

Abstract
All herpesviruses encode a homolog of the herpes simplex virus type-1 UL11 tegument protein. Deletion of UL11 disrupts virus envelopment, causes capsid accumulation within the cytoplasm, and reduces virus release. UL11 requires acylation with myristate and palmitate for membrane binding, lipid raft trafficking, and accumulation at the site of virus envelopment. Thus, it was predicted that acylation of UL11 would be necessary for efficient virion production, similar to HIV-1 Gag which requires myristylation for virus production. Accordingly, recombinant viruses were created to express UL11 derivatives that are not acylated, are partially acylated, or contain foreign acylation signals. Unexpectedly, the non-acylated UL11 rescued some growth defects of a UL11-null mutant, even though the unmodified protein was unstable. Furthermore, a myristylated and palmitylated chimera did not fully rescue the null-virus. These results suggest UL11 maintains some function(s) when not membrane-bound, and the sequence context of the acylations is important for UL11 function.
Keywords: UL11, Herpes simplex, Myristate, Palmitate, Recombinant virus, BAC
Introduction
The UL11 tegument protein of herpes simplex virus type-1 (HSV-1) is conserved among all herpesviruses, and each homolog contains amino acid motifs that allow covalent modifications with two fatty acids, myristate and palmitate. Without these modifications, HSV-1 UL11 lacks all membrane binding activity and is free in the cytoplasm (Baird et al., 2008; Loomis et al., 2001). Myristylation occurs co-translationally on an N-terminal glycine, which is exposed following removal of the initiator methionine (MacLean et al., 1989; Resh, 1999). Palmitylation occurs on at least one of three cysteines only after the myristylated protein binds to the cytoplasmic face of cellular membranes (Loomis et al., 2001). Thus, a mutant of UL11 that lacks the glycine residue is also defective for palmitylation, even though the cysteine residues are present (Loomis et al., 2001). Dual acylation is necessary for the trafficking of the protein through lipid rafts (or detergent resistant membranes, DRMs) (Baird et al., 2008); however, it remains unknown why UL11 travels this pathway. In any case, at steady state conditions, UL11 accumulates on membranes of the trans-Golgi network (TGN), the site where virion envelopment occurs to create the infectious particle (Mettenleiter, 2004; Mettenleiter et al., 2006).
At the TGN, the envelopment process may be promoted by bridging interactions between the membrane-bound UL11 and the capsid-bound tegument protein UL16 (Loomis et al., 2003). This hypothesis is supported by two lines of evidence. First, UL16 can directly interact with UL11 in vitro (Yeh et al., 2008). Second, all UL11-null herpesviruses have defects during virion envelopment (Baines and Roizman, 1992; Britt et al., 2004; Fulmer et al., 2007; Kopp et al., 2003; Kopp et al., 2004; MacLean et al., 1992; Schimmer and Neubauer, 2003; Silva et al., 2003; Silva et al., 2005). As a result of the envelopment defects, capsids accumulate within the cytoplasm of the infected cells and fewer virions are released into the extracellular space.
In retrovirology, it is well known that fatty acids are required to anchor the budding machinery to membranes during virion formation. In the case of HIV-1, for example, myristate-minus Gag polyproteins fail to interact with membranes and consequently are severely defective for virus production (Gottlinger et al., 1989). Similarly, when HSV-1 UL11 acylation mutants have to compete with the wild-type UL11 for incorporation into virus particles in a transfection-infection assay, only the fully-acylated form is efficiently packaged (Loomis et al., 2006). Given these observations, it was predicted that the ability of UL11 to function properly and promote virion envelopment would require the acyl modifications. Unexpectedly, this was not the case, and the expression of a non-acylated UL11 rescued some growth defects of a UL11-null virus. Furthermore, a UL11-chimera that contains foreign myristylation and palmitylation signals at the N-terminus failed to fully rescue a UL11-null virus, suggesting acylations alone are not sufficient for protein function and the sequence context of the modifications is also critical.
Results
Relocation of the UL11-coding sequence
The acylated residues of UL11 reside within its first few amino acids, but the coding sequence for these residues overlaps the reading frame of the essential UL12 gene (Fig. 1A). Consequently, it is not possible to delete the 5’-coding region of UL11, or to make substitutions of the N-terminal acylation signals of UL11, without disrupting the function of UL12. Indeed, removal of the entire UL11-coding sequence, including the UL12 overlap, resulted in a non-viable virus (data not shown). The lethality of this deletion can be attributed to the truncation of UL12 since a mutation of the UL11 start codon (with a silent alteration in UL12) produces a virus that retains viability (Leege et al., 2009).
FIG. 1.
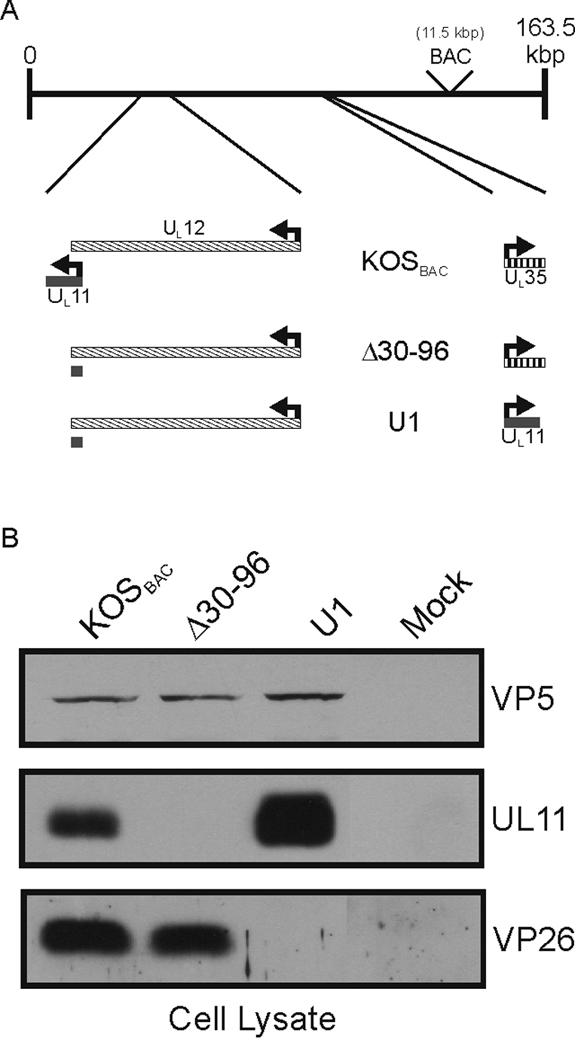
Relocalization of the UL11-coding sequence. (A) Stepwise strategy used to move the UL11-coding sequence into the UL35 locus. A recombinant of the KOS strain of HSV-1 that contains a bacterial artificial chromosome (“BAC,” 11.5 kbp) inserted between UL37 and UL38 was used (“KOSBAC”). In mutant Δ30–96, all the nucleotides that do not overlap with the essential UL12 gene were deleted, leaving the codons for the N-terminal 29 amino acids of UL11. Next, the codons at the UL35 locus were replaced with the entire UL11-coding sequence, creating mutant U1. (B) Vero cells were infected with the indicated viruses and harvested 24 h later. Equal numbers of infected cells were lysed in sample buffer, separated by SDS-PAGE, and proteins were transferred to nitrocellulose. The indicated proteins were detected by immunoblotting with the corresponding antibodies.
