

Abstract
The capacity of nicotine to affect the behavior of non-neuronal cells through neuronal nicotinic acetylcholine receptors (nAChRs) has been the subject of considerable recent attention. Previously, we showed that exposure to nicotine activates the nuclear factor of activated T cells (NFAT) transcription factor in lymphocytes and endothelial cells, leading to alterations in cellular growth and vascular endothelial growth factor production. Here, we extend these studies to document effects of nicotine on lymphocyte survival. The data show that nicotine induces paradoxical effects that might alternatively enforce survival or trigger apoptosis, suggesting that depending on timing and context, nicotine might act both as a survival factor or as an inducer of apoptosis in normal or transformed lymphocytes, and possibly other non-neuronal cells. In addition, our results show that, while having overlapping functions, low and high affinity nAChRs also transmit signals that promote distinct outcomes in lymphocytes. The sum of our data suggests that selective modulation of nAChRs might be useful to regulate lymphocyte activation and survival in health and disease.
Keywords: nicotine, lymphocytes, acetylcholine receptors, calcium, apoptosis, RNA interference
Introduction
More than 5,000 bioactive compounds are generated by tobacco pyrolysis, and most remain in the mouth, throat, and lungs. Nicotine is among the few tobacco toxicants that achieve high systemic distribution. This compound is responsible for the addictive effects of tobacco, but it has generally been considered to be otherwise “safe,” and so is the mainstay of tobacco cessation programs. Still, the possibility that nicotine-containing products used for tobacco cessation may contribute to cancer and other diseases has not been extensively studied and recent data suggest that nicotine may contribute to tumor progression (Cooke, 2007; Catassi et al., 2008). A potential mechanism to explain this relationship is nicotine-dependent modulation of apoptosis, but experiments evaluating the effects of nicotine on apoptosis in non-neuronal cells have yielded contradictory results. For example, a direct relationship between exposure to nicotine and apoptosis of primary human cells and cultured cell lines is documented, including reports showing that lymphocytes from smokers were more susceptible to apoptosis than lymphocytes from non-smokers (Mariggio et al., 2001; Wu et al., 2002) and that nicotine treatment increased expression of Fas ligand (FasL1) (Suzuki et al., 1999). On the other hand, nicotine also has been shown to prevent or delay apoptosis of normal and transformed human and mouse cells induced by factors as diverse as tumor necrosis factor-α, ultraviolet light (UV), chemotherapeutic drugs, opiates, and corticosteroids (Maneckjee and Minna, 1990; Wright et al., 1993; Aoshiba et al., 1996; Tohgi et al., 2000; Garrido et al., 2001; Hakki et al., 2001; Sugano et al., 2001), possibly by inhibiting caspases (Garrido et al., 2001; Hakki et al., 2001), by activating the Akt pathway (West et al., 2003), by upregulating expression of Survivin (Dasgupta et al., 2006) or by activating phosphorylation of Bcl-2, through the action of PKCα or of Erk mitogen activated protein kinases (Mai et al., 2003).
These disparate effects of nicotine on cellular survival could be explained by the fact that this compound binds to high- and low-affinity receptors with distinct functional consequences in cells. Specifically, nicotinic acetylcholine receptors (nAChR) are members of the ligand-gated ion channel superfamily of receptors that include neuronal serotonin receptors, γ-amino butyric acid receptors, and glycine receptors (Lindstrom, 1997; Conti-Fine et al., 2000; Grutter and Changeux, 2001). Binding of acetylcholine (ACh), nicotine or other agonists to nAChRs leads to an allosteric change with conformational transition to the open-channel state, resulting in membrane depolarization and influx of calcium from the extracellular environment (Grutter and Changeux, 2001). Sixteen genes encoding nAChR subunits that are expressed ubiquitously have been identified in the mammalian genome (Leonard and Bertrand, 2001; Changeux and Edelstein, 2005). In both neuronal and non-neuronal cells, nAChR subunits assemble as heteropentamers (thought to consist of three α subunits and two ϐ subunits) to form high-affinity receptors, or homopentamers (consisting of five α7-receptors) to form low-affinity receptors.
The cholinergic signaling system is among the oldest and most highly conserved in evolution. Nicotine is only one among many compounds that plants produce as natural defense systems to target cholinergic receptors, leading to death of insects and other herbivorous predators (George et al., 2000). While reasonably effective to balance insect predation, at relatively small doses nicotine is not lethal for humans. Instead it stimulates addictive behaviors that have led to major health problems in our society. The nAChR signaling pathway has clear behavioral outcomes in the brain, and functional outcomes in neuromuscular communication. Yet, its evolutionary persistence in non-neuronal (and non-muscle) cells can only be explained if it serves an essential physiological function. ACh production and release have been previously quantified in leukocytes, and may play an important role in immunomodulation (Kawashima and Fujii, 2000). As apoptosis is the fate for many or most activated leukocytes, the innate and adaptive immune systems have acquired a variety of mechanisms that support survival during immune and inflammatory responses, and it is reasonable to anticipate that these mechanisms took advantage of pre-existing signaling pathways, including nicotinic and muscarinic cholinergic signaling. The selective advantage of multiple subunits that can form high-affinity and low-affinity nAChRs is unclear, but it is likely that these receptors play redundant functions (or at least compensate for one another) in cells, as targeted deletion of single receptors in mice is not lethal (Cordero-Erausquin et al., 2000; Fowler et al., 2008). Yet, the diversity of nicotine receptors could readily lend itself for adaptation to various functions, among which could be modulation of apoptosis. Such functional duality has been documented for other ion channels that can promote survival or apoptosis under different conditions or in distinct environments (Lang et al., 2005).
We chose to evaluate the effects of nicotine on apoptosis in cultured human lymphocytes, and specifically, how distinct nicotine receptor subunits may mediate these effects. We used two investigational models: peripheral blood T cells and the transformed Jurkat T cell leukemia line. Experiments to assess T cell receptor-mediated responses in Jurkat cells are informative, and these cells are especially receptive for genetic manipulation (Abraham and Weiss, 2004). Our data suggest that nicotine is able to modulate lymphocyte function directly, as well as through the interaction of lymphocytes with their microenvironment, accounting in part for its pleotropic influence on cellular survival.
Results
Exposure to nicotine mobilizes calcium, a key lymphocyte regulator
Various studies show nicotine modulates lymphocyte function (Petro et al., 1992; Geng et al., 1995; Petro et al., 1999; Middlebrook et al., 2002; Frazer-Abel et al., 2004). The calcium mobilization that follows binding of nAChR presents a possible initial element of nicotine-induced effects on cell growth. We evaluated changes in intracellular calcium (Cai2+) in primary human T cells (Frazer-Abel et al., 2004) and in cultured Jurkat T cells treated with nicotine (0–50 μM) and soluble anti-CD3 (10 ng/ml). Cai2+ was measured flow cytometrically in Indo-1-loaded cells by the ratio of emission at 405 nm (chelated form of the dye) over emission at 480 nm (free form of the dye). Figure 1a shows stimulation of Jurkat cells with anti-CD3 showed a characteristic rise in Cai2+ within ~1 min followed by a slowly decreasing plateau. Pre-treatment with nicotine caused dose-dependent bimodal alterations in Cai2+: when nicotine was present at concentrations between 0.05 and 50 nM, the calcium response was characterized by a slow, steady rise that lasted >5 min, and was still evident when anti-CD3 was added. At concentrations >5 nM, the response was characterized by a rapid calcium spike within <1 min of addition, which became the dominant response when the concentration reached 500 nm and was followed by a return to basal levels. Intriguingly, incubation of T cells in the presence of nicotine at μM doses blunted the calcium response seen upon stimulation with anti-CD3. This had functional consequences for activation: not only did exposure to nicotine reduce expression of CDK4 in primary T cells and Jurkat cells (Frazer-Abel et al., 2004), but it also destabilized cyclin D2. Figure 1b shows this was achieved in Jurkat cells at least in part by a 2 to 3.5-fold increase of polyubiquitinated complexes that direct the protein to the proteasome. The native cyclin D2 molecule has an apparent molecular weight of 34 kDa. Addition of a single ubiquitin molecule reduces its electrophoretic mobility to an apparent molecular weight of ~41 kDa, and polyubiquitination further reduces this in increments of ~7 kDa, as evident by the bands that are visible by immunoblotting Cyclin D2 immunoprecipitates with anti-ubiquitin antibodies (Figure 1b). The effect of nicotine is evident by the relative increase in polyubiquitinated cyclin D2, as compared to monoubiquitinated cyclin D2 in cells treated with nicotine. This effect was consistent with previous results showing specific changes in CDK4 and cyclin D2 (see Cyclin D2 immunoblot in the absence of proteasome inhibitors in Figure 1b); in contrast, nicotine does not impact CDK6 levels in primary T cells or in Jurkat cells (Frazer-Abel et al., 2004). Nicotine also had no effect on accumulation of monoubiquitinated CDK6 and there was no appreciable polyubiquitinated CDK6 in Jurkat cells (data not shown). In contrast to its effect on CDK4 and Cyclin D2, nicotine led to a ~5-fold increase in the expression of p27 that was most appreciable in primary T cells stimulated with soluble anti-CD3, (Figure 1c), potentially contributing to the inability of these cells to achieve a competent state progress through G1 and into the S phase (Frazer-Abel et al., 2004).
