improved compared to WT OleD.
Donor specificity of P67T/I112K/A242V
The sugar nucleotide donor promiscuity of WT and mutant P67T/I112K/A242V OleD was probed using RP-HPLC analysis with a set of 20 potential ‘unnatural’ UDP-donors as surrogates for UDP-Glc (5) in the presence of 1 as acceptor (Figure 4A). This set was comprised of unnatural sugar nucleotides generated via chemoenzymatic synthesis, representing alterations of the sugar at C1″, C2″, C3″, C4″ or C6″ [6, 22, 23]. Putative product identities were confirmed by LC-MS (Supplementary Figure 2 and Supplementary Table 1). Of the 21 sugar nucleotides tested, only UDP-Glc (5), UDP-xylose (11), UDP-6-deoxy-glucose (12), and UDP-4,6-dideoxy-glucose (18) led to detectable product with WT OleD, ranging from ~0.1–3% conversion in 3 h (Figure 4B). In contrast, the optimized mutant P67T/I112K/A242V accepted 10 of 21 sugar nucleotide donors examined, 6 of which were not detectable substrates of WT OleD, with improvements in conversion ranging from 10–375 fold (Figure 4B and Supplementary Table 1).
Figure 4. Probing the donor specificity of WT OleD and mutant prodigy.

(A) The set of UDP-sugar donors used to probe specificity. Dashed-boxed donors were detectable substrates for both WT and mutant P67T/I112T/A242V OleD, while solid boxed donors were substrates only for P67T/I112T/A242V. (B) Successful conversion rates (%) after 3 h with WT or P67T/I112T/A242V OleD using 50 μM acceptor 1 and 250 μM UDP-sugar donors (the reactions containing 28 were incubated for 18 h). (C) Improvement of donor promiscuity with increasing proficiency of OleD variants.
Proficiency and promiscuity
A subset of donors (5, 11, 15, 21, 22), representing diverse sugar modifications, was employed to further probe the donor promiscuity of several of OleD variants using 1 as acceptor. The variants selected included WT, the optimal triple mutant P67T/I112K/A242V (Figure 4B), the previously described P67T/S132F/A242V [16], and the scaffold for saturation mutagenesis (P67T/I112T/A242V) to represent a ‘family’ of mutant OleDs with gradual improvements in proficiency toward the substrate pair 1/5. This analysis revealed WT OleD to accept only 2 of the donor subset (2 and 11, Figures 4A–C) while the mutant P67T/S132F/A242V was ~5-fold improved toward donor 5, slightly enhanced with 11 and also accepted 15 (albeit poorly) (Figure 4C). Replacing S132F with I112T in this triple mutant P67T/S132F/A242V further improved activity toward 5, 11, and 15 (Figure 4C). The final optimal mutant (P67T/I112K/A242V) afforded by saturation mutagenesis displayed further improvements toward 5, 11, 15 and detectable turnover with 22 (Figures 4A–C). None of the mutants displayed any detectable activity toward 21.
Discussion
Directed evolution has proven an effective tool for altering the specificity of enzymes and presents a possible solution to overcome the strict specificity of certain glycosyltransferases such as NovM [31]. However, the success of directed evolution is distinctly dependent upon the availability of a suitable high-throughput screen (HTS) or selection for the desired activity. This is especially problematic in the context of glycosyltransferases, given the huge variety of glycosyl donors and acceptors utilized by this large family of enzymes [13, 32]. For example, in the case of novobiocin, the aglycon 1 and any corresponding glycoside (e.g. 7) are indistinguishable spectrophotometrically. Interestingly, the recently successful directed evolution of the macrolide GT OleD, based upon a simple fluorescent acceptor surrogate (4), led to the discovery of an OleD variant which displayed a modest (5-fold) improvement in 1 glucosylation [16]. This pioneering study served not only as a potential starting point for circumventing the stringency of the native noviosyltransferase NovM, but also suggested that the amino acid positions identified (‘hot spots’) as contributing to GT proficiency and/or promiscuity via the high throughput fluorescence-based screen (using 4), may also to contribute to utilization of other variant substrates (e.g. 1). Notably, while positions identified by random mutagenesis and screening/selection for a desired activity have often been further optimized by saturation mutagenesis/recombination [33], the application of the secondary saturation mutagenesis/recombination to optimize a completely distinct activity (e.g. utilization of a very different non-natural substrate) has not been reported.