To circumvent the overlap problem, the UL11-coding sequence was relocated within a copy of the HSV-1 KOS-strain genome carried on a bacterial artificial chromosome (“BAC,” Fig. 1A). The position of the BAC sequence within the genome does not alter the growth properties HSV-1 (Fig. 3B) (Gierasch et al., 2006). The first step in the gene rearrangement was to delete the portion of UL11 that does not overlap with UL12, creating the UL11-null mutant Δ30–96. Deletion of the non-overlapping sequence was chosen over a substitution of the UL11 start codon for two reasons. One, previous UL11-null viruses have been constructed in this manner, allowing a direct comparison of these results to published results (Baines and Roizman, 1992; Fulmer et al., 2007; MacLean et al., 1992). And two, a spontaneous (and undesired) reversion is less likely to occur with the deletion. Next, the open reading frame of UL35 was replaced with the entire UL11-coding sequence to create a recombinant named U1 (Fig. 1A). The UL35 gene, which encodes the VP26 capsid protein, was chosen because it: 1) is not needed for growth in cell culture (the VP26-null mutant is reduced only 50% for virus production) (Desai et al., 1998), 2) does not overlap any other reading frames, 3) is approximately the same size as UL11 (~100 amino acids), and 4) is expressed late during an infection, like UL11 (McNabb and Courtney, 1992). Both Δ30–96 and U1 retain the first 29 codons of the native UL11 locus (Fig. 1A and Table 1); however, no peptide was detectable from either the cell lysates or purified virions when examined by either immunoblotting or radiolabeling and immunoprecipitation (data not shown). The lack of a detectable peptide was consistent with previous UL11-null viruses, which also left a short piece of the reading frame intact (Baines and Roizman, 1992; Fulmer et al., 2007; MacLean et al., 1992).
FIG. 3.
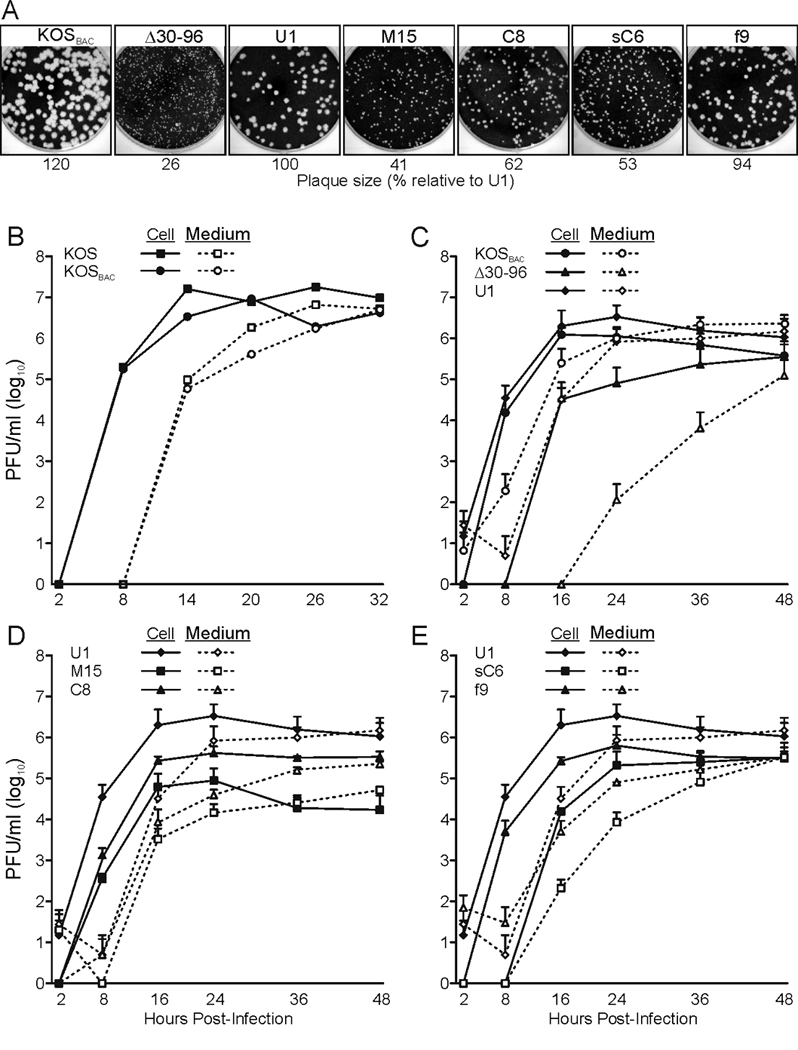
Growth properties of the recombinant viruses. (A) Plaque sizes of UL11 mutants. Confluent monolayers of Vero cells were infected with the indicated viruses, and 4 days later, the cells were stained with crystal violet and imaged. Plaque size was determined by measuring 10 randomly selected plaques and represented as a percent relative to U1. (B–E) Single-step growth curve analyses of the mutants. Vero cells were infected with indicated virus and then acid-washed to inactivate any input virus that had not fused with the cellular membrane. At the indicated times, samples were collected, and virus titers were determined by plaque assay. The results of at least two independent experiments are shown for each mutant. (B) Comparison of the wild-type KOS strain and the recombinant carrying the bacterial artificial chromosome (“KOSBAC”). (C) Comparison of mutants that lack full-length UL11 expression (“Δ30–96”) or express UL11 from the UL35 locus (“U1”). (D and E) Analysis of recombinant viruses that encode UL11-derivatives with altered acylation signals. Solid lines denote PFU associated with the cells, whereas dashed lines represent PFU released into the medium.
Table 1.
Names, loci of interest, and acylations of recombinant viruses.
| Virus Name |
Parent Virus |
UL11 Locus | UL35 Locus | Myristate | Palmitate |
|---|---|---|---|---|---|
| KOS | - | UL11 | UL35 | X | X |
| KOSBAC | KOS | UL11 | UL35 | X | X |
| Δ30–96 | KOSBAC | Δ30–96 | UL35 | ||
| U1 | Δ30–96 | Δ30–96 | UL11 | X | X |
| M15 | Δ30–96 | Δ30–96 | Myr(−) | ||
| C8 | Δ30–96 | Δ30–96 | CCC(−) | X | |
| sC6 | Δ30–96 | Δ30–96 | sCCC(−) | X | |
| f9 | Δ30–96 | Δ30–96 | fUL11 | X | X |
| stopM15 | M15 | stopΔ30–96 | Myr(−) | ||
| MG16 | Δ30–96 | Δ30–96 | Myr(−)GFP |
The name of each recombinant virus, the parent virus from which it was constructed, and the ORF present at each locus (UL11 or UL35) are indicated. The acylation state of the UL11-variant is also indicated. “KOS” and “KOSBAC” are genetically identical, except for the absence or presence of the bacterial artificial chromosome, respectively.
