

Abstract
Understanding the structural organization and distribution of proteins in biological cells is of fundamental importance in biomedical research. The use of conventional fluorescent microscopy for this purpose is limited due to its relatively low spatial resolution compared to the size of a single protein molecule. Atomic force microscopy (AFM), on the other hand, allows one to achieve single-protein resolution by scanning the cell surface using a specialized ligand-coated AFM tip. However, because this method relies on short-range interactions, it is limited to the detection of binding sites that are directly accessible to the AFM tip. We developed a method based on magnetic (long-range) interactions and applied it to investigate the structural organization and distribution of endothelin receptors on the surface of smooth muscle cells. Endothelin receptors were labeled with 50-nm superparamagnetic microbeads and then imaged with magnetic AFM. Considering its high spatial resolution and ability to “see” magnetically labeled proteins at a distance of up to 150 nm, this approach may become an important tool for investigating the dynamics of individual proteins both on the cell membrane and in the submembrane space.
Introduction
Membrane surface proteins play a pivotal role in cellular function. It is well accepted that most of the components of the cell membrane, e.g., ion channels, receptors, and exchangers, have a complex multimeric structure and are often localized in specialized membrane regions such as lipid rafts and caveolae. The investigation of the structure and distribution of membrane proteins and their complexes on the surface of living cells, however, presents an enormous challenge and currently is chiefly limited to fluorescence imaging techniques. Such techniques involve the labeling of protein macromolecules with specific antibodies, which are then targeted by fluorescent probes (1). Recent advances in fluorescence microscopy, combined with the development of genetically coded fluorescent proteins (2) and quantum dots (3), have significantly improved our understanding of the functional and temporal dynamics of intracellular proteins. However, imaging of individual proteins remains beyond the resolution of optical instruments (4). This is due to the inherent limitations of optical instruments, whose resolution is restricted by the wavelength of the light. The invention of the atomic force microscope has opened up a novel approach for studying individual proteins, their topography, and protein-protein interactions at the nanoscale level. It is currently the only technique that can provide nanometer resolution under the physiological conditions required for living cells. Atomic force microscopy (AFM) imaging has revealed fine structures of bacteriorhodopsins in isolated bacterial membranes (5) (for a review, see Frederix et al. (6)), nuclear pore complexes in the nuclear envelope (7–9), gap junctions (10), and receptors (11,12) overexpressed in mammalian cell lines. Also, multimeric structures of purified isolated receptors and ion channel proteins have been demonstrated by AFM imaging (13–18). The recent development of simultaneous topography and recognition imaging (19,20) has significantly increased the lateral resolution, allowing visualization of individual protein molecules. Despite this progress, however, the identification of specific proteins on the surface of intact cells by AFM remains a challenging task and relies entirely on direct interactions between the functionalized (ligand-coated) AFM probe and a corresponding protein target. However, the functionalized AFM tip usually has a relatively short lifetime due to the instability of attached functional ligands (for review, see Hinterdorfer and Dufrêne (21)). In recent studies using magnetically coated AFM cantilevers, interactions between streptavidin molecules attached to a self-assembled monolayer or to a modified glass surface and biotin-coated magnetic nanoparticles were demonstrated using the magnetic mode of AFM (MFM) (22,23). In these two studies, magnetic nanoparticles were either chemically derived (22) or isolated from magnetotactic bacteria (23). Here, we further developed this new methodological approach and applied it to investigate the distribution of endothelin (ET) receptors on the surface of intact rat aortic smooth muscle cells (SMCs). We used an innovative labeling approach to specifically tag ET-1, a potent and highly specific endogenous agonist of ET receptors, with superparamagnetic microbeads (∼50 nm in diameter). Magnetically labeled receptors were then detected and imaged by AFM and MFM. The choice of cell type (ET-1 and ET receptors) for this study was dictated by a several considerations. From a functional point of view, the ET receptors (which mainly consist of two subtypes, ETA and ETB) play an important role in the cardiovascular system under physiological conditions and in various disease states (24). Biochemical, ligand-binding, and functional studies have demonstrated that vascular SMCs endogenously express both subtypes of the ET receptors, with the ETA subtype being generally dominant (25). However, the distribution and organization of receptors on the surface of intact vascular cells at the molecular level have only been studied in the vascular wall of the capybara basilar artery using electron microscopy (26). Another important consideration was the highly specific and potent binding of ET-1 with the receptor and its extremely slow dissociation from the receptor, which make interactions between the ET receptor and ET-1 virtually irreversible (27). These features of the agonist-receptor interaction should facilitate preparation of ET-1-treated cells for AFM and MFM imaging without a significant loss of the agonist-receptor complexes. Also, because ET-1 is a short 21-amino acid peptide containing two primary amines, at a lysine-9 residue and a cysteine-1 residue at the N-terminus, it can be biotinylated using a standard procedure and then targeted with anti-biotin-coated superparamagnetic nanoparticles for MFM imaging. Finally, since ET-1 binds equally well to both ET receptor subtypes, its usage would maximize the number of endogenous agonist-receptor complexes available on the cell surface for high-resolution MFM imaging.