Of the mutations identified in the initial directed evolution study using fluorescent surrogate 4 (P67T, I112T, S132F, A242V, G340W) [16], the first four individually contributed to improved glycosylation of 1 with I112T as the most active single mutant. It should be mentioned that, given the low mutation rates and limited library sizes typically employed in directed evolution strategies, it is uncommon to find multiple functional mutations in the same clone. Yet, both P67T and I112T, originally discovered as a combination in the single clone 8B3, improved activity with 1. Subsequent combinations of P67T, S132F, and A242V, on an invariant I112T background, led to catalyst P67T/I112T/A242V which displayed a 22-fold improvement in specific activity toward 1 compared to WT OleD. Saturation mutagenesis at Pro-67, Ile-112, and Ala-242 on the same background revealed additional gains via incorporation of polar/charged amino acids at position 112 (I112T or I112K) and increasing hydrophobic steric bulk at residue 242 (A242L). In contrast, variation of Pro-67 failed to improve the desired activity under the conditions employed. While this preliminary analysis is consistent with P67T as the optimal substitution, it is also possible that other mutations at Pro-67 simply did not improve the rate of glucosylation at the concentration of acceptor/donor used (i.e. mutations could improve Km but not kcat). Interestingly, a final combination of the optimized mutations (I112K and A242L) was slightly detrimental to activity under the assay conditions used, as P67T/I112K/A242L was less active than both P67T/I112K/A242V and P67T/I112T/A242L. Yet, the final optimized variant (P67T/I112K/A242V) displayed a 150-fold improved specific activity toward 1 compared to WT OleD. Steady-state kinetic analysis illustrated P67T/I112K/A242V to be 200- or 300-fold more efficient (in terms of kcat/Km) with UDP-Glc (5) or acceptor 1, respectively, compared to WT OleD, and this change reflected improvements in both kcat and Km for donor and acceptor.
Using the high-throughput fluorescence-based ‘surrogate’ screen to first identify functional ‘hot spots’ notably limited the low-throughput catalyst optimization to screening only ~300 colonies via HPLC. As a comparison, analysis of the OleD-macrolide complex structure implicates at least 32 residues (including the acceptor binding pocket and ‘loop N3’) key to forming the static acceptor binding site (Figure 5) in an ‘open’ conformation [34]. Thus, to simply assess the single best amino acid at each of the 32 structure-designated positions via saturation mutagenesis would require screening >3000 colonies (32×100 for ~95% coverage) by HPLC. Among the ‘hot spots’ identified via the fluorescence-based screen, the OleD structure reveals Ile-112 to be intimately associated with acceptor binding (Figure 5) and the previous mutation of an equivalent residue (Ile-117) in oleandomycin GT OleI (45% identity with OleD) reduced kcat/Km ~100-fold [34]. Pro-67 is part of the substrate-binding ‘loop N3’ (amino acids 60–76) following β-sheet 3 in the N-terminal domain (Figure 5) and a similarly located proline has been implicated in controlling substrate specificity of urdamycin GTs [35]. Structural analysis does not predict the synergistic/antagonistic influences of S132 and/or the A242 optimal substitutions for activity toward 1/5 and may also exclude certain dynamic elements/residues critical for catalysis. Thus, while both structure-guided and directed-evolution approaches may ultimately arrive at many of the same mutations, the ‘hot spot’-focused strategy described herein may present a more streamlined approach for catalyst optimization.
Figure 5. OleD active site structure.