The mutant genomes were transfected into Vero cells to produce viruses for further study. As expected, Δ30–96 expressed VP26 but not UL11, whereas U1 expressed UL11 but not VP26 (Fig. 1B). Also, the UL11 produced from U1 was incorporated into virus particles at levels comparable to wild-type KOSBAC (Fig. 2), even though UL11 was expressed at much higher levels in the U1-infected cells (Fig. 1B and 2). Similar to KOS (the wild-type strain without any “BAC” sequence), extracellular virions of KOSBAC contained UL16 (Fig. 2 and data not shown), a tegument protein which is both soluble and capsid-bound within the cytoplasm of infected cells (Meckes, Jr. and Wills, 2007). However, UL16 was greatly reduced for packaging into Δ30–96 virions but incorporated at wild-type levels into U1 particles (Fig. 2), indicating that UL11, but not VP26, is needed for efficient incorporation of UL16.
FIG. 2.
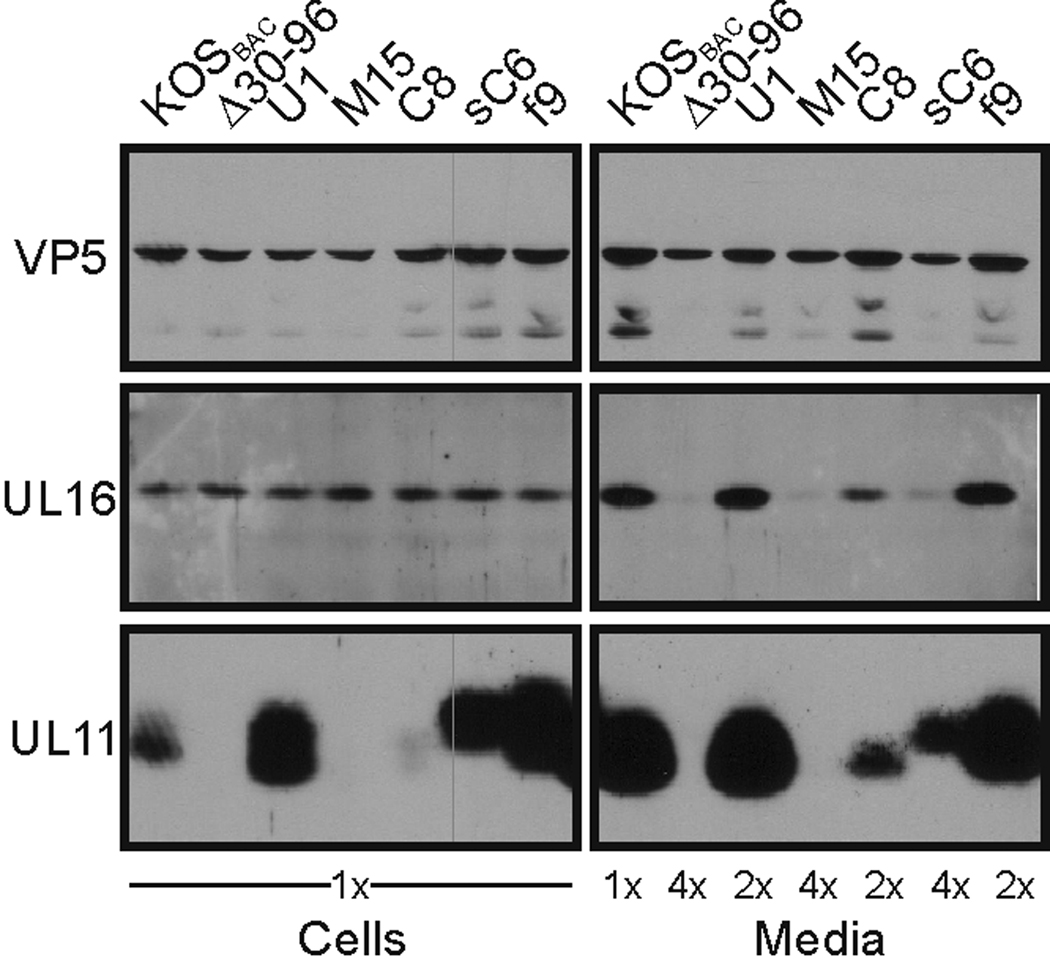
UL11 expression and virion incorporation. Vero cells were infected with the indicated viruses, and 24 h later, extracellular virions were collected from the media by centrifugation through a 30% (w/v) sucrose cushion. The virions and infected-cell lysates were separated by SDS-PAGE and transferred to nitrocellulose. The indicated proteins were detected using corresponding antibodies. Equal numbers of infected cells were loaded with VP5, the major capsid protein, serving as the loading control. To achieve approximately equal VP5 levels for the “Media” samples, additional cells (indicated as “2×” or “4×”) were infected, and the collected media were combined.
To examine the replication properties of the mutants, single-step growth curves and plaque sizes were examined. Previously described UL11-null viruses have noticeable growth defects 24 h post infection (Baines and Roizman, 1992; Fulmer et al., 2007; MacLean et al., 1992), and Δ30–96 behaved identically to such clones (Fig. 3C). However, the growth defects were far less noticeable when the infection was given more time. This can be seen by carefully comparing the growth curves, which show that Δ30–96 was delayed many hours for the initial production of cell-associated virus, and consequently, a delayed release of virus into the medium was seen (Fig. 3C). Moreover, Δ30–96 had a slower rate of release compared to KOSBAC (Fig. 3C, compare slopes of the “Medium” lines), despite a rate of virus production in the cell that was similar to KOSBAC once it began (Fig. 3C, compare slopes of the “Cell” lines). After 24 h of infection, these defects resulted in nearly a 4-log reduction in virus titer in the medium, a result similar to what has been seen previously (Baines and Roizman, 1992). However, after 48 h, the titer of Δ30–96 in the medium was only down about 1-log compared to KOSBAC. Another indicator of the growth defect associated with Δ30–96 was that the plaques it produced were much smaller than those of KOSBAC (Fig. 3A).
The effects of expressing UL11 from the UL35 locus were examined next (virus U1), and as expected, nearly all of the growth defects of Δ30–96 were eliminated. Only minor growth differences between KOSBAC and U1 were evident in both its growth curve (Fig. 3C, Cell and Medium) and plaque size (Fig. 3A). These small defects were expected based on studies of a VP26-null virus, which was reduced only 2-fold in its ability to produce infectious virions compared to the wild type (Desai et al., 1998).
UL11 acylation mutants
The main goal of these experiments was to examine the importance of fatty acid modifications for the functions of UL11 during virus replication. It was predicted that acylation would be required for virus production, as is the case for retroviruses such as HIV-1. Thus, only expression of a fully-acylated UL11 was expected to rescue mutant virus Δ30–96, whereas expression of partially- or non-acylated UL11 derivatives would not. To test this hypothesis, recombinant viruses were constructed to express UL11-acylation mutants from the UL35 locus. Using Δ30–96 as the parent, the UL35-coding sequence was replaced with the previously described UL11 alleles that encode either a myristylation mutant [Myr(−), glycine deletion] or a palmitylation mutant [CCC(−), alanine substitution] (Fig. 4A) (Baird et al., 2008; Loomis et al., 2001; Loomis et al., 2006), creating the recombinant viruses M15 and C8, respectively (Table 1). Previous studies of these UL11 alleles in transfected cells showed that myristylation is a prerequisite for palmitylation, and thus the Myr(−) derivative is not acylated at all and fails to bind membranes; the CCC(−) mutant, on the other hand, encodes a protein that is myristylated but not palmitylated and retains some membrane binding activity (Baird et al., 2008; Loomis et al., 2001; Loomis et al., 2006). These properties of the two UL11 mutants were also found when they were expressed in the context of the recombinant viruses, M15 and C8 (Fig. 5).