Figure 1. Dose-dependent calcium mobilization in human T cells induced by nicotine has functional consequences.

Panel a shows Jurkat cells treated with nicotine at the indicated concentrations for 5 min prior to the addition of anti-CD3 (10 ng/ml). Alterations in Cai2+ were measured using a MoFlo flow cytometer. Data on the X-axis represent nicotine dose (nM), on the Y-axis, time (sec) and on the Z-axis (Calcium flux) they are expressed as the product of excitation X proportion of responding cells. Panel b shows Jurkat cells cultured as indicated in the presence or absence of proteasome inhibitors Lactacystin and MG132. Cyclin D2 complexes were immunoprecipitated using anti-cyclin D2 antibody and immunoblotted with an anti-ubiquitin antibody (top). Monoubiquitinated Cyclin D2 complexes migrate with an apparent MW of 41 kDa; polyubiquitinated complexes migrate with an apparent MW of ~90 kDa (compared to the 34 kDa native protein). The ratio of polyubiquitinated to monoubiquitinated Cyclin D2 was 2.5-fold and 2-fold greater, respectively, in cells treated with nicotine or with anti-CD3 than in unstimulated cells. The effects of nicotine and anti-CD3 were additive, with the ratio increasing to 3.2-fold over untreated cells when both compounds were used together. The lower immunoblot shows levels of Cyclin D2 in whole cell lysates from cells stimulated in an identical manner without proteasome inhibitors. Panel c shows the levels of the p27 CDK inhibitor in whole cell lysates from primary T cells stimulated in an identical manner in the absence of proteasome inhibitors. The steady state levels of p27 were significantly different (5-fold greater) in cells treated with nicotine and anti-CD3 together, or with ionomycin than in untreated cells. Ionomycin was included in the experiments shown in panel b and panel c to control for non-specific effects of calcium mobilization, and ϐ-actin immunoblots are included as loading controls.
Primary and immortalized lymphocytes express nAChRs
It is possible that the distinct calcium responses, and by extension, the activation of specific signaling pathways, were related to activation of different receptor types. To verify expression of nAChR expression in lymphocytes, we used RT-PCR to amplify mRNA isolated from primary T cells from 10 healthy donors. Figure 2 shows qualitative mRNA expression for α4- and ϐ4-nAChR subunits (panel a), which are required for assembly of both major types of high affinity receptors (α3/ϐ4 and α4/ϐ2 heteropentamers). Message for both receptors was expressed in all subjects; on the other hand, mRNA encoding the α7-nAChR subunit that forms the low affinity receptor, was at or below the level of RT-PCR detection in primary T cells from normal subjects, but it reached a detectable threshold upon stimulation by anti-CD3 and/or nicotine (panel b). Unlike our results in primary T cells, immortalized Jurkat T leukemia cells had detectable levels of α7-nAChR mRNA, and both Jurkat and HL-60 myelogenous leukemia cells expressed α7-nAChR protein constitutively (panel c), suggesting this subunit is sensitive to upregulation by events that drive or sustain proliferation.
Figure 2. Nicotinic acetylcholine receptor expression in human T lymphocytes.

The expression of messenger RNA (a and b) for α4-, ϐ4-, and α7-nAChR subunits, and protein for the α7-subunit (c) was examined in human peripheral blood T cells and in Jurkat T cells using RT-PCR and immunoblotting, respectively. Panel a shows expression of α4- (418 bp amplification product) and ϐ4- (472 bp amplification product) subunits in lymphocytes from ten healthy, adult non-smokers, as well as ϐ-actin as a loading control (note that data for ϐ4-nAChR and for ϐ-actin are compiled from two gels, representing the indicated donors); panel b shows expression of the α7-subunit (122 bp amplification product) in peripheral blood T cells from one representative healthy non-smoker and in Jurkat T cells. ϐ-actin expression in the same samples was used to confirm integrity of the RNA and equivalent loading; panel c shows protein expression of the α7-nAChR in HL-60 and Jurkat cells. A ϐ-actin immunoblot from the same samples is shown as a loading control.
Nicotine increases cell death in activated primary lymphocytes
We next examined how nicotine influenced survival of primary human lymphocytes and cell lines. Nicotine did not effectively reduce cell proliferation of Jurkat T cells or IL-2-dependent Kit-225 T cells over a period of 30 days in culture (10 passages) at concentrations ranging from 10 nM to 100 μM. Normal human T cells do not proliferate spontaneously, but remain viable in culture without stimulation for several days. Consistent with previous studies (Yoshida et al., 1998; Mariggio et al., 2001), exposure to less than, or equal to, 200 μM nicotine (replenished daily into the media for up to a week) also did not affect viability based on trypan blue exclusion or by uptake of 7-AAD in unstimulated peripheral blood lymphocytes (data not shown) or in UV-treated Jurkat cells (see below). Yet, the effect of nicotine on activated lymphocytes has been examined in less detail. To test the hypothesis that nicotine signals modulate lymphocyte proliferation and survival, we added increasing concentrations of nicotine to T cells cultured with or without anti-CD3. There was a modest trend to increasing death in activated T cells (from ~10% to ~18 %) that were pre-incubated with nicotine for 30 min prior to stimulation, and that remained exposed to nicotine for the duration of the experiment, as determined by uptake of 7-AAD after 48–55 hr in culture (Figure 3).
Figure 3. Increased numbers of dead cells are present in activated T cells cultured in the presence of nicotine.

Peripheral blood T cells were incubated with or without nicotine for 30 min prior to competence induction with anti-CD3 (10 ng/ml) as indicated and cultured for 48–55 hr without additional stimuli. Loss of cell viability was determined flow cytometrically by uptake of 7-AAD and is indicated as the percent of total events analyzed. The fold-change in dead cells (normalized to 1.00 for unstimulated cells without exposure to nicotine) is shown above each bar. Data represent means (±S.D) from three independent experiments.
Nicotine promotes pro-apoptotic and anti-apoptotic events in lymphocytes
Several mechanisms could account for the reduced lymphocyte viability seen in the presence of nicotine. For example, nicotine activates nuclear factor of activated T cells (NFAT) transcription factors in human T cells (Frazer-Abel et al., 2004), suggesting transcriptional NFAT targets such as FasL, the physiological ligand for the Fas (CD95) “death receptor”, might be upregulated in response to nicotine. Prior experimental data support this notion: FasL was reportedly elevated in primary lymphocytes from smokers (Suzuki et al., 1999); thus, we examined the effect of nicotine on FasL expression in primary human lymphocytes (from non-smokers). Predictably, FasL mRNA was undetectable in unstimulated T cells and was induced in T cells that were rendered competent with soluble anti-CD3 and cultured for 72 hr (Figure 4), although this does not lead to proliferation (Frazer-Abel et al., 2004). Interestingly, nicotine alone did not promote robust FasL gene expression, but when nicotine-treated cells were subsequently stimulated with anti-CD3, steady state levels of FasL mRNA were consistently increased.
Figure 4. Nicotine induces FasL and Survivin gene expression in normal human peripheral blood T cells.