Materials and Methods
Isolation of rat aortic SMCs and primary cell culture
Thoracic aortas were taken from male Wistar rats (250–300 g) after they were humanely killed by cervical dislocation in accordance with the UK Home Office legislation and guidelines. The thoracic aorta was cleaned of connective tissue and cut into pieces (∼2 mm2) in a cold HEPES-buffered saline solution (HBS) of the following composition (in mM): 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, 10 glucose. Single SMCs were obtained by means of a modified enzymatic isolation procedure described previously (28). Pieces of tissue were placed in cold Ca2+, Mg2+-free HBS containing 0.1 mg/mL bovine serum albumin (BSA, fraction V; Sigma, Gillingham, UK) for 10 min and then incubated for 20 min at 37°C in a fresh 2 mL of Ca2+, Mg2+-free HBS digestion solution containing 1 mg/mL BSA, 2 mg/mL collagenase type XI, 1 mg/mL papain, 1 mg/mL dithiothreitol, and 0.5 mg/mL trypsin inhibitor (all from Sigma), followed by another 20 min incubation at 37°C in a fresh digestion solution. Pieces of tissue were then gently triturated in fresh Ca2+, Mg2+-free HBS to yield cells. The cell suspension was centrifuged at 1100 × g for 12 min and then the cell pellet was resuspended in 5 mL of fresh HBS or Dulbecco's modified Eagle's medium (DMEM)/F-12 media (Invitrogen, Paisley, UK) for primary cell culture (29). The DMEM/F-12 also contained 30 μg/mL penicillin-streptomycin and 0.25 μg/mL fungizone (all from Invitrogen). For AFM and MFM imaging, cells were plated on 9 mm sterile glass coverslips and cultured for 48 h in a humidified cell CO2 incubator (LEEC, Nottingham, UK).
Cell biotinylation
SMCs were treated for 30 min at room temperature (RT) with freshly prepared biotinylation solution containing nine volumes of phosphate-buffered saline (PBS; Sigma), one volume of 1 M sodium bicarbonate and 0.5 mg/mL water soluble biotin-XX sulfosuccinimidyl ester (SSE) (Invitrogen), pH = 8.0. The cells were gently agitated (every 5 min for freshly isolated cell suspension) or rotated at 20 times/min (primary cultured on microscope coverslips). Biotinylation was terminated by washing the cells four times with fresh PBS, one time with 100 mM glycine in PBS, and two times with ice-cold PBS. Primary cultured SMCs were fixed with 3% paraformaldehyde (PFA) (30 min) and then washed with 0.1 mg/mL BSA in PBS (10 min, RT) before the biotinylation procedure was performed. Freshly isolated cells were first biotinylated, then concentrated by centrifugation at 1100 × g for 12 min during the termination procedure, and plated on glass coverslips before the fixation procedure was performed as described for cultured SMCs.
Biotinylation of ET-1 and its bioavailability
Biotinylation of ET-1 (100 μg; Sigma) was performed with the Biotin-XX Microscale Protein Labeling kit (B30010; Invitrogen) according to the manufacturer's instructions, except that centrifugation at 14,000 × g for 90 s (instead of the recommended 16,000 × g for 15 s) was used to purify biotinylated ET-1 (bET-1) from unbound biotin-XX SSE. Also, 100 mM of glycine were added to the filtered solution to stop remaining reactions and quench unbound labeling reagent (30). The concentration of protein was measured with the use of Bradford reagent. bET-1 was aliquoted and stored at −20°C.