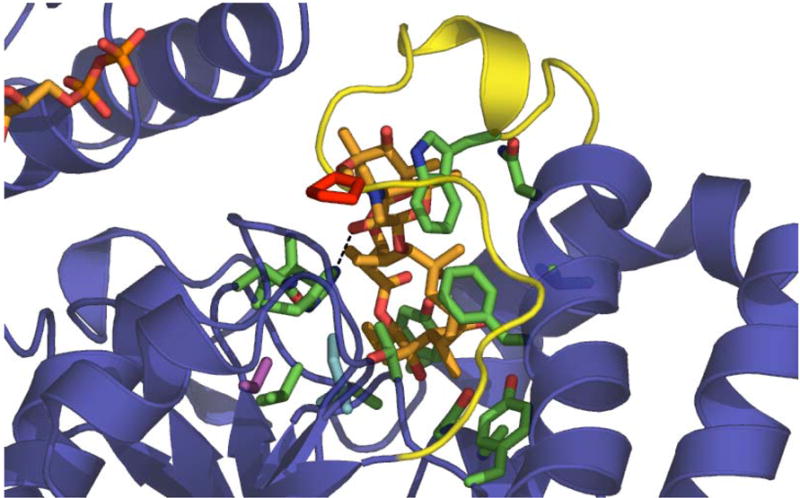
The key residues delineated in this study are highlighted within the previously reported active site structure of OleD bound to oleandomycin and NDP (PDB file 21YF). Color designations – substrates, orange; Pro-67, red; Ile-112, cyan; Ser-132, magenta; loop N3, yellow; dashed line, H-bond between the catalytic His-25 and acceptor sugar-OH. Residues in green are those that form the acceptor binding pocket, which is largely hydrophobic.
It has been previously suggested that naturally occurring GTs with high turnover numbers will be more promiscuous [36]. The availability of a unique family of OleD variants displaying a gradient of catalytic efficiencies from this study allowed a more direct test of this hypothesis. As illustrated in Figure 4C, enhancements in NDP-sugar donor promiscuity indeed paralleled the improvements in catalyst proficiency product culminating in a catalyst capable of accepting 11 of 21 UDP-donors tested (compared to only trace conversion of 4 UDP-donors by WT OleD). Although this study is consistent with the notion that more proficient GTs are generally promiscuous, it is also important to note that several naturally occurring GTs display very high efficiency yet remain exquisitely selective. For example, WT NovM with its natural substrates TDP-L-noviose (2) and 1 is ~3 orders of magnitude more proficient than the triple mutant P67T/I112K/A242V with 1 and 5, based upon kcat/Km, but displays a remarkably narrow substrate specificity range [11, 12]. On the other hand, P67T/I112K/A242V is ~1 order of magnitude more efficient (in terms of kcat/Km toward 1 and 5) than WT NovM toward an alternative donor, TDP-6-deoxy-glucose [11], and approaches the catalytic efficiency of other natural product GTs such as the relatively promiscuous teicoplanin GT tGtfb [39].
With respect to the importance of aminocoumarin glycosylation, the 2″-, 3″-, and 4″-noviose moieties are critical for maintaining antibacterial activity of 8 [24–26] while removal of the 8 3-carbomoyl moiety of noviose increases Hsp90 inhibition ≥ 70-fold [27, 28]. Glycosyl-modified 8 analogs also provide potent neuroprotective activities [29]. Yet, while the potential value of varying aminocoumarin glycosylation clearly exists, pathway engineering and semi-synthetic efforts to date have led to fairly conservative glycosyl modifications [30]. The present study sets the stage to greatly expand upon the availability of differentially-glycosylated aminocoumarins. For example, glucoside 7 and the glycosides derived from donors 15, 18, 19, 22, 23, 24 and 28 from this proof of concept study have not been previously described. Access to these compounds may further the therapeutic development of aminocoumarins and could also be used to interrogate the specificity of late stage aminocoumarin biosynthetic modifying enzymes, such as the acyltransferase CouN7 [37, 38], toward further diversification. Utilization of UDP-6-azido-glucose (15) as a new substrate also presents the potential for further downstream chemo-selective diversification [6].
Significance
Using the macrolide glycosyltransferase OleD as a model, this study unveils a general enzyme optimization strategy (hot spot saturation mutagenesis) applicable to reactions limited by amenable high throughput screens. Specifically, the application of a high throughput screen (based upon the fluorescent acceptor umbelliferone) was used to identify key amino acid ‘hot spots’ that contribute to GT proficiency and/or promiscuity. Saturation mutagenesis of the corresponding hot spots facilitated the utilization of a lower throughput screen (based upon the acceptor novobiocic acid) to provide OleD prodigy capable of efficiently catalyzing the production of a novel set of differentially glycosylated aminocoumarins - an important class of natural product with known antibiotic, anticancer and anti-neurodegenerative activities. A systematic comparison of OleD variants from this study also revealed the first direct correlation between catalyst proficiency and increased donor promiscuity. While this work demonstrates a platform for the rapid generation of new glycosylation catalysts, the concept of hot spot saturation mutagenesis as applied herein should be broadly applicable in the context of new catalyst development.