FIG. 4.

UL11 mutants. (A) Diagram of the wild-type and mutant forms of UL11 as expressed from the UL35 locus. The sites of myristylation (G) and palmitylation (CCC) are indicated with jagged lines denoting the fatty acid modifications. N-terminal extensions corresponding to the first 10 amino acids of v-Src [sCCC(−)] or Fyn (fUL11) are indicated with gray lines. Fatty-acid modifications are indicated and “+” indicates basic residues. “stop” marks the location of a 2-nucleotide change that created a stop codon in mutant Δ30–96 without altering the UL12 protein, and the dotted line represents the remaining UL11-coding sequence downstream from the introduced stop codon. The solid circle represents the GFP-tag on the C-terminus of Myr(−)GFP (B) Alignment of the UL11- and UL12-coding sequences. The two nucleotide substitution that introduces a stop codon in the UL11 sequence without altering UL12 is indicated.
FIG. 5.
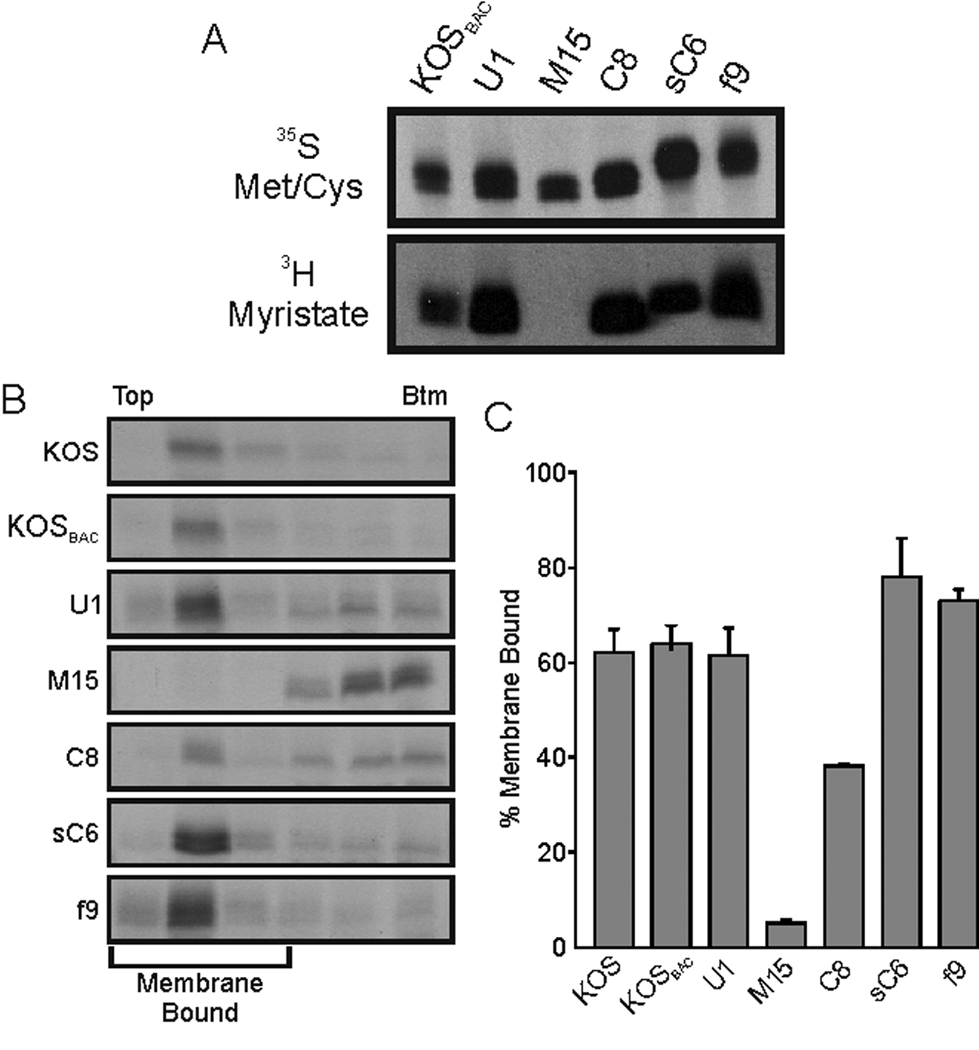
Myristylation and membrane-binding properties of UL11 derivatives. (A) Vero cells were infected with the indicated viruses and radiolabeled with either [35S]methionine/cysteine for 2.5 h or [3H]myristic acid for 30 min. All labeling periods were concluded at 9 hpi, at which time cell lysates were prepared. The UL11 derivatives were immunoprecipitated, resolved by SDS-PAGE, and visualized by autoradiography. (B) In parallel experiments, cells were labeled for 2.5 h with [35S]methionine/cysteine, scraped from the plates, and osmotically disrupted. The ability of the UL11 mutants to float to the upper regions of sucrose step-gradients during centrifugation was monitored. Representative autoradiographs are shown with the tops and bottoms of the gradients indicated. (C) Phosphorimager analysis was used to quantitate the amount of membrane-bound UL11 (top three fractions) relative to the total. The results from at least two independent experiments are shown.
Examination of the mutant proteins expressed by M15 and C8 revealed that the stability of UL11 in the infected cell was influenced by its acylation. That is, cells infected with either virus produced UL11-variants that were efficiently radiolabeled and immunoprecipitated (Fig. 5A, 35S bands); however, examination of steady-state levels by immunoblotting revealed that the non-acylated protein from M15 was not detectable and the partially-acylated protein from C8 was barely detectable (Fig. 2, Cells). Similarly, extracellular virions from the M15 infection contained no UL11 while those produced by mutant C8 contained only a little in immunoblot assays (Fig. 2, Media). Furthermore, incorporation of UL16 into virions varied with the expression of the different UL11 variants. Compared to the parental U1 virus, M15 virions had virtually no UL16 whereas those of C8 had reduced levels of this protein (Fig. 2, Media); again suggesting that UL11 somehow influences the packaging of its binding partner.
The growth curves of M15 and C8 showed that expression of Myr(−) and CCC(−) resulted in different amounts of infectious virus released into the medium at 48 h post infection, a difference that varied with the acylations of UL11 (Fig. 3D). As such, a growth hierarchy was evident for the recombinants as follows: U1 > C8 > M15 ≈ Δ30–96. However, earlier time points of the same growth curves clearly show that M15 did not behave like Δ30–96, but actually grew better. M15 did not have the large lag-period of virus production that Δ30–96 did (compared to U1) in either the cell-associated or the medium samples (Figs. 3C and D). Additionally, the plaques produced by M15 were ~50% larger than those of Δ30–96 (Fig. 3A). The ability of Myr(−) to relieve some of the growth defects of Δ30–96 was surprising because Myr(−) is not acylated and had severely reduced protein stability, virion incorporation, and membrane binding.