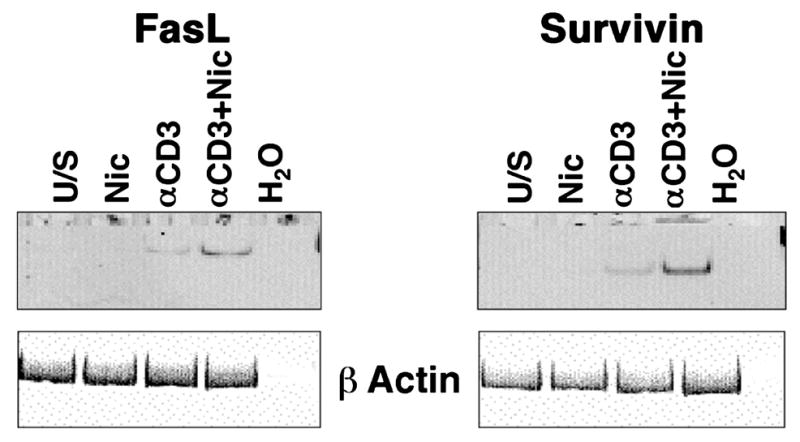
Human peripheral blood T cells were incubated with or without nicotine for 30 min prior to competence induction as indicated and cultured for 72 hr without additional stimuli. Expression of FasL and Survivin was examined using RT-PCR under conditions of linear amplification. Amplification products for each gene were 239 and 199 bp, respectively. The same reaction omitting input cDNA (H2O) was used as a negative control. Expression of ϐ-actin (317 bp amplification product) was used to ensure integrity of the RNA. The experiment was repeated in at least 3 donors for each gene with similar results.
Despite this increase in FasL expression, the observation that nicotine did not promote extensive cell death (i.e., <20% of cells underwent apoptosis) could be a quantitative phenomenon, or it could be due to the simultaneous upregulation of survival signals. Nicotine was reported to induce Survivin expression in lung cancer cells (Dasgupta et al., 2006), possibly accounting for their resistance to chemotherapy agents. Unlike tumors and cell lines, primary lymphocytes did not express Survivin mRNA (Figure 4), but the gene was inducible in cells rendered competent by stimulation with soluble anti-CD3. It is worth reiterating that induction of “competence” (transition to the G0/G1 boundary) does not result in proliferation, (Modiano et al., 1999), so upregulation of Survivin does not reflect progression of these T cells into mitosis when the gene would normally be expressed at high levels. As was true for FasL, nicotine alone did not promote robust Survivin gene expression, but in cells stimulated by anti-CD3, nicotine increased the steady state levels of Survivin mRNA.
Consistent with the immortalized phenotype, Survivin and another potent anti-apoptotic protein, Bcl-2, were expressed constitutively in asynchronously growing Jurkat cells (Figure 5). In these cells, nicotine treatment did not alter the expression of either protein; however, nicotine reproducibly increased total caspase activity as measured by cleavage of a fluorescent ZVAD substrate (Figure 6). Nevertheless, nicotine did not significantly affect the net balance of apoptosis (basal or inducible) in Jurkat cell as measured by measured by DNA fragmentation or by PARP cleavage (Figure 6). In fact, nicotine treatment alone did not consistently lead to accumulation of cleaved Caspase-3 in Jurkat cells, and the accumulation of cleaved Caspase-3 in cells exposed to UV irradiation or treated with soluble FasL also was unaffected by nicotine. On average, expression of Survivin and Bcl-2 also was not significantly different in nicotine-treated Jurkat cells with or without UV exposure, which was unlike the observed reduction in the steady state levels of CDK4 (Figure 5). Together, the data suggest the nicotine-associated activity detected by ZVAD cleavage is independent of Caspase-3 (Frost and Sinclair, 2000; Frost et al., 2001; Kane et al., 2004; Medina-Palazon et al., 2004; Rezvani et al., 2007; Tambyrajah et al., 2007), or alternatively, that, at least under certain circumstances, activation of caspases might be dissociated from apoptosis in Jurkat cells.
Figure 5. Effect of nicotine on expression of Survivin, Bcl-2, and CDK4 in UV-treated Jurkat cells.

Jurkat cells were treated by exposure to UV light (2 min) with or without nicotine as indicated and cultured for 4 hours without additional stimuli. Steady state levels of Survivin, Bcl-2, and CDK4 were examined by immunoblotting. ϐ-actin was used as a loading control. The strips shown are from one representative experiment of at least 5 done for each target protein.
Figure 6. Nicotine upregulates caspase activity, but does not tilt the net balance apoptosis in Jurkat cells exposed to UV irradiation.

Jurkat cells were treated by exposure to UV light (2 min) with or without nicotine as indicated and cultured for 4 hours without additional stimuli. (A) Pan-caspase activity was analysed fluorimetrically using a FITC-labeled VAD-fmk conjugate. Data show the means (±S.E.M.) of 5 experiments were relative fluorescence was normalized to a maximum of 1.0. (B) DNA fragmentation was measured flow cytometrically using the Apo-BrDU Tunel assay kit. Data show the means (±S.E.M.) of 3 experiments were the maximum rate of apoptosis (97% for UV + nicotine-treated cells) was normalized to 1.0. The inset shows immunoblots from whole cell lysates were evaluating cleavage of PARP (a prototypical Caspase-3 substrate). ϐ-actin was used as a loading control.
Distinct nAChRs have unique and overlapping effects of in lymphocytes
Various studies have used selective chemical inhibitors of nAChRs to address the function of the distinct receptors. While these experiments can be informative, they also have inherent disadvantages. First, chemical ligands can lack selectivity for the nAChR subunits, and second, even compounds that selectively bind a single subunit (notably, α-bungarotoxin for α7 subunits) can have mixed agonist/antagonist effects on the receptor. To avoid the bias of chemical inhibitors, we chose to use a genetic approach to assess the role of high- and low-affinity nAChRs in cellular survival. We designed small hairpin RNA (shRNA) constructs to reduce ϐ4- and α7-nAChR subunits (Table 2). To address potential off-target effects, we designed double point mutant controls for each shRNA (Table 2). Transfection of Jurkat cells with an irrelevant shRNA molecule (designed to knock down PTEN (Lin et al., 2007) had no effect on nAChR expression (data not shown). In contrast, transient transfection of the ϐ4- and α7-shRNA constructs attenuated expression of their respective targets (Figure 7a), although in these transient transfection experiments, the ϐ4-nAChR shRNA significantly reduced expression of the α7-nAChR subunit (Figure 7a top panel). The incomplete attenuation of the ϐ4 subunit in the same ϐ4-nAChR shRNA-transfected cells (Figure 7a middle panel) could have been related to transfection efficiency. We believe this is unlikely, as we normally achieved transfection of >75% of Jurkat cells, and even though the relative expression of GFP, used as a transfection indicator, was not normally distributed in the samples, there was no difference in the overall GFP expression among transfected samples. There also was no difference in the relative survival of these cells at the time we measured gene expression (48 hr), suggesting the effect might be due to the relative efficiency with which the shRNA molecule targets the ϐ4 subunit for degradation. The attenuation of the α7 subunit in cells that were transfected with ϐ4-nAChR shRNA could have been due to off-target effects, or to a requirement for ϐ4-nAChR signaling to sustain expression of the α7 subunit. To address these possibilities, we examined nAChR expression in Jurkat cells after stringent selection on puromycin-containing media. Figure 7b shows that after selection, the expression of the respective subunit was significantly reduced in each transfectant, and cells transfected with the ϐ4-nAChR shRNA did not maintain an α7-knockdown phenotype. We thus examined how attenuation of the ϐ4- and α7-nAChR subunits affected nicotine-dependent calcium mobilization in the stable knockdown cells. As shown in Figure 1, nicotine induced calcium mobilization in Jurkat cells. The relatively modest effects of nicotine were probably related to both the total influx of calcium through the nAChR channel and to the number of cells responding, which was generally <20% of the total population (compared to stimulation by anti-CD3, which led to a response by >85% of the cells). Predictably, both ϐ4-nAChR and α7-nAChR knockdown cells had blunted calcium mobilization (both as measured by total calcium response and by the number of nicotine-responsive cells, which was no different from the baseline seen in untreated cells) in response to stimulation by nicotine. However, these cells also showed blunted calcium responses to anti-CD3 with α7-nAChR knockdown cells showing the more severe phenotype (Figure 7c and d), and in these cells, the addition of nicotine did not further reduce this calcium response to anti-CD3.
Table 2.
Target Sequences and Controls for RNA Inhibition
| Gene | siRNA construct | Target Sequence |
|---|---|---|
| CHRNA7 (α7-nAChR) | Alpha-7.5 (position 326) | AACCAGACATTCTTCTCTATA |
| CHRNA7 negative control | Aplha-7.5 DPM | AACCTGACATTCTTAACATATA |
| CHRNB4 (ϐ4-nAChR) | Beta-4.3 (position 351) | TGTCTGGCTGAAACAGGAAT |
| CHRNB4 negative control | Beta-4.3 DPM | TCTCTGGGTGAAACAGGAAT |
Figure 7. Attenuation of nAChRs in Jurkat cells using RNA interference.