Experimental procedures
General
Bacterial strain E. coli BL21(DE3)pLysS was from Stratagene. NovaBlue was from Novagen. Plasmid pET28/OleD was a generous gift from Prof Hung-Wen Liu (University of Texas-Austin, Austin, USA) and pET28a was from Novagen. All other chemicals were reagent-grade purchased from Fluka, New England Biolabs, or Sigma, unless otherwise stated. Primers were ordered from Integrated DNA Technologies (Coralville, IA). Novobiocic acid (1) was prepared as previously described from novobiocin [11]. Product standard 4-methyl-umbelliferyl-7-O-β-D-glucoside (6), and UDP-Glc (5) were from Sigma. Analytical HPLC was performed on a Rainin Dynamax SD-2/410 system connected to a Rainin Dynamax UV-DII absorbance detector. Mass spectra were obtained using electrospray ionization on an Agilent 1100 HPLC-MSD SL quadropole mass spectrometer connected to a UV/Vis diode array detector. For LC-MS analysis, quenched reaction mixtures were analyzed by analytical reverse-phase HPLC with a 250 mm × 4.6 mm Gemini 5μ C18 column (Phenomenex, Torrance, CA) using a gradient of 10–90% CH3CN in 0.1% formic acid/H2O in 20 min at 1ml/min, with detection at 254 nm. Structural characterization was performed by NMR spectroscopy using a Varian UNITYINOVA 500 MHz instrument (Palo Alto, CA) in conjunction with a Protasis/MRM CapNMR capillary probe (Savoy, IL). The spectrum was referenced to DMSO-d6 at 2.50 ppm.
Site-directed mutagenesis
Site-specific OleD variants were constructed using the Stratagene QuikChange II Site-Directed Mutagenesis Kit, as described by the manufacturer. Constructs were confirmed to carry the correct mutation(s) via DNA sequencing.
Saturation library preparation
The saturation mutagenesis libraries ‘P67X’, ‘I112X’, and ‘A242X’ were constructed using the Stratagene QuikChange II Site-Directed Mutagenesis Kit, as described by the manufacturer using the mutant P67T/I112T/A242V as template. For each library, plasmid DNA (digested with DpnI) was transformed into Novablue chemical competent cells and the transformants grown overnight at 37 °C on LB agar supplemented with 50 μg/ml kanamycin. Colonies from each library were subsequently pooled and used to inoculate 5 ml of LB medium supplemented with 50 μg/ml kanamycin for overnight growth at 37°C with shaking (350 rpm). Using standard mini-prep purification, the mixed-population plasmid prep was prepared from each 5 ml culture and transformed into E. coli BL21(DE3)pLysS and the transformants grown overnight at 37 °C on LB agar supplemented with 50 μg/ml kanamycin.
For protein expression, individual colonies from plates containing BL21(DE3)pLysS transformants were subsequently used to inoculate wells of a 96-deep well microtitre plate wherein each well contained 1 ml of LB medium supplemented with 50 μg/ml kanamycin. Culture plates were tightly sealed with AeraSeal™ breathable film (Research Products International Corp.). After cell growth at 37°C for 18 h with shaking at 350 rpm, 100 μl of each culture was transferred to a fresh deep-well plate containing 1 ml of LB medium supplemented with 50 μg/ml kanamycin. The original plate was sealed and stored at 4°C, or a glycerol copy made by mixing 100 μl of each culture with 100 μl 50% (v/v) glycerol and storing at −80°C. The freshly inoculated plate was incubated at 37°C for 2–3 h with shaking at 350 rpm. Protein expression was induced at OD600 ~0.7, and isopropyl β-D-thiogalactoside (IPTG) was added to a final concentration of 0.4 mM and the plate incubated for 18 h at 18°C. Cells were harvested by centrifugation at 3000 g for 10 min at 4°C, the cell pellets thoroughly resuspended in chilled 50 mM Tris-HCl (pH 8.0) containing 10 mg/ml lysozyme (Sigma), and the plates were subjected to a single freeze/thaw cycle to lyse the cells. After thawing, cell debris was collected by centrifugation at 3000 g for 20 min at 4°C and 50 μl of the cleared supernatant used for enzyme assay.