Why did non-myristylated UL11 enhance the growth of Δ30–96?
At least three possibilities existed for the enhanced growth properties of M15: 1) The 29-amino acid, N-terminal peptide is produced from the UL11 locus, interacts with full-length Myr(−) expressed from the UL35 locus, and enables the non-myristylated protein to bind membranes, 2) M15 has a greater specific infectivity than Δ30–96, and 3) The Myr(−) protein can provide its function even at very low steady-state levels by binding to some other protein in a manner that is not dependent on its ability to bind membranes.
The first two possibilities were readily testable. To test the trans-complementation hypothesis, a stop codon was introduced into the UL11 locus to eliminate expression of the peptide, creating mutant virus stopM15 (to create an allele named stop Δ30–96; Fig. 4A and Table 1). Importantly, this change is silent in the essential UL12 gene (Fig. 4B). M15 and stopM15 grew with similar properties, which indicates that an acylated-UL11 peptide was not acting in trans to complement Myr(−) expressed from the UL35 locus (Fig. 6A). To test the specific-infectivity hypothesis, quantitative PCR (qPCR) was used, and this revealed that the ratio of genome-containing particles to PFU for M15 was not significantly different from Δ30–96 or the parental virus (Fig. 6B).
FIG. 6.
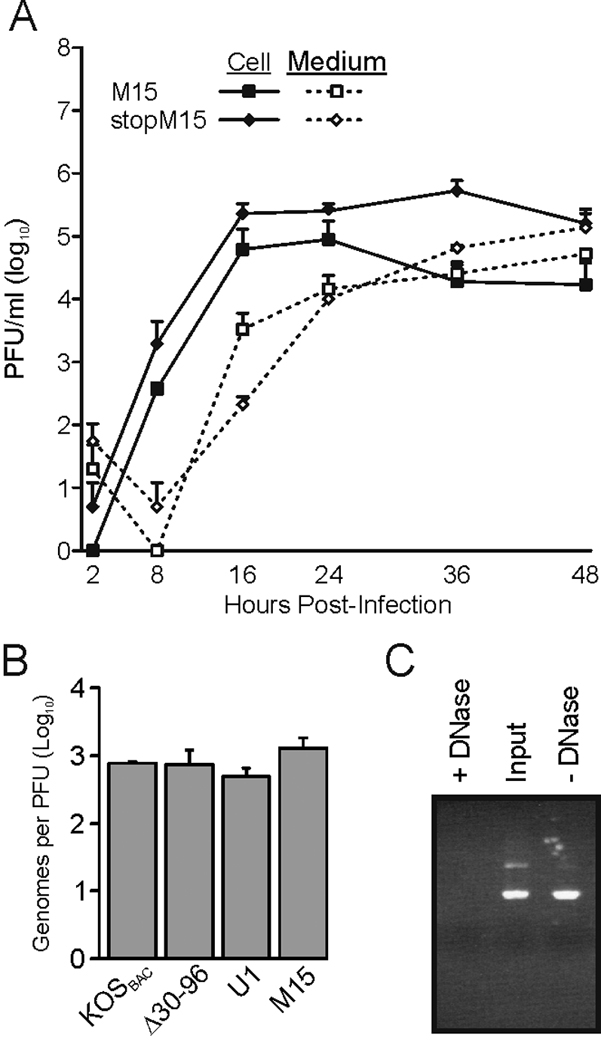
Further characterization of mutant M15. (A) The growth properties of M15 and stopM15 were compared, revealing that the 29-amino-acid, N-terminal peptide does not trans-compliment. The results from two independent experiments are shown. (B) To compare the specific infectivities of the indicated mutant virus stocks, samples were treated with DNase to eliminate any DNA that was not contained in virions, and viral DNA was subsequently purified. The numbers of genomes present in each sample were measured by quantitative PCR and normalized for PFUs. The limit of detection was 100 copies per PCR reaction. The results from at least two independent experiments are shown. (C) To ensure that the DNase treatments were effective, random PCR products were incubated with (+) or without (−) the enzyme. “Input” is the initial amount of PCR product in each sample. A representative agarose gel stained with ethidium bromide is shown.
The third possibility for the enhance growth of M15 over Δ30–96 was that the Myr(−) protein retained some ability to interact with other proteins, viral or cellular. This is difficult to examine directly because the protein is highly unstable. However, previous studies using transfections showed that fusion of GFP to the C-terminus of Myr(−) [Myr(−)GFP] stabilizes the protein so that it can be readily detected at steady state levels (Loomis et al., 2006). Therefore, it was examined whether expression of the more stable Myr(−)GFP might enhance the growth of Δ30–96 compared to the untagged protein. To create such a virus, the full-length Myr(−)GFP-coding sequence was used to replace the UL35-coding sequence of Δ30–96, making the virus MG16 (Fig. 4 and Table 1). Critically, the new virus MG16 was created independently from M15 in a separate recombineering reaction. And, as expected, when Myr(−)GFP was expressed by MG16, the tagged protein was detectable at steady-state levels, unlike its untagged counterpart produced by M15 (Fig. 7A, “Cells”); however, the increased stability of Myr(−)GFP did not increase the amount of UL11-protein packaged into virions (Fig. 7A, “Media”). To compare the abilities of the untagged and tagged proteins to rescue growth of Δ30–96, single-step growth curves of M15 and MG16 were performed (Fig. 7B). As seen by the “Medium” lines (dashed), MG16 did not release infectious virus into the medium any better than M15. However, the “Cell” lines (solid) suggest that MG16 may have produced more cell-associated virus than M15.
FIG. 7.
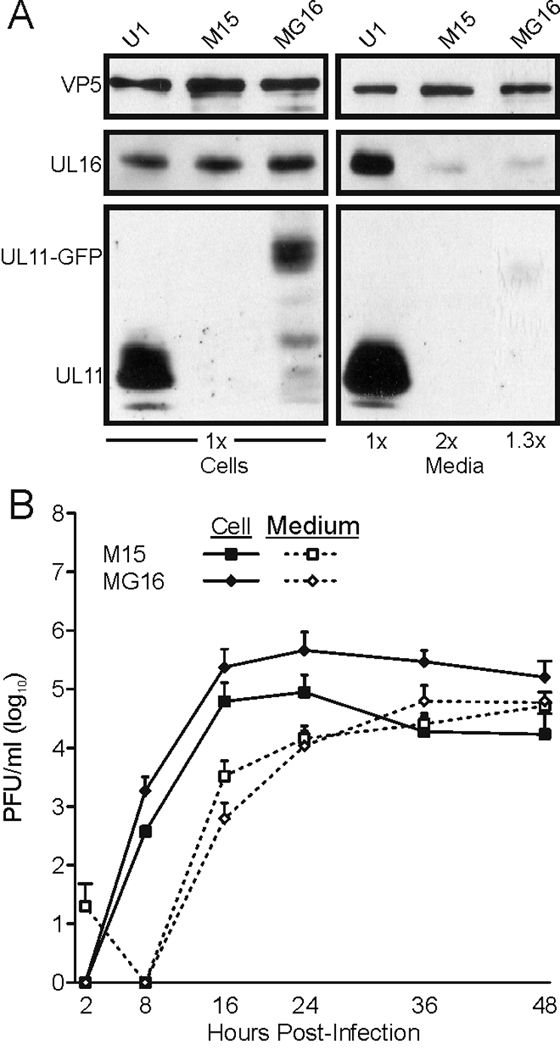
Addition of GFP stabilizes Myr(−)UL11 but does not enhance growth of the recombinant virus. Vero cells were infected with the indicated viruses. Cell and virus samples were harvested, and proteins were separated by SDS-PAGE. The indicated proteins were detected using the corresponding antibodies. (B) Single step growth curves were performed as described in the legend to Fig. 3.