Panel a shows the effect of transient transfection of shRNA constructs for ϐ4- (Beta-4.5) or α7- (Alpha-7.13) nAChR subunits in Jurkat T cells. Controls were designed for each shRNA by creating double point mutant oligonucleotides (see Table 2). Amplification products for each subunit and for ϐ-actin (as a control for RNA integrity and loading) shown in the figure are from the same experiment. Control shRNA vectors are indicated by the abbreviation DMC (double point mutant control). Panel b illustrates the nAChR expression phenotypes in Jurkat T cells after puromycin selection. Expression of nAChR mRNA in both transiently and stably transfected cells was examined by RT-PCR under conditions of linear amplification (KD = “knockdown”). Panels c and d show calcium mobilization in the nAChR-knockdown Jurkat cells. Control Jurkat cells (“WT”), α7-KD (“a7shRNA”), or ϐ4-KD cells (“ϐ4shRNA) cells were left untreated or treated with nicotine (10 μM) as indicated. After 5 min, cells were allowed to remain as previously treated, or were stimulated by addition of anti-CD3. Alterations in intracellular ionized calcium (Cai++) were measured using a MoFlo flow cytometer. Y-axis values represent Indo-1 emission ratios at 405 nm/480 nm. Basal ionized calcium levels were ~150 nM.
Given the observation that nicotine failed to protect Jurkat cells from apoptotic stimuli, along with the observation that it might potentiate caspase activity, we sought to determine if attenuation of nAChR subunits affected lymphocyte survival. Predictably, soluble FasL induced Jurkat cell apoptosis (measured in this case by appearance of Caspase-3 cleavage products), and this was unaffected by exposure to soluble anti-CD3 or nicotine (Figure 8). Intriguingly, reduction of the α7-nAChR subunit enhanced both basal and FasL-mediated cleavage of Caspase-3 in Jurkat cells (Figure 8). Caspase-3 cleavage products were detectable in untreatedα7-nAChR knockdown cells, while treatment with soluble FasL induced a reduction in the levels of pro-Caspase-3 with accumulation of additional cleaved Caspase-3. Cleavage of Caspase-3 in response to soluble FasL was neither inhibited nor enhanced by nicotine, and likewise, treatment with anti-CD3 antibodies did not consistently prevent Caspase-3 cleavage in these cells. Attenuation of the ϐ4-nAChR subunit did not consistently show a comparable, significant enhancement of Caspase-3 cleavage, but in these cells, we reproducibly observed multiple cleaved Caspase-3 products. These results are consistent with our observations regarding the survival of these cells in culture. Both the α7- and the ϐ4-nAChR knockdown cells grew more fastidiously. Within 3–6 months, cells seemed to revert to a wild type phenotype with detectable levels of the targeted nAChR, indicating selective pressures allowed them to circumvent the effects of the shRNA. This is not unique to these cells, as we have seen similar effects in other cell types where essential survival factors are knocked down using the pSuper shRNA system (A. Jackson et al, manuscript in preparation). Curiously, myeloid leukemia cell lines died rapidly when transfected with shRNAs against either nAChR subunit (but not when transfected with the control shRNAs), suggesting that they did not tolerate loss of nAChRs. Because the increased tendency to die upon loss of nAChRs may have been a function of the malignant phenotype, we examined how attenuation of nAChR subunits affected survival of primary human lymphocytes. Primary T cells were transfected by nucleofection with dsRed as a fluorescent tracer, concomitantly with control shRNA, α7-subunit shRNA ϐ4-subunit shRNA, or both α7-and ϐ4-subunit shRNAs. Survival of transfected cells (dsRed+) was quantified flow cytometrically. Survival and transfection efficiencies immediately after the procedure and after 24 hr were not significantly different between any conditions. However, after 48 hr there were fewer surviving dsRed+ cells in the α7-subunit KD cells and in the double KD cells, and after 72 hr in culture, the differences were significant between both nAChR-KD cells and controls (Figure 9). More interestingly, no detectable dsRed+ cells remained 72 hr after simultaneous knockdown of both subunits (and therefore high- and low-affinity nAChRs), suggesting that these receptors transmit tonic and inducible signals that promote lymphocyte survival.
Figure 8. Effect of nAChR loss on Jurkat cell apoptosis.

Control Jurkat cells (WT),α7-nAChR knockdowns (α7-KD) or ϐ4-nAChR-knockdowns (ϐ4-KD) were cultured in the presence or absence of soluble FasL (sFasL) for 4 hr as indicated, with or without a 15-min pre-exposure to nicotine (1 μM) and/or concomitant exposure to anti-CD3 (10 ng/ml). Cleavage of pro-Caspase-3, examined by immunoblotting, was used as a measure of apoptosis. ϐ-actin was used as a loading control. The data show one representative experiment of three done.
Figure 9. Effect of nAChR loss on survival of normal human peripheral blood T cells.

T cells were transfected with dsRed and the shRNA constructs indicated. Transfection efficiencies were comparable for all conditions after 24 hr. The number of dsRed+ cells remaining in the cultures was evaluated by flow cytometry after 72 hr. Mean percent (range) dsRed+ cells in triplicate transfections were: none (background), 2.6% (2.4–2.8); Control, 31.7%(25–34), ϐ4-nAChR shRNA, 28%(26–30); α7-nAChR shRNA, 17.4%(15–19); ϐ4-nAChR shRNA+α7 nAChR shRNA, 1.8%(1.3–2.5).
Discussion
Nicotine has pleotropic effects within and outside the nervous system that are incompletely understood. This is one of the few tobacco-derived compounds that achieve systemic pharmacological levels (Gritz et al., 1981; Rose et al., 1984; Rose et al., 1985; Benowitz et al., 1988; Schneider et al., 1996; Jarvik et al., 2000), and its effects on the central nervous system are largely responsible for the addictive properties of tobacco products. Outside of these effects, nicotine has been perceived to be rather innocuous, so much so that systemic nicotine treatment is one of the mainstays of tobacco cessation programs as the active principle in a variety of devices such as gum, patches, and inhalers (Benowitz and Gourlay, 1997).
Recently, four independent genome wide association studies found that the risk of lung cancer in smokers was strongly associated with a marker in chromosome 15 that is coincident with the α3-nAChR subunit (Amos et al., 2008; Berrettini et al., 2008; Hung et al., 2008; Thorgeirsson et al., 2008), which partners with the ϐ4 subunit to form one of the high affinity receptors (Leonard and Bertrand, 2001). These groups favored different hypotheses to explain the association. For example Thorgeirsson et al (Thorgeirsson et al., 2008) suggested a functional difference in the amount of tobacco consumed could explain the risk (in other words, the polymorphism might dictate addictive potential), while Hung et al (Hung et al., 2008) suggested a functional role in transformation in the lung. Our data suggest that altered inflammation or anti-tumor immunity could be a third, non-mutually exclusive mechanism that contribute to this association.
We tested a series of consecutive hypotheses to address inherent contradictions in the literature regarding the effects of nicotine on apoptosis of non-neuronal cells, and specifically white blood cells. Initially, we tested the hypothesis that leukocytes express functional nicotine receptors, and may thus be useful surrogates to study systemic effects of nicotine. We confirmed and extended previous results showing the expression of subunits that form high-affinity and low affinity nAChR in human PBMC, primary T cells, and transformed lymphoma and leukemia cell lines (Petro et al., 1992; Geng et al., 1995; Petro et al., 1999; Middlebrook et al., 2002; Frazer-Abel et al., 2004). We also showed that nAChRs are functional, based on tubocurarine and hexamethonium-sensitive calcium mobilization in response to nicotine (Frazer-Abel et al., 2004); so, while some in vivo effects may be attributable to central effects of nicotine on the hypothalamic-pituitary axis (with consequent release of endogenous corticosteroids) (Sopori, 2002), nicotine can unquestionably influence lymphocyte activation in vitro.