For crude extract assays, cleared supernatant was mixed with an equal volume (50 μl) of 50 mM Tris-HCl (pH 8.0) containing 10 mM MgCl2, 200 μM 1, and 1.0 mM 5 using a Biomek FX Liquid Handling Workstation (Beckman Coulter, Fullerton, CA). Upon mixing, the reactions were incubated for 3 h at 30°C, at which point, the crude reaction mixture was mixed with an equal volume of MeOH, and centrifuged at 3000 g for 20 min at 4°C. Aliquots of each product mixture (40 μl) were analyzed by RP-HPLC as described below for determination of specific activity.
Protein expression and purification
For characterization of specific OleD variants, single colonies were used to inoculate 3 ml LB medium supplemented with 50 μg/ml kanamycin and cultured overnight at 37°C. The entire starter culture was then transferred to 1 liter LB medium supplemented with 50 μg/ml kanamycin and grown at 37°C until the OD600 was ~0.7, IPTG to a final concentration of 0.4 mM was added and the flask incubated for 18 h at 18°C. Cell pellets were collected by centrifugation at 10,000 g and 4°C for 20 min, resuspended into 10 ml 20 mM phosphate buffer, pH 7.4, containing 0.5 M NaCl and 10 mM imidazole and were lysed by sonication. Cell debris was removed by centrifugation at 10,000 g and 4°C for 30 min and the cleared supernatant immediately applied to 2 ml of nickel-nitrilotriacetic acid (Ni-NTA) resin (QIAgen Valencia, CA), pre-equilibrated with the lysis buffer. Protein was allowed to bind for 30 min at 4°C with gentle agitation, and the resin washed 4 times with 50 ml each lysis buffer. Finally, the enzyme was eluted by incubation of the resin with 2 ml lysis buffer containing 100 mM imidazole for 10 min at 4°C with gentle agitation. The purified enzyme was applied to a PD-10 desalting column (Amersham Biosciences AB) equilibrated with 50 mM Tris-HCl (pH 8.0) and eluted as described by the manufacturer. Protein aliquots were immediately flash frozen in liquid nitrogen and stored at −80°C. Protein purity was verified by SDS-PAGE. Protein quantification was carried out using the Bradford Protein Assay (Bio-Rad, Hercules CA).
Determination of specific activity with 1 or 4 as acceptor
Enzyme reactions were carried out in a total volume of 100 μl 50 mM Tris-HCl (pH8.0) containing 5 mM MgCl2, 200 μM 1 or 4, 5 mM 5, and typically 10–50 μg pure enzyme. At a suitable time interval during which rate of product formation was linear with time (typically 2–20 min), reactions were quenched with 100 μl MeOH, and the samples centrifuged at 10,000 g for 10 min. Supernatants were analyzed by analytical reverse-phase HPLC with a Gemini 5μ C18 column (Phenomenex) using a gradient of 10–90% CH3CN in 0.1% trifluoroacetic acid (TFA)/H2O in 20 min, with detection at 254 nm. Under these conditions, substrates 1 (tR=25.5 min) or 4 (tR=9.95 min) and the glucoside products 7 (tR=18.8 min) or 6 (tR=14.5 min) were readily resolved. The identity of 7 was confirmed by LC-MS and NMR while the dentity of glucoside 6 was confirmed by co-elution with a commercial standard and LC-MS analysis. HPLC peak areas were integrated, and the product concentrations calculated as a percent of the total peak area. Specific activity was expressed as nmols product formed/min/mg protein.