Fusion of foreign acylation signals to UL11
The fatty acids on UL11 appear to be required for efficient virion biogenesis; however, it is quite clear that they are also critical for membrane-binding (Baird et al., 2008; Loomis et al., 2001). Therefore, it is possible that UL11 simply requires membrane binding, but not the specific combination of myristylation and palmitylation, for its function. To test this hypothesis, the coding sequence for a previously-described chimera of UL11 that binds membranes efficiently and independently of palmitylation, sCCC(−) (Baird et al., 2008), was inserted into the UL35 locus to create virus sC6 (Fig. 4A and Table 1). sCCC(−) has the 10-amino-acid membrane-binding domain of the Rous sarcoma virus oncoprotein (v-Src) fused to the N-terminus of CCC(−). For sCCC(−), membrane binding is mediated only by the myristate and basic residues of the v-Src peptide. In another construct, fUL11, the 10-amino-acid membrane-binding domain of the cellular Fyn protein was added to the N-terminus of UL11 (Fig. 4A). This chimera contains sites for myristylation and palmitylation within the Fyn peptide but also retains the UL11 palmitylation site (Baird et al., 2008; Loomis et al., 2001). Insertion of the fUL11-coding sequence into UL35 created virus f9 (Table 1).
Following infection of cells with either sC6 or f9, the chimeric-UL11 proteins were found to be expressed at high steady-state levels (Fig. 2) and myristylated (Fig. 5A). Both chimeras appeared to bind membranes better than the wild type (Figs. 5B and C), but only the myristylated and palmitylated fUL11 construct was incorporated into virions as efficiently as the wild-type protein produced by virus U1 (Fig. 2). Thus, it seems that the dual acylation of UL11 may be required for packaging, with other mechanisms of membrane binding being inferior; however, sC6 and f9 mutants have N-terminal sequence differences that could also account for the differences in their ability to be incorporated and support viral growth. Moreover, the amount of UL16 incorporated into sC6 and f9 particles was again dependent on the packaging levels of the UL11-variants (Fig. 2).
To measure the ability of the chimeric proteins to rescue the Δ30–96 mutant, the plaque sizes and growth curves of sC6 and f9 were examined. Expression of either sCCC(−) or fUL11 increased the plaque size of Δ30–96, but only fUL11 restored the plaques to the size of U1 (Fig. 3A). However, examination of the sC6 and f9 growth curves revealed that expression of the dually-acylated fUL11 was not functionally equivalent to wild-type UL11 (Fig. 3E). That is, virus f9 did not produce cell-associated virus or release virus into the medium as efficiently as U1; but, expression of fUL11 did rescue growth of Δ30–96 better than (16–24 h) or equal to (48 h ) sCCC(−), implying dual acylation is better than mono acylation.
The most distinct and unexpected result obtained with the chimeras was a delay in the production of infectious sC6 within the cell without a delay of virus release into the medium (Fig. 3E). That is, this mutant has the unusual property of producing infectious virus within the cell at the same time virus is released into the medium.
Discussion
The experiments in this study were designed to examine the importance of UL11 acylation for protein function during envelopment and release of HSV-1 particles. It was predicted that the modifications would be required, and that expression of a non-acylated UL11 variant would not rescue the growth of the UL11-null virus, Δ30–96. However, the data presented here show that expression of the non-acylated protein (virus M15) partially rescued the growth defects seen with Δ30–96, despite being highly unstable, implying that some function was maintained. Furthermore, not all combinations of myristylation and palmitylation are sufficient for UL11 function because expression of the dually-acylated fUL11 chimera (virus f9) did not completely rescue Δ30–96. This suggests the context of the acylations is also critical for UL11 function.
Why did the non-acylated Myr(−) protein enhance growth of Δ30–96 even though does not bind membranes, does not accumulate at the TGN or in DRMs, and is not stable? One possible explanation is that Myr(−) may interact with an unknown protein, either cellular or viral, to promote the enhanced growth properties of M15. Bacterially-produced UL11, which is not acylated, interacts with UL16 (Loomis et al., 2003; Yeh et al., 2008); hence, Myr(−) may interact with either soluble or capsid-bound UL16 to influence interactions between the capsid and the TGN-membrane that eventually lead to the formation of the viral membrane during final envelopment. Alternatively, UL11 also interacts in some manner with the tails of the glycoproteins gD and gE (Farnsworth et al., 2007) and perhaps the unmyristlated protein can promote virus growth through those interactions. If so, then only small amounts must be needed because only 5% of Myr(−) floats with membranes, versus >60% of dually-acylated UL11.
To stabilize, and therefore increase the chances of interactions between Myr(−) and other proteins, a GFP tag was fused to the C-terminus of the non-acylated protein. As expected, this protein was readily detectable at steady state levels. Though the increase in protein concentration had no affect on incorporation of the UL11-variant into virus particles and release of virus into the medium, an increase of cell-associated virus was observed. These results suggest that expression of Myr(−)GFP promotes the formation of infectious virions within the cell better than the untagged Myr(−). However, the MG16 virions are not efficiently released into the medium, supporting the hypothesis that UL11 has a post-envelopment function, such as transport of virus-containing vesicles to the cell periphery.
Comparing the growth data of viruses MG16 and M15 also demonstrates no secondary mutations are present within either viral genome. Since viruses M15 and MG16 were created totally independent of each other, yet behave quite similarly (despite the great differences in protein stability), it is unlikely that a secondary mutation is present in M15 that promoted the unexpected growth. Furthermore, because Δ30–96 (the parent virus to both M15 and MG16, Table 1) behaves like previously constructed UL11-null viruses (Baines and Roizman, 1992; Fulmer et al., 2007; MacLean et al., 1992), it is unlikely that a mutation in the viral genome was present in this clone that could ultimately have caused the unexpected growth of M15.
In addition to requiring fatty acids for full function, the data presented here show that the sequence context of the fatty acids is important. Recombinant f9 encodes a UL11 derivative with an acylation pattern similar to wild-type UL11 and behaves similarly to UL11 with respect to membrane binding, DRM association, and virion incorporation. However, despite these similarities, virus f9 did not grow as efficiently as the U1 parent, although expression of fUL11 did rescue growth of Δ30–96 better than all the other UL11 mutants. The growth defects seen with virus f9 may have occurred because the addition of the peptide altered the structure of UL11, thereby inhibiting proper interactions with other proteins. However, the interaction with UL16 seems to be normal based on the high levels of packaging of this binding partner observed for the f9 virus. Also, it is possible that the basic residues within the Fyn peptide altered the ability of fUL11 to be released from membranes. That is, wild-type UL11 relies on myristate and palmitate for membrane binding, and the latter modification is reversible, leaving only a myristyl group. Since the membrane-binding strength of myristate alone is low, non-palmitylated UL11 would “slip” on and off membranes readily. In contrast, the Fyn peptide contains basic residues, which bind acidic phospholipids (Sigal et al., 1994), and these could enable fUL11 to irreversibly bind to membranes. While it remains to be seen whether UL11 exhibits dynamic interactions with membranes, the enhanced growth properties of viruses M15 (unmodified UL11) and C8 (only myristylated) relative to the parental Δ30–96 (null) virus are intriguing.