We used two complementary models to test nicotine response in human T cells. One was primary peripheral blood T cells; the other was the Jurkat T cell leukemia line. Nicotine responses appear to be conserved across both normal T cells and Jurkat cells, although the dose response might be left shifted in Jurkat cells (responses plateau at 100 nM to 1 μM (Frazer-Abel et al., 2004)), perhaps due to constitutive expression of the α7-nAChR subunit in Jurkat cells. In addition, despite these conserved signaling pathways, potential cell cycle effects noted in primary peripheral blood T cells (Frazer-Abel et al., 2004) may be dissociated from nicotine responses in Jurkat cells, which proliferate spontaneously, and the same could be true for pro-apoptotic responses that require PTEN, a tumor suppressor protein that is inactive in Jurkat cell lines.
The elevations in Cai2+ induced by nicotine were smaller in magnitude than those induced by anti-CD3 in both normal T cells and Jurkat cells, not because of fewer cells responding, but rather because individual cells registered lower Cai2+ concentrations (Frazer-Abel et al., 2004). Increasing the nicotine concentration also led to reduced calcium mobilization in response to anti-CD3. This could be due to exhaustion of intracellular stores, abrogation of antigen receptor signals, a decrease in the number of responding cells, or desensitization of nAChRs, which may contribute to the antigen receptor mediated extracellular calcium flux. It is unlikely that the blunted response was due to exhaustion of intracellular stores or to cell death. The magnitude of the calcium flux induced by nicotine is rather small and most likely due to influx of extracellular calcium, and measures of viability were unaffected under all conditions of acute exposure to nicotine at concentrations >100 μM in both normal peripheral blood T cells and Jurkat cells. Similarly, transmission of T cell antigen receptor signals was evident by accumulation of early response proteins such as CDK6 and IL-2 receptors (data not shown). Our data show fewer cells mobilized calcium in response to anti-CD3 at the highest nicotine concentration tested (72% at 50 μM vs. 88% without nicotine), but there was no difference in the number of responding cells at all other concentrations, suggesting that between 50 nM and 10 μM, nicotine blunted anti-CD3-dependent calcium mobilization equally in all the cells. Thus, we favor the explanation that nAChRs, which can undergo desensitization upon nicotine binding, contribute to the observed calcium influx that follows stimulation through the antigen receptor. The effects of nicotine are distinct from those of ionomycin, vis-a-vis the observation that nicotine alone induced Cyclin D2 polyubiquitination (which was not seen in ionomycin-treated cells), but it did not lead to significant increases in p27 in the absence of concurrent stimulation by anti-CD3 (whereas p27 levels were significantly increased in cells treated with ionomycin). This suggests that different patterns of calcium mobilization have distinct functional consequences or that nicotine activates additional pathways that are independent of calcium mobilization.
Jurkat cells are commonly used to examine T-cell signaling events (Abraham and Weiss, 2004), as early responses to antigen receptor stimulation are largely preserved and they are more tractable than primary cells for genetic manipulation. The use of RNA interference to attenuate specific subunits in immortalized Jurkat cells then allowed us to interpret the calcium mobilization data more clearly: we predicted nAChR knockdown cells would be unable to mobilize calcium in response to nicotine. Indeed, ϐ4-nAChR and α7-nAChR knockdowns had blunted calcium fluxes in response to stimulation by either nicotine or anti-CD3. In other words, not only were basal calcium fluxes in response to anti-CD3 reduced in ϐ4-nAChR knockdown cells, but also nicotine-treated cells showed reduced calcium mobilization when stimulated with anti-CD3. The calcium response to anti-CD3 was even more severely blunted in the α7-nAChR knockdown cells, and pre-incubation with nicotine did not reduce this response further.
Together, these results suggest that lymphocytes require both high- and low-affinity nAChR to achieve physiological levels of calcium flux upon triggering the antigen receptor/CD3 complex, although the low affinity α7-receptors may account for a greater component of the response in Jurkat cells. Furthermore, both sets of receptors appear to become desensitized when previously bound by nicotine. That is to say, we propose that ACh contributes to the normal calcium flux associated with lymphocyte activation, and nicotine desensitization of the receptors diminishes this effect, providing an explanation for why certain antagonists fail to reverse the effects of nicotine, as they would block the ability to ACh to bind the receptors in stimulated cells. This phenomenon is well characterized in neurons, where agonist binding to the nAChR leads to transient desensitization, an effect that may contribute to addiction (Leonard and Bertrand, 2001).
The dose response profiles also are informative. The Kd for nicotine binding of the high affinity α3/ϐ4, and α4/ϐ2 heteropentameric receptors in human leukocytes is ~3 – 6 nM (Benhammou et al., 2000). While the low affinity α7 homopentameric receptor also binds nicotine at nM concentrations, its half-maximal activation requires ~40 μM nicotine (Leonard and Bertrand, 2001), suggesting high affinity receptors probably are responsible for the slow, steady, sustained rise in calcium seen at low nicotine concentrations, and low affinity receptors, which desensitize rapidly (Quick and Lester, 2002; Wang and Sun, 2005), are probably responsible for the transient spike seen at higher nicotine concentrations. Together, our data support the hypothesis that human leukocytes harbor functional high affinity and low affinity nicotinic receptors.
We next tested the hypothesis that nicotine modulates survival of these cells. Previous experiments evaluating the effects of nicotine on apoptosis in non-neuronal cells have yielded contradictory results. The literature is replete of examples, but data from Arredondo et al (Arredondo et al., 2002; Arredondo et al., 2003) are illustrative. The investigators described a nicotine-induced, α7nAChR-dependent Ca2+ influx in keratinocytes exposed to 10μM nicotine for 24h that led to increased expression of p21, cyclin D1, PCNA, Ki-67 and Bcl-2. However, expression of Caspase-3 also increased. The authors hence presumed nicotine probably activates compensatory death and survival mechanisms in keratinocytes.
Our data uncover a similar paradox. Treatment with nicotine potentiated FasL expression in activated lymphocytes and induced the appearance of a caspase or caspase-like activity, suggesting it could facilitate apoptosis both of target cells that bear nicotinic receptors and of neighboring cells in their local microenvironment. At the same time, nicotine induced Survivin expression in primary T cells upon stimulation that promoted transition across the G0/G1 boundary. Nicotine-mediated upregulation of Survivin is not restricted to T cells, and it may account for chemoresistance seen in some tobacco-related malignancies (Dasgupta et al., 2006), although this response was not equally robust in Jurkat cells, perhaps because these cells have constitutively elevated Survivin expression. There may be crosstalk between these factors that leads cells to escape from apoptosis; for example, increased Survivin expression led to a concomitant increase in FasL expression in colon cancer cells which allowed them to kill immune effector cells by engaging Fas receptors on their surface, and thus evade their cytotoxic effects and consequent apoptosis (Asanuma et al., 2004). The net balance of apoptosis in primary cells and in Jurkat cells does not seem to be significantly affected by nicotine, although this compound may increase “fragility” of primary T cells that are activated by antigen-receptor signals and may activate caspases in Jurkat cells, although this elevated caspase activity seems to be independent of Caspase-3. Recent data suggest that nicotine can modulate proteasomal activity, including effects on chymotrypsin-like and caspase-like activities (Rezvani et al., 2007; Tambyrajah et al., 2007). Nicotine also appears to modulate proteasomal activity in lymphocytes, at least reducing degradation of cyclin D2. The accumulation of p27 in these cells, on the other hand, could reflect increased expression, reduced turnover, or a combination thereof.
Finally, we tested the hypothesis that high affinity and low affinity nAChRs contribute to these responses differently, possibly explaining conflicting data from myriad functional studies. Our data suggest this is likely, but nAChRs also may have overlapping functions. Attenuation of the α7-nAChR subunit rendered cells fragile and stable knockdowns only grew fastidiously in culture with eventual loss of the knockdown phenotype (death). The phenotype caused by reduction of the ϐ4-subunit was significantly less dramatic, but the cells did show delayed proliferation and a modest increase in their sensitivity to apoptosis as compared to wild type Jurkat cells. What is more, myeloid leukemia cell lines died rapidly when transfected with shRNAs against either nAChR subunit (but not when transfected with other shRNAs), suggesting these cells are even less tolerant of nAChR loss. The increased tendency to die upon attenuation of nAChRs was not simply a function of the malignant phenotype, as it was recapitulated in primary T cells where silencing of the α7-subunit alone introduced a selective survival disadvantage, and silencing of both the α7- and the ϐ4-subunit seemed to be lethal, suggesting these receptors transmit both tonic and inducible signals that promote cellular survival.