Scale-up and characterization of glucoside 7
The preparative reaction to synthesize the novobiocyl glucoside 7 was accomplished at room temperature in a total volume of 10 mL in 50 mM Tris-HCl (pH 8.0) containing 5 mM MgCl2, 10 mg novobiocic acid (1), 60 mg UDP-Glc (5), and 5 mg purified OleD mutant P67T/I112T/A242V. The reaction was incubated for 48 h after which protein was removed by adding 30 mL cold MeOH and centrifugation at 10,000 g and 4°C for 30 min. The supernatant was concentrated, lyophilized, and resuspended in 1 mL DMSO and the sample was purified by HPLC using a Gemini 5μ C18 column (Phenomenex) using a gradient of 10–90% CH3CN in 0.1% trifluoroacetic acid (TFA)/H2O in 20 min, with detection at 254 nm. The product fractions were pooled and lyophilized. 1H NMR (1:2 DMSO-d6:CD3OD) δ 7.69 (br s, 1 H, H5′), 7.66 (br s, 1 H, H2), 7.50 (m, 1 H, H6), 7.09 (br d, J = 7.1 Hz, 1 H, H6′), 6.69 (m, 1 H, H5), 5.24 (br s, 1 H, H8), 4.90 (d, J = 6.9 Hz, 1 H, H1″), 3.75 (d, J = 11.5 Hz, 1 H, H6a″), 3.56 (m, 1 H, H6b″), 3.36 (m, 4 H, H2″–5″), 3.27 (m, 2 H, H7), 2.09 (s, 3 H, H11′), 1.62 (s, 6 H, H10,11). The anomeric coupling constant and NOESY highlighted below (see also Supplemental Figure 1) are consistent with the C7-β-glucoside 7.
Determination of kinetic parameters
Enzyme assays were carried out in a total volume of 100 μl 50 mM Tris-HCl (pH 8.0) containing 5 mM MgCl2, and typically 10–50 μg pure enzyme. Kinetic parameters kcat and Km were determined with both 1 and 5 as variable substrates. For the determination of Km for 1, 5 was constant at 5 mM and 1 was varied between 0.025 and 5 mM. For the determination of Km for 5, 1 was constant at 5 mM and 5 was varied between 0.05 and 25 mM. Each experiment was performed in triplicate. Aliquots (100 μl) were removed between 0 and 30 min, at which time product formation was still linear with respect to time, and quenched with 100 μl of ice-cold MeOH, and centrifuged at 10,000 g for 10 min. Supernatants were analyzed by analytical reverse-phase HPLC as described above. The substrate 1 and the glucoside product 7 HPLC peak areas were integrated, and the product concentration calculated as a percent of the total peak area. Initial velocities were fitted to the Michaelis-Menten equation using Sigma Plot.
Donor specificity
To assess donor specificity, assays contained 50 μM acceptor 1 and ~500 μM putative NDP-sugar donor in a total assay volume of 50 μl. For this study, the NDP-sugars were used directly from RmlA-catalyzed reactions [6, 22, 23]. Although 11, 14, and 24 were also commercially available, low levels of contamination with UDP-Glc in the commercial reagents complicated product analysis by LC-MS (data not shown) and therefore, the RmlA-derived 11, 14, and 24 were utilized. Reactions were incubated at 25°C for 3 h. Aliquots (25 μl) were quenched with 25 μl of ice-cold MeOH, and centrifuged at 10,000 g for 10 min. Supernatants were analyzed by analytical reverse-phase HPLC as described above. HPLC peak areas were integrated, and the product concentration calculated as a percent of the total peak area. All products were characterized by LC-MS.
Supplementary Material
Acknowledgments
We are grateful to the School of Pharmacy Analytical Instrumentation Center for analytical support. This work was supported in part by National Institutes of Health Grants AI52218 and U19 CA113297. J.S.T is a UW HI Romnes Fellow.