The growth properties of the virus sC6 were unique and unexpected. Unlike all the other recombinants, the lag period between production of this virus within the cell and its release into the medium was eliminated (or reduced to less than 8 hours). If UL11 is involved with transport of virus-containing vesicles to the plasma membrane, then it is possible that the sCCC(−) chimera somehow accelerates the rate of egress. For example, because this highly-expressed chimera does not traffic to DRMs (Baird et al., 2008) and is poorly packaged into virions (Fig. 2), it may traffic to a subcellular location where it is better able to provide an egress function, as opposed to an envelopment function. Alternatively, the Src peptide is well known to be an efficient plasma membrane-targeting signal, and it is conceivable that the modified UL11 protein promotes final envelopment at the cell surface. This model seems less likely since no evidence was found for it when sC6-infected cells were examined by electron microscopy (data not shown).
Another striking observation from these studies was the clear correlation between packaging of UL11 and UL16. A similar observation has been seen for a UL11-null mutant of pseudorabies virus (Klupp et al., 2005). This might be explained by either of two models. First, there is a population of UL16 that is capsid-bound (Meckes, Jr. and Wills, 2007), and it is possible that an interaction with UL11 is needed for this population to stay on capsids and to be packaged. Second, the population of capsid-bound UL16 may only represent a small fraction of the UL16 that is found within virions, and the bulk of UL16 might be recruited by UL11 during envelopment. Further studies are needed to better understand the interplay between UL11 and UL16 during virion assembly.
Materials and Methods
Cells and viruses
Vero cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen) supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin (Gibco, 15140-148). All infections were performed in DMEM supplemented with 2% FBS, 25mM HEPES buffer, glutamine (0.3ug/ml), penicillin, and streptomycin. All viruses were derived from the KOS strain of HSV-1 (Smith, 1964).
Recombinant viruses
“KOSBAC” is a bacterial artificial chromosome that contains the HSV-1 KOS strain genome (Gierasch et al., 2006). Recombinant HSV-1 clones were made via “recombineering” methods with a galK-based positive/negative selection system (Warming et al., 2005). All UL11-coding sequences inserted into the UL35 locus were PCR amplified from plasmids that have been described previously (Baird et al., 2008; Loomis et al., 2001; Loomis et al., 2003; Loomis et al., 2006)
All viral clones were analyzed for DNA integrity by two methods. First, the locus of interest was PCR amplified and sequenced. Second, total DNA was isolated for each virus and monitored for large deletions and rearrangements (data not shown). The bacterial sequence (“BAC”) was left in the HSV-1 genome for all recombinants.
Following recombineering, high-quality viral DNA was obtained from the bacteria using the Qiagen Large Construct kit (cat. no. 12462). Purified DNA was transfected into Vero cells (35-mm dishes, ~50% confluent) using Lipofectamine 2000 (Invitrogen, cat. no. 11668-019). 5 days post-transfection, the cells were scraped off the plates, frozen/thawed 3 times, and cell debris was removed by centrifugation. New monolayers of Vero cells were infected with the cleared lysate to produce a viral stock. DNA was purified from the resulting virus stocks using the Qiagen DNeasy Blood and Tissue Kit (Cat no. 69504) and the locus of interest was sequenced to verify the mutations. Virus stock preparations were also titered, as follows. Vero cells were infected in duplicate wells for 1 hour at 37°C. After incubation, the cells were washed with either 1% FBS in PBS (“PBS titer”) or an acid wash (135 mM NaCl, 10 mM KCl, 40 mM citric acid, pH 3.0) followed by 1%FBS in PBS (“acid titer”). Cells were overlayed with 0.5% (w/v) methylcellulose for 4 days, stained with crystal violet, and plaques counted.
Antibodies
UL11-specific antibodies were developed in rabbits and have been described previously (Loomis et al., 2003). Antibodies against VP5 and VP26 were kindly provided by Richard Courtney, The Pennsylvania State University.
Growth curves
Vero cells (5×105/well) were infected with specified virus at a multiplicity of infection (MOI) of 1 (based on “acid titer”) at 37°C. After 1h of adsorption, free virus was aspirated and cells were washed sequentially with an acid wash (135 mM NaCl, 10 mM KCl, 40 mM citric acid, pH 3.0) and 1%FBS in PBS, then overlayed with 1ml DMEM containing 2% FBS. At indicated times post infection, medium and cells were harvested from a single well. Medium was cleared of cells, and frozen. Cells were resuspended in PBS, washed 1× with PBS, and freeze/thawed 3 times to release virus. Each sample (“Medium” and “Cell”) was then titered on Vero cells as detailed above.
Membrane flotation
Membranes were isolated using a previously described flotation protocol (Baird et al., 2008; Spearman et al., 1997). Briefly, Vero cells (1.2×107) were infected at an MOI of 10 (“PBS” titer). At 5.5 hpi, cells were starved in DMEM lacking methionine and cysteine for 30 min. Post-starvation, cells were radiolabeled with an L-[35S] methionine-cysteine mix (300uCi/plate, >1000 Ci/mmol) for 2.5 h, scraped off the plates, and washed in cold NTE (10mM Tris-HCl, pH 7.4, 100 mM NaCl, 1mM EDTA). After pelleting, cells were resuspended and swollen in hypotonic lysis buffer (10 mM Tris-HCl, pH 7.4, 0.2 mM MgCl2) on ice for 30 min. Cells were lysed at 4°C by 30 strokes with a dounce homogenizer and then centrifuged at low speed. Post-nuclear supernatants were mixed with 65% (w/w) sucrose (58% final, 2.0 ml total), placed in the bottom of a Beckman SW55 Ti tube, and sequentially overlaid with 2.0 ml of 45% sucrose and 1.0 ml of 2.5% sucrose. All sucrose solutions were made in NTE buffer. The samples were centrifuged for 18 h at 200,000 ×g and 4°C in a Beckman ultracentrifuge, and six equal-volume fractions were collected from the top. UL11 was immunoprecipitated using rabbit anti-UL11 antibodies, separated by SDS-PAGE, and quantitated by phosphorimager.
Incorporation assay
Virion incorporation was monitored using a published protocol (Loomis et al., 2006), modified as described below. Vero cells were infected at an MOI of 1 (“PBS” titer) and 24 hpi, medium was collected and cleared of cellular debris. Virions were pelleted from the supernatant by centrifugation through a 30% (w/v) sucrose cushion (1 ml) at 4°C using a Beckman SW41 Ti at 83,500 ×g for 1 h. Infected-cell lysates were scraped into PBS, pelleted, and resuspended in sample buffer. Samples were then sonicated at maximum power for 3 min. Cell lysates and collected virions were separated by SDS-PAGE and analyzed by Western blot using specified antibodies. For the “Cells” samples, lysates of equal numbers of cells were loaded on the gel for each mutant, and this resulted in approximately equal amounts of VP5 (the major capsid protein). To achieve approximately equal VP5 levels for the “Media” samples, some mutants required that additional cells be infected and the collected media were combined.