In summary, we show here that nicotine, at concentrations found in the circulation of habitual users of tobacco products and nicotine cessation devices, can modulate lymphocyte function and might influence survival. Our data provide a framework and a set of tools that will be useful to address specific mechanisms that mediate the apparent paradoxically effects of nicotine to simultaneously promote and oppose apoptosis in lymphocytes and other non-neuronal cells. We believe these effects are context-specific, and might implicate nicotine directly in the pathogenesis of cancers that arise outside of the aerodigestive tract (e.g., leukemia). Our data also suggest that nicotine might modulate inflammation in the tumor microenvironment and anti-tumor immune responses, both of which we now know are important contributors to the biological behavior and natural history of human cancers. It is also worth noting that in our experiments, nicotine did not affect the levels or kinetics of immunomodulating and inflammatory cytokines (A. Pierce et al, unpublished). Thus, we favor the theory that it is the effects of nicotine on survival, and not its effect on cytokine responses that explain how this ubiquitous alkaloid alters pro-inflammatory environments that influence tumor progression.
Material and Methods
Cells and cell culture
Procedures using human cells were reviewed and approved by the Colorado Multiple Institutional Review Board. Whole blood or apheresis residues were obtained from healthy adults with informed consent. Primary T lymphocytes and immortalized Jurkat and Kit-225 human T cell leukemias and HL-60 human myelogenous leukemia cells were prepared and maintained as described (Khare et al., 2003; Frazer-Abel et al., 2004). Transfections were done one day after fresh passage of cells using electroporation with an Amaxa nucleofector (Lonza, Cologne, Germany) with the Human T cell Nucleofector Kit on setting U-14 for primary T cells and the Cell line Nucleofector Kit V on settings S-18/X-005 for Jurkat T cells as per the manufacturers recommendations. The transfection efficiency using this system was >75% for Jurkat cells and ~30% for primary T cells. Cells used for experiments were used at a concentration 1–5×106/ml in 6 well plates. Nicotine was prepared daily by dissolving nicotine tartrate salt in media immediately prior to addition to the cells. For apoptosis induction, cells were exposed to UV irradiation for 2 minutes, or to recombinant soluble FasL (10 ng/ml) used in the presence of a cross-linker as recommended by the manufacturer (Alexis Biochemicals, Plymouth Meeting, PA) and incubated for 4 hours at 37°C in a humidified 5% CO2 atmosphere prior to harvesting for analysis.
Immunoprecipitation and immunoblotting
Cyclin D2 complexes were immunoprecipitated from Jurkat cells using a monoclonal antibody directed against the C-terminal domain (Santa Cruz Biotechnology, Santa Cruz, CA, USA) as described (Modiano et al., 1994). Immunoprecipitates were separated electrophoretically and immunoblotted with an antibody directed against ubiquitin (Santa Cruz). Immunoblotting on whole cell lysates was done generally as described (Jubala et al., 2005) using antibodies against p27, the pro-survival proteins Survivin and Bcl-2, pro-Caspase-3 and cleaved Caspase-3 (all from Santa Cruz), α7-nAChR (Abcam, Cambridge, MA), cleaved PARP (Cell Signaling Technology, Danvers, MA), and ϐ-actin (mouse monoclonal anti-human, 1:5000, Sigma). For detection of multiple proteins on the same blot, membranes were stripped for 25 min at 40°C in 100mM 2-mercaptoethanol, 2%SDS, 62.5mM Tris-HCl pH 6.7 and reprobed.
Calcium mobilization
Calcium mobilization was preformed as described (Frazer-Abel et al., 2004). Briefly, cells were loaded with 2μg/ml Indo-1-acetoxymethylester (Indo-1) a fluorescent calcium chelator (Calbiochem, San Diego, CA). After washing the alterations in calcium flux were monitored on a MoFlo flow cytometer (Beckman Coulter, Miami, FL).
Detection of apoptosis and cell death
Apoptosis was measured using several distinct methods. Membrane integrity (intact membranes are indicative of viable cells) was measured flow cytometrically by uptake of the fluorescent vital dye 7-amino actinomycin D (7AAD). Loss of membrane asymmetry was assessed flow cytometrically by Annexin V binding. Propagation of apoptosis signals was determined by caspase activation using a fluorescent substrate and specificity verified by use of a competitive pan-caspase inhibitor (VAD-fmk, Promega, Madison, WI). Finally, assessment of nucleosomal cleavage was confirmed using the APO-BrdU TUNEL kit (Invitrogen-Molecular Probes, Eugene, OR) according manufacturer instructions. At least 10,000 events were collected for flow cytometric analyses using the FC500 flow cytometer (Beckman Coulter). Doublets were excluded from the analysis using the peak versus integral gating method. Each experiment was repeated 3–5 times; means (±S.D. or S.E.M., as appropriate) for all replicates or data from a representative experiment are shown as indicated in the figure legends.
Gene expression
Gene expression was measured as described previously (Benhammou et al., 2000; Frazer-Abel et al., 2004). The primer sequences and condition used to amplify each of the genes of interest are listed in Table 1. Messenger RNAs for nAChR subunits in lymphocytes are present in low abundance, so the following modifications were used to optimize amplification of PCR products: for the α4-nAChR subunit, a pre-amplification cycle included a denaturing step at 96°C for 3 min, 5 cycles of amplification using 96°C for 1min for denaturation, 46°C for 30 sec for annealing, and 72°C for 1 min for extension, and 5 additional cycles using 96°C for 1min for denaturation, 48°C for 30 sec for annealing, and 72°C for 1 min for extension prior to 30 cycles using the conditions listed in Table 1. For the ϐ4-nAChR subunit, the pre-amplification cycle included a denaturing step at 96°C for 3 min, and 5 cycles of amplification using 96°C for 1min for denaturation, 48°C for 30 sec for annealing, and 72°C for 1 min for extension followed by 35 cycles using the conditions listed in Table 1. The second set of oligonucleotide PCR primers used to amplify the ϐ4-nAChR subunit required an initial denaturation step at 95°C for 5 min followed by 35 cycles using the conditions listed in Table 1. For the α7-nAChR subunit, pre-amplification only required an initial denaturing step at 96°C for 10 min followed by 30 cycles using the conditions listed in Table 1.
Table 1.
Primers and RT-PCR Conditions
| Gene | Sense Primer | Antisense Primer | PCR Conditions | Amplification Product (bp) |
|---|---|---|---|---|
| Denaturing (Temp/Time) | ||||
| Annealing (Temp/Time) | ||||
| Extension (Temp/Time) | ||||
| CHRNA4 (α4-nAChR) | TGCACATGCAAGAAGGAGCC | CCACAGAGTCCAGGGAGAAGC | 95°C/30 sec | 391 |
| 53°C/30 sec | ||||
| 72°C/30 sec | ||||
| CHRNA7 (α7-nAChR) | TTTACAGTGGAATGTGTCAGAATATCC | TGTGGAATGTGGCGTCAAAG | 96°C/30 sec | 237 |
| 58°C/30 sec | ||||
| 72°C/1 min | ||||
| CHRNA7 (α7-nAChR) | CACCGTCTACTTCTCCCTGAGCCTCCTG | ACGTTAGTGTGGAATGTGGCGTCAAAGC | 96°C/30 sec | 237 |
| 58°C/30 sec | ||||
| 72°C/1 min | ||||
| CHRNB4 (ϐ4-nAChR) | CGGCGAGAAGATGACACTGTG | AGAGGACCGCAGCCAGAAAT | 95°C/30 sec | 472 |
| 53°C/30 sec | ||||
| 72°C/30 sec | ||||
| CHRNB4 (ϐ4-nAChR) | ATGGTGCTGGTCACCTTCTC | ATGAAGCTGACGCCCTCTAA | 95°C/1 min | 380 |
| 55°C/1 min | ||||
| 72°C/1 min | ||||
| FASLG (Fas ligand) | Proprietary (R&D Systems RDP-58) | Proprietary (R&D Systems RDP-58) | 94°C/45 sec | 239 |
| 55°C/45 sec | ||||
| 72°C/45 sec | ||||
| BIRC5 (Survivin) | Proprietary (R&D Systems RDP-204-025) | Proprietary (R&D Systems RDP-204–025) | 94°C/45 sec | 199 |
| 55°C/45 sec | ||||
| 72°C/45 sec | ||||
| ϐ-actin | ATGTTTGAGACCTTCAACACCC | GCCATCTCTTGCTCGAAGTCCA | 95°C/1 min | 317 |
| 60°C/1 min | ||||
| 72°C/2 min |
Design of vectors for RNA inhibition
We used the pSuper platform (OligoEngine, Seattle, WA) to clone nAChR short hairpin RNA molecules (shRNAs) as described (Lin et al., 2007). Small inhibitory RNA sequences and controls (Table 2) were designed from the human CHRNA7 and CHRNAB4 gene sequences according to previously described algorithms (Lin et al., 2007). Sequences were selected based on efficacy to knock down the respective target genes in Jurkat cells in transient transfection experiments, with off-target effects assessed using controls designed by double point mutations in each sequence where the resulting oligonucleotides did not show perfect matches for any mammalian sequence using basic blast (NCBI, National Library of Medicine, NIH). Small hairpin RNA vectors were designed by cloning the oligonucleotides into the BglII/HindIII sites of the pSuper-puro vector. Stable cell lines integrating shRNA vectors were selected in puromycin containing media.