References
- 1.Thorson JS, Vogt T. Glycosylated natural products. In: Wong C-H, editor. Carbohydrate-Based Drug Discovery. Weinheim: Wiley-VCH; 2002. pp. 685–712. [Google Scholar]
- 2.Weymouth-Wilson AC. The role of carbohydrates in biologically active natural products. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed A, Peters NR, Fitzgerald MK, Watson JA, Jr, Hoffmann FM, Thorson JS. Colchicine glycorandomization influences cytotoxicity and mechanism of action. J Am Chem Soc. 2006;128:14224–14225. doi: 10.1021/ja064686s. [DOI] [PubMed] [Google Scholar]
- 4.Langenhan JM, Griffith BR, Thorson JS. Neoglycorandomization and chemoenzymatic glycorandomization: two complementary tools for natural product diversification. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]
- 5.Griffith BR, Langenhan JM, Thorson JS. ‘Sweetening’ natural products via glycorandomization. Curr Opin Biotechnol. 2005;16:622–630. doi: 10.1016/j.copbio.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Fu X, Albermann C, Jiang J, Liao J, Zhang C, Thorson JS. Antibiotic optimization via in vitro glycorandomization. Nat Biotechnol. 2003;21:1467–1469. doi: 10.1038/nbt909. [DOI] [PubMed] [Google Scholar]
- 7.Zhang C, Albermann C, Fu X, Thorson JS. The in vitro characterization of the iterative avermectin glycosyltransferase AveBI reveals reaction reversibility and sugar nucleotide flexibility. J Am Chem Soc. 2006;128:16420–16421. doi: 10.1021/ja065950k. [DOI] [PubMed] [Google Scholar]
- 8.Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
- 9.Borisova SA, Zhang C, Takahashi H, Zhang H, Wong AW, Thorson JS, Liu HW. Substrate specificity of the macrolide-glycosylating enzyme pair DesVII/DesVIII: opportunities, limitations, and mechanistic hypotheses. Angew Chem Int Ed Engl. 2006;45:2748–2753. doi: 10.1002/anie.200503195. [DOI] [PubMed] [Google Scholar]
- 10.Yuan Y, Chung HS, Leimkuhler C, Walsh CT, Kahne D, Walker S. In vitro reconstitution of EryCIII activity for the preparation of unnatural macrolides. J Am Chem Soc. 2005;127:14128–14129. doi: 10.1021/ja053704n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albermann C, Soriano A, Jiang J, Vollmer H, Biggins JB, Barton WA, Lesniak J, Nikolov DB, Thorson JS. Substrate specificity of NovM: implications for novobiocin biosynthesis and glycorandomization. Org Lett. 2003;5:933–936. doi: 10.1021/ol0341086. [DOI] [PubMed] [Google Scholar]
- 12.Freel Meyers CL, Oberthur M, Anderson JW, Kahne D, Walsh CT. Initial characterization of novobiocic acid noviosyl transferase activity of NovM in biosynthesis of the antibiotic novobiocin. Biochemistry. 2003;42:4179–4189. doi: 10.1021/bi0340088. [DOI] [PubMed] [Google Scholar]
- 13.Williams GJ, Zhang C, Thorson JS. Natural product glycosyltransferases: Properties and applications. Adv Enzymol Relat Areas Mol Biol. 2008;76 doi: 10.1002/9780470392881.ch2. in press. [DOI] [PubMed] [Google Scholar]
- 14.Hancock SM, Vaughan MD, Withers SG. Engineering of glycosidases and glycosyltransferases. Curr Opin Chem Biol. 2006;10:509–519. doi: 10.1016/j.cbpa.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Brazier-Hicks M, Offen WA, Gershater MC, Revett TJ, Lim EK, Bowles DJ, Davies GJ, Edwards R. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0706421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams GJ, Zhang C, Thorson JS. Directed evolution of a natural product glycosyltransferase. Nat Chem Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]
- 17.Williams GJ, Thorson JS. A high-throughput fluorescence-based glycosyltransferase screen and its application in glycosyltransferase directed-evolution. Nat Prot. 2008 doi: 10.1038/nprot.2007.538. in press. [DOI] [PubMed] [Google Scholar]
- 18.Maxwell A, Lawson DM. The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr Top Med Chem. 2003;3:283–303. doi: 10.2174/1568026033452500. [DOI] [PubMed] [Google Scholar]
- 19.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 20.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 21.Firth AE, Patrick WM. Statistics of protein library construction. Bioinformatics. 2005;21:3314–3315. doi: 10.1093/bioinformatics/bti516. [DOI] [PubMed] [Google Scholar]