Myristylation of UL11
Vero cells were seeded into parallel plates: a 100-mm (3×106) and a 35-mm (6×105). The next day, the cells were infected with designated virus at an MOI of 10 (“PBS” titer). At 6 hpi, the 35-mm dishes were starved for 30 min, then radiolabeled with an L-[35S] methionine-cysteine mix (300 uCi/ml, >1000 Ci/mmol) for 2.5 h (3 h total, 6 – 9 hpi). Concurrent with the final 0.5 h of the 35S label (8.5 – 9 hpi), the 100-mm dishes were radiolabeled with [3H] myristic acid (300 uCi/ml, 30 Ci/mmol) for 30 min in serum-free DMEM. After labeling, cells were mixed with RIPA buffer, and the UL11 proteins were immunoprecipitated with polyclonal anti-UL11 antiserum. Immunoprecipitated proteins were separated by SDS-PAGE. Gels were treated with EN3HANCE (PerkinElmer) as per manufacturer’s instructions prior to drying and exposure to autoradiography film.
Quantitative PCR analysis
Using prepared virus stocks, DNA was extracted from 1×107 PFU (“PBS” titer). Virus was mixed with PBS, layered onto a sucrose cushion (30%, w/v), and centrifuged at 4°C in a Beckman TLA100.3 at 83,500 ×g for 1 h. Viral pellets were resuspended in water and treated with DNase I (0.1U/µl; New England Biolabs, M0303) for 30 min at 37°C. The enzyme was inactivated by incubation at 75°C for 10 min. Samples were pelleted again as above, but without a sucrose cushion. Next, virions were resuspended in PBS, and DNA isolated using the Qiagen DNeasy Blood and Tissue Kit (Cat no. 69504). Quantification of viral DNA copy number was made using an Opticon 2 real-time PCR machine (BioRad). Amplification was performed using iQ SYBR Green Supermix (BioRad, 170–8880). HSV-1 specific primers (forward, 5’-CACAGGCGGGACACCAGC; reverse, 5’-CCTCCGCAATCCCAAGATTC) were utilized to amplify a 97 bp fragment from the U L13 gene. The HSV-genome copy number was determined against a standard curve constructed by serial dilution of DNA isolated from the WT virus, KOSBAC. Limit of detection for viral DNA was 100 copies per PCR reaction (20µl).
Acknowledgements
Thanks to the Penn State College of Medicine Molecular Genetics Core Facility for sequencing all recombinant viruses. Also, thanks to David Meckes, Jake Marsh, and Pei-Chun Yeh for their helpful discussions during the course of these studies. This work was supported by NIH grant AI071286 awarded to J.W.W. J.L.S. was supported by a training grant from the NIH (CA60395).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baines JD, Roizman B. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 1992;66:5168–5174. doi: 10.1128/jvi.66.8.5168-5174.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NL, Yeh PC, Courtney RJ, Wills JW. Sequences in the UL11 tegument protein of herpes simplex virus that control association with detergent-resistant membranes. Virology. 2008;374:315–321. doi: 10.1016/j.virol.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt WJ, Jarvis M, Seo JY, Drummond D, Nelson J. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J. Virol. 2004;78:539–543. doi: 10.1128/JVI.78.1.539-543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, DeLuca NA, Person S. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology. 1998;247:115–124. doi: 10.1006/viro.1998.9230. [DOI] [PubMed] [Google Scholar]
- Farnsworth A, Wisner TW, Johnson DC. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 2007;81:319–331. doi: 10.1128/JVI.01842-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulmer PA, Melancon JM, Baines JD, Kousoulas KG. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 2007;81:3097–3108. doi: 10.1128/JVI.02201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods. 2006;135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. 1989;86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 2005;79:1510–1522. doi: 10.1128/JVI.79.3.1510-1522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M, Granzow H, Fuchs W, Klupp B, Mettenleiter TC. Simultaneous deletion of pseudorabies virus tegument protein UL11 and glycoprotein M severely impairs secondary envelopment. J. Virol. 2004;78:3024–3034. doi: 10.1128/JVI.78.6.3024-3034.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J. Virol. 2003;77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leege T, Fuchs W, Granzow H, Kopp M, Klupp BG, Mettenleiter TC. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 2009;83:896–907. doi: 10.1128/JVI.01842-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Bowzard JB, Courtney RJ, Wills JW. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2001;75:12209–12219. doi: 10.1128/JVI.75.24.12209-12219.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, Wills JW. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2003;77:11417–11424. doi: 10.1128/JVI.77.21.11417-11424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis JS, Courtney RJ, Wills JW. Packaging determinants in the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2006;80:10534–10541. doi: 10.1128/JVI.01172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean CA, Clark B, McGeoch DJ. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 1989;70(Pt 12):3147–3157. doi: 10.1099/0022-1317-70-12-3147. [DOI] [PubMed] [Google Scholar]
- MacLean CA, Dolan A, Jamieson FE, McGeoch DJ. The myristylated virion proteins of herpes simplex virus type 1: investigation of their role in the virus life cycle. J. Gen. Virol. 1992;73(Pt 3):539–547. doi: 10.1099/0022-1317-73-3-539. [DOI] [PubMed] [Google Scholar]
- McNabb DS, Courtney RJ. Identification and characterization of the herpes simplex virus type 1 virion protein encoded by the UL35 open reading frame. J. Virol. 1992;66:2653–2663. doi: 10.1128/jvi.66.5.2653-2663.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckes DG, Jr, Wills JW. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 2007;81:13028–13036. doi: 10.1128/JVI.01306-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC. Budding events in herpesvirus morphogenesis. Virus Res. 2004;106:167–180. doi: 10.1016/j.virusres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 2006;9:423–429. doi: 10.1016/j.mib.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- Schimmer C, Neubauer A. The equine herpesvirus 1 UL11 gene product localizes to the trans-golgi network and is involved in cell-to-cell spread. Virology. 2003;308:23–36. doi: 10.1016/s0042-6822(02)00060-0. [DOI] [PubMed] [Google Scholar]
- Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD. Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids. Proc. Natl. Acad. Sci. 1994;91:12253–12257. doi: 10.1073/pnas.91.25.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Schroer J, Shenk T. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. 2005;102:2081–2086. doi: 10.1073/pnas.0409597102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Yu QC, Enquist L, Shenk T. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 2003;77:10594–10605. doi: 10.1128/JVI.77.19.10594-10605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KO. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 1964;115:814–816. doi: 10.3181/00379727-115-29045. [DOI] [PubMed] [Google Scholar]
- Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 1997;71:6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh PC, Meckes DG, Jr, Wills JW. Analysis of the Interaction between the UL11 and UL16 Tegument Proteins of Herpes Simplex Virus. J. Virol. 2008;82:10693–10700. doi: 10.1128/JVI.01230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]