Statistics
Data were analyzed using GraphPad Prism version 4.0 c (San Diego, CA). Statistical significance was determined as p ≤ 0.05, as measured by unpaired 2-tailed Student’s t-test.
Image manipulations
Brightness and contrast for the composite blot images were optimized using Adobe Photoshop CS3 (Adobe, San Jose, CA). Donor samples evaluating expression of α4-nAChR were run in two separate gels. Images from gels were cropped and joined at D7–D8.
Acknowledgments
The authors wish to thank Drs. Sherry Leonard, Mariella DeBiasi, and Beth Tamburini for helpful discussions, and Evan Pushchak, Daniel Davila, Heathers Brungardt-Meylemans, Lori Gardner, and Dr. Angela Pierce for technical assistance.
Footnotes
Abbreviations used in this manuscript: FasL, Fas ligand; UV, ultraviolet; nAChR, nicotinic acetylcholine receptor; ACh, acetylcholine; Cai2+, intracellular ionized calcium; NFAT, nuclear factor of activated T cells; shRNA, small hairpin RNA
Conflict of Interest: The authors declare that there are no conflicts of interest.
Supported by a grant from the Philip Morris External Research Program, grants R55CA086432, P30CA046934, and P30CA077598 from the National Cancer Institute, and RSG-02-173-01-LIB from the American Cancer Society. AAFA was supported in part by a University of Colorado Cancer Center Fellowship and by grant F32AI050392-01A1 from the National Institute of Allergy and Infectious Diseases, SR is supported by grant T32RR018719 from the National Center for Research Resources. DK was supported by USDA Challenge grant Summer Research Scholarship. Philanthropic funds from the Monfort Family Foundation (to the University of Colorado Cancer Center), the Minnesota Medical Foundation, and the University of Minnesota Foundation were used to support part of this research. JFM is supported in part by the Alvin S. and June Perlman Chair in Animal Oncology at the University of Minnesota.
References
- Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–308. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoshiba K, Nagai A, Yasui S, Konno K. Nicotine prolongs neutrophil survival by suppressing apoptosis. J Lab Clin Med. 1996;127:186–194. doi: 10.1016/s0022-2143(96)90077-3. [DOI] [PubMed] [Google Scholar]
- Arredondo J, Hall LL, Ndoye A, Nguyen VT, Chernyavsky AI, Bercovich D, Orr-Urtreger A, Beaudet AL, Grando SA. Central role of fibroblast alpha3 nicotinic acetylcholine receptor in mediating cutaneous effects of nicotine. Lab Invest. 2003;83:207–225. doi: 10.1097/01.lab.0000053917.46614.12. [DOI] [PubMed] [Google Scholar]
- Arredondo J, Nguyen VT, Chernyavsky AI, Bercovich D, Orr-Urtreger A, Kummer W, Lips K, Vetter DE, Grando SA. Central role of alpha7 nicotinic receptor in differentiation of the stratified squamous epithelium. J Cell Biol. 2002;159:325–336. doi: 10.1083/jcb.200206096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma K, Tsuji N, Endoh T, Yagihashi A, Watanabe N. Survivin enhances Fas ligand expression via up-regulation of specificity protein 1-mediated gene transcription in colon cancer cells. J Immunol. 2004;172:3922–3929. doi: 10.4049/jimmunol.172.6.3922. [DOI] [PubMed] [Google Scholar]
- Benhammou K, Lee M, Strook M, Sullivan B, Logel J, Raschen K, Gotti C, Leonard S. [(3)H]Nicotine binding in peripheral blood cells of smokers is correlated with the number of cigarettes smoked per day. Neuropharmacology. 2000;39:2818–2829. doi: 10.1016/s0028-3908(00)00153-2. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Gourlay SG. Cardiovascular toxicity of nicotine: implications for nicotine replacement therapy. J Am Coll Cardiol. 1997;29:1422–1431. doi: 10.1016/s0735-1097(97)00079-x. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Porchet H, Sheiner L, Jacob Pd. Nicotine absorption and cardiovascular effects with smokeless tobacco use: comparison with cigarettes and nicotine gum. Clin Pharmacol Ther. 1988;44:23–28. doi: 10.1038/clpt.1988.107. [DOI] [PubMed] [Google Scholar]
- Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–373. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catassi A, Servent D, Paleari L, Cesario A, Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–231. doi: 10.1016/j.mrrev.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Edelstein SJ. Nicotinic acetylcholine receptors: from molecular biology to cognition. Odile Jacob Publishing Corp; New York: 2005. [Google Scholar]
- Conti-Fine BM, Navaneetham D, Lei S, Maus AD. Neuronal nicotinic receptors in non-neuronal cells: new mediators of tobacco toxicity? Eur J Pharmacol. 2000;393:279–294. doi: 10.1016/s0014-2999(00)00036-4. [DOI] [PubMed] [Google Scholar]
- Cooke JP. Angiogenesis and the role of the endothelial nicotinic acetylcholine receptor. Life Sci. 2007;80:2347–2351. doi: 10.1016/j.lfs.2007.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Erausquin M, Marubio LM, Klink R, Changeux JP. Nicotinic receptor function: new perspectives from knockout mice. Trends Pharmacol Sci. 2000;21:211–217. doi: 10.1016/s0165-6147(00)01489-9. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A. 2006;103:6332–6337. doi: 10.1073/pnas.0509313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CD, Arends MA, Kenny PJ. Subtypes of nicotinic acetylcholine receptors in nicotine reward, dependence, and withdrawal: evidence from genetically modified mice. Behav Pharmacol. 2008;19:461–484. doi: 10.1097/FBP.0b013e32830c360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer-Abel AA, Baksh S, Fosmire SP, Willis D, Pierce AM, Meylemans H, Linthicum DS, Burakoff SJ, Coons T, Bellgrau D, Modiano JF. Nicotine activates NFATc2 and prevents cell cycle entry in T cells. J Pharmacol Exp Ther. 2004;311:758–769. doi: 10.1124/jpet.104.070060. [DOI] [PubMed] [Google Scholar]
- Frost V, Al-Mehairi S, Sinclair AJ. Exploitation of a non-apoptotic caspase to regulate the abundance of the cdkI p27(KIP1) in transformed lymphoid cells. Oncogene. 2001;20:2737–2748. doi: 10.1038/sj.onc.1204367. [DOI] [PubMed] [Google Scholar]
- Frost V, Sinclair AJ. p27KIP1 is down-regulated by two different mechanisms in human lymphoid cells undergoing apoptosis. Oncogene. 2000;19:3115–3120. doi: 10.1038/sj.onc.1203657. [DOI] [PubMed] [Google Scholar]
- Garrido R, Mattson MP, Hennig B, Toborek M. Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome c release and apoptosis of cultured spinal cord neurons. J Neurochem. 2001;76:1395–1403. doi: 10.1046/j.1471-4159.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- Geng Y, Savage SM, Johnson LJ, Seagrave J, Sopori ML. Effects of nicotine on the immune response. I. Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol Applied Pharmacol. 1995;135:268–278. doi: 10.1006/taap.1995.1233. [DOI] [PubMed] [Google Scholar]
- George J, Bais HP, Ravishankar GA. Biotechnological production of plant-based insecticides. Crit Rev Biotechnol. 2000;20:49–77. doi: 10.1080/07388550091144186. [DOI] [PubMed] [Google Scholar]
- Gritz ER, Baer-Weiss V, Benowitz NL, Van Vunakis H, Jarvik ME. Plasma nicotine and cotinine concentrations in habitual smokeless tobacco users. Clin Pharmacol Ther. 1981;30:201–209. doi: 10.1038/clpt.1981.149. [DOI] [PubMed] [Google Scholar]
- Grutter T, Changeux JP. Nicotinic receptors in wonderland. Trends Biochem Sci. 2001;26:459–463. doi: 10.1016/s0968-0004(01)01921-1. [DOI] [PubMed] [Google Scholar]