- 22.Barton WA, Biggins JB, Jiang J, Thorson JS, Nikolov DB. Expanding pyrimidine diphosphosugar libraries via structure-based nucleotidylyltransferase engineering. Proc Natl Acad Sci U S A. 2002;99:13397–13402. doi: 10.1073/pnas.192468299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J, Albermann C, Thorson JS. Application of the nucleotidylyltransferase Ep toward the chemoenzymatic synthesis of dTDP-desosamine analogues. Chembiochem. 2003;4:443–446. doi: 10.1002/cbic.200200566. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Heide L, Li SM. New aminocournarin antibiotics formed by a combined mutational and chemoenzymatic approach utilizing the carbamoyltransferase novN. Chem Biol. 2004;11:655–662. doi: 10.1016/j.chembiol.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 25.Freitag A, Galm U, Li SM, Heide L. New aminocoumarin antibiotics from a cloQ-defective mutant of the clorobiocin producer Streptomyces roseochromogenes DS12.976. J Antibiot (Tokyo) 2004;57:205–209. doi: 10.7164/antibiotics.57.205. [DOI] [PubMed] [Google Scholar]
- 26.Galm U, Dessoy MA, Schmidt J, Wessjohann LA, Heide L. In vitro and in vivo production of new aminocoumarins by a combined biochemical, genetic, and synthetic approach. Chem Biol. 2004;11:173–183. doi: 10.1016/j.chembiol.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 27.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS. Hsp90 inhibitors identified from a library of novobiocin analogues. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 28.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BS. Novobiocin: Redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 29.Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BSJ. A non-toxic Hsp90 inhibitor protects neurons from A beta-induced toxicity. Bioorganic & Medicinal Chemistry Letters. 2007;17:1984–1990. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Galm U, Heller S, Shapiro S, Page M, Li SM, Heide L. Antimicrobial and DNA gyrase-inhibitory activities of novel clorobiocin derivatives produced by mutasynthesis. Antimicrobial Agents And Chemotherapy. 2004;48:1307–1312. doi: 10.1128/AAC.48.4.1307-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin-Pitel SB, Zhao H. Recent advances in biocatalysis by directed enzyme evolution. Comb Chem High Throughput Screen. 2006;9:247–257. doi: 10.2174/138620706776843183. [DOI] [PubMed] [Google Scholar]
- 32.Hu Y, Walker S. Remarkable structural similarities between diverse glycosyltransferases. Chem Biol. 2002;9:1287–1296. doi: 10.1016/s1074-5521(02)00295-8. [DOI] [PubMed] [Google Scholar]
- 33.Chica RA, Doucet N, Pelletier JN. Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr Opin Biotechnol. 2005;16:378–384. doi: 10.1016/j.copbio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Bolam DN, Roberts S, Proctor MR, Turkenburg JP, Dodson EJ, Martinez-Fleites C, Yang M, Davis BG, Davies GJ, Gilbert HJ. The crystal structure of two macrolide glycosyltransferases provides a blueprint for host cell antibiotic immunity. Proc Natl Acad Sci USA. 2007;104:5336–5341. doi: 10.1073/pnas.0607897104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmeister D, Wilkinson B, Foster G, Sidebottom PJ, Ichinose K, Bechthold A. Engineered urdamycin glycosyltransferases are broadened and altered in substrate specificity. Chem Biol. 2002;9:287–295. doi: 10.1016/s1074-5521(02)00114-x. [DOI] [PubMed] [Google Scholar]
- 36.Oberthűr M, Leimkuhler C, Kruger RG, Lu W, Walsh CT, Kahne D. A systematic investigation of the synthetic utility of glycopeptide glycosyltransferases. J Am Chem Soc. 2005;127:10747–10752. doi: 10.1021/ja052945s. [DOI] [PubMed] [Google Scholar]
- 37.Balibar CJ, Garneau-Tsodikova S, Walsh CT. Covalent CouN7 enzyme intermediate for acyl group shuttling in aminocoumarin biosynthesis. Chem Biol. 2007;14:679–690. doi: 10.1016/j.chembiol.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Fridman M, Balibar CJ, Lupoli T, Kahne D, Walsh CT, Garneau-Tsodikova S. Chemoenzymatic formation of novel aminocoumarin antibiotics by the enzymes CouN1 and CouN7. Biochemistry. 2007;46:8462–8471. doi: 10.1021/bi700433v. [DOI] [PubMed] [Google Scholar]
- 39.Howard-Jones AR, Kruger RG, Lu W, Tao J, Leimkuhler C, Kahne D, Walsh CT. Kinetic analysis of teicoplanin glycosyltransferases and acyltransferase reveal ordered tailoring of aglycone scaffold to reconstitute mature teicoplanin. J Am Chem Soc. 2007 doi: 10.1021/ja0735857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.