- Hakki A, Pennypacker K, Eidizadeh S, Friedman H, Pross S. Nicotine inhibition of apoptosis in murine immune cells. Exp Biol Med. 2001;226:947–953. doi: 10.1177/153537020122601011. [DOI] [PubMed] [Google Scholar]
- Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- Jarvik ME, Madsen DC, Olmstead RE, Iwamoto-Schaap PN, Elins JL, Benowitz NL. Nicotine blood levels and subjective craving for cigarettes. Pharmacol Biochem Behav. 2000;66:553–558. doi: 10.1016/s0091-3057(00)00261-6. [DOI] [PubMed] [Google Scholar]
- Jubala CM, Wojcieszyn JW, Valli VEO, Getzy DM, Fosmire SP, Coffey D, Bellgrau D, Modiano JF. CD20 expression in normal canine B cells and in canine non-Hodgkin’s lymphoma. Vet Pathol. 2005;42:468–476. doi: 10.1354/vp.42-4-468. [DOI] [PubMed] [Google Scholar]
- Kane JK, Konu O, Ma JZ, Li MD. Nicotine coregulates multiple pathways involved in protein modification/degradation in rat brain. Brain Res Mol Brain Res. 2004;132:181–191. doi: 10.1016/j.molbrainres.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther. 2000;86:29–48. doi: 10.1016/s0163-7258(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Khare S, Banai Y, Gokulan K, Smith R, 3rd, Linthicum DS, Modiano JF. Early changes in metabolism of leukemic cell lines upon induction of apoptosis by cytotoxic drugs. Eur J Pharmacol. 2003;465:23–30. doi: 10.1016/s0014-2999(03)01425-0. [DOI] [PubMed] [Google Scholar]
- Lang F, Foller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, Huber SM. Ion channels in cell proliferation and apoptotic cell death. J Membr Biol. 2005;205:147–157. doi: 10.1007/s00232-005-0780-5. [DOI] [PubMed] [Google Scholar]
- Leonard S, Bertrand D. Neuronal nicotinic receptors: from structure to function. Nicotine Tob Res. 2001;3:203–223. doi: 10.1080/14622200110050213. [DOI] [PubMed] [Google Scholar]
- Lin PY, Fosmire SP, Park SH, Park JY, Baksh S, Modiano JF, Weiss RH. Attenuation of PTEN increases p21 stability and cytosolic localization in kidney cancer cells: a potential mechanism of apoptosis resistance. Mol Cancer. 2007;6:16. doi: 10.1186/1476-4598-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- Mai H, May WS, Gao F, Jin Z, Deng X. A functional role for nicotine in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 2003;278:1886–1891. doi: 10.1074/jbc.M209044200. [DOI] [PubMed] [Google Scholar]
- Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA. 1990;87:3294–3298. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariggio MA, Guida L, Laforgia A, Santacroce R, Curci E, Montemurro P, Fumarulo R. Nicotine effects on polymorphonuclear cell apoptosis and lipopolysaccharide-induced monocyte functions. A possible role in periodontal disease? J Periodontal Res. 2001;36:32–39. doi: 10.1034/j.1600-0765.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- Medina-Palazon C, Bernard E, Frost V, Morley S, Sinclair AJ. KIPase activity is a novel caspase-like activity associated with cell proliferation. Eur J Biochem. 2004;271:2716–2723. doi: 10.1111/j.1432-1033.2004.04200.x. [DOI] [PubMed] [Google Scholar]
- Middlebrook AJ, Martina C, Chang Y, Lukas RJ, DeLuca D. Effects of nicotine exposure on T cell development in fetal thymus organ culture: arrest of T cell maturation. J Immunol. 2002;169:2915–2924. doi: 10.4049/jimmunol.169.6.2915. [DOI] [PubMed] [Google Scholar]
- Modiano JF, Domenico J, Szepesi A, Lucas JJ, Gelfand EW. Differential requirements for interleukin-2 distinguish the expression and activity of the cyclin-dependent kinases Cdk4 and Cdk2 in human T cells. J Biol Chem. 1994;269:32972–32978. [PubMed] [Google Scholar]
- Modiano JF, Mayor J, Ball C, Chitko-McKown CG, Sakata N, Domenico-Hahn J, Lucas JJ, Gelfand EW. Quantitative and qualitative signals determine T-cell cycle entry and progression. Cell Immunol. 1999;197:19–29. doi: 10.1006/cimm.1999.1563. [DOI] [PubMed] [Google Scholar]
- Petro TM, Peterson DS, Fung YK. Nicotine enhances interleukin production of rat splenic T lymphocytes. Immunopharmacol Immunotoxicol. 1992;14:463–475. doi: 10.3109/08923979209005405. [DOI] [PubMed] [Google Scholar]
- Petro TM, Schwartzbach SD, Zhang S. Smokeless tobacco and nicotine bring about excessive cytokine responses of murine memory T-cells. Int J Immunopharmacol. 1999;21:103–114. doi: 10.1016/s0192-0561(98)00070-8. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Shim D, De Biasi M. Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J Neurosci. 2007;27:10508–10519. doi: 10.1523/JNEUROSCI.3353-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Herskovic JE, Trilling Y, Jarvik ME. Transdermal nicotine reduces cigarette craving and nicotine preference. Clin Pharmacol Ther. 1985;38:450–456. doi: 10.1038/clpt.1985.203. [DOI] [PubMed] [Google Scholar]
- Rose JE, Jarvik ME, Rose KD. Transdermal administration of nicotine. Drug Alcohol Depend. 1984;13:209–213. doi: 10.1016/0376-8716(84)90061-9. [DOI] [PubMed] [Google Scholar]
- Schneider NG, Lunell E, Olmstead RE, Fagerstrom KO. Clinical pharmacokinetics of nasal nicotine delivery. A review and comparison to other nicotine systems. Clin Pharmacokinet. 1996;31:65–80. doi: 10.2165/00003088-199631010-00005. [DOI] [PubMed] [Google Scholar]
- Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- Sugano N, Minegishi T, Kawamoto K, Ito K. Nicotine inhibits UV-induced activation of the apoptotic pathway. Toxicol Lett. 2001;125:61–65. doi: 10.1016/s0378-4274(01)00416-7. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Wakisaka S, Takeba Y, Mihara S, Sakane T. Effects of cigarette smoking on Fas/Fas ligand expression of human lymphocytes. Cell Immunol. 1999;192:48–53. doi: 10.1006/cimm.1998.1432. [DOI] [PubMed] [Google Scholar]
- Tambyrajah WS, Bowler LD, Medina-Palazon C, Sinclair AJ. Cell cycle-dependent caspase-like activity that cleaves p27(KIP1) is the beta(1) subunit of the 20S proteasome. Arch Biochem Biophys. 2007;466:186–193. doi: 10.1016/j.abb.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KK, de Vegt F, Mulders PF, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, Stefansson K. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgi H, Utsugisawa K, Nagane Y. Protective effect of nicotine through nicotinic acetylcholine receptor alpha 7 on hypoxia-induced membrane disintegration and DNA fragmentation of cultured PC12 cells. Neurosci Lett. 2000;285:91–94. doi: 10.1016/s0304-3940(00)01026-0. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun X. Desensitized nicotinic receptors in brain. Brain Res Brain Res Rev. 2005;48:420–437. doi: 10.1016/j.brainresrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, Harris C, Belinsky S, Dennis PA. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111:81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SC, Zhong J, Zheng H, Larrick JW. Nicotine inhibition of apoptosis suggests a role in tumor promotion. FASEB J. 1993;7:1045–1051. [PubMed] [Google Scholar]
- Wu YP, Kita K, Suzuki N. Involvement of human heat shock protein 90 alpha in nicotine-induced apoptosis. Int J Cancer. 2002;100:37–42. doi: 10.1002/ijc.10449. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Sakagami H, Yamanaka Y, Amano Y, Yamaguchi M, Yamamura M, Fukuchi K, Gomi K, Ohata H, Momose K, Takeda M. Induction of DNA fragmentation by nicotine in human myelogenous leukemic cell lines. Anticancer Res. 1998;18:2507–2511. [PubMed] [Google Scholar]