The functional bioavailability of bET-1 was verified in intact rat small mesenteric arteries (third branch) using a small vessel wire myograph (methodological details are given in the Supporting Material). The results clearly demonstrate that the biotinylation procedure did not impair the ability of ET-1 to constrict blood vessels (see Fig. S1).
Cell labeling with bET-1
Fixed primary cultured SMCs were incubated with 200 nM of biotinylated ET-1 (bET-1) in PBS (15 min, RT), washed several times with fresh PBS, and treated with either fluorescein isothiocyanate (FITC) for confocal imaging or superparamagnetic microbeads for AFM/MFM imaging. Fixation of cells was used to minimize potential internalization of activated ET receptors (31–33).
FITC labeling and confocal imaging
After biotinylation and fixation were achieved, cells were incubated with FITC-conjugated anti-biotin rabbit antibody (1:150 in PBS containing 0.1 mg/mL BSA) for 90 min. To ensure that acetone dehydration (which was used for cultured SMCs treated with superparamagnetic microbeads for AFM/MFM imaging) did not affect cell labeling, cultured SMCs (biotinylated or treated with bET-1) were initially incubated in ice-cold acetone for 10 min, allowed to dry, and then incubated with anti-biotin rabbit antibody-FITC as described above. FITC-labeled cells were visualized with a Zeiss LSM 510 or LSM 300 Olympus confocal microscope equipped with a Plan-Apochromat 63×/1.4 oil DIC or Plan-Neofluar 40×/1.3 oil objective. An excitation wavelength of 488 nm and emission bandwidth of 505–530 nm were used to record FITC fluorescence. Control cell samples were prepared in the same way, except that the biotinylation procedure was excluded, and imaged under the same settings. Images were analyzed using the software program LSM 510 v. 4.0 (Carl Zeiss, Welwyn Garden City, UK) or Fluoview v. 5.0 (Olympus, Southend-on-Sea, UK).
Cell labeling with superparamagnetic microbeads and AFM/MFM imaging
Biotinylated or bET-1-labeled cultured aortic SMCs were gently rotated with anti-biotin-coated superparamagnetic microbeads (MACS, Miltenyi Biotec, Woking, UK; 1:10 dilution of the supplied stock solution in 2 mL of PBS containing 0.5% BSA and 2 mM EDTA) for 30 min (RT) and then washed four times with fresh PBS. To ensure firm binding of the microbeads to biotinylated cells or to bET-1 bound to the receptors, a postfixation procedure was applied that included treatment of cells with 4% PFA and 2% glutaraldehyde in PBS (15 min, RT) followed by washing several times with PBS and washing for 5 min with MilliQ water. Cells were then dehydrated with a gradually increasing concentration of ethanol in water (25%, 50%, 75%, and 100%) and in a 50% acetone/ethanol mixture (each for 5 min), followed by a 10 min incubation in pure acetone and air-drying. The cell dehydration procedure and AFM/MFM imaging were performed in a clean-room environment with controlled temperature (23°C) and humidity (45%). It is worthwhile to note that commercially purchased magnetic anti-biotin-coated microbeads, which were used for labeling in our experiments, have a magnetic (iron oxide) core and a total diameter of ∼50 nm according to their specifications. This was independently confirmed by scanning electron microscopy (not shown). Of importance, it was previously demonstrated that these microbeads are superparamagnetic (34), i.e., their magnetic moments change randomly in time at RT in the absence of applied magnetic fields. This is because the energy required to change the direction of magnetization of the superparamagnetic nanoparticle is smaller than the thermal energy kT. Therefore, MFM imaging was performed on the top of a permanent magnet that produces a vertical magnetic field of 85 Oe to polarize the superparamagnetic microbeads and increase the strength of the magnetic interaction between the MFM tip and microbeads, thus increasing the signal/noise ratio.
AFM and MFM imaging was performed using silicon cantilevers coated with a 100 nm layer of cobalt-chromium alloy (high-moment MFM probes; Asylum Research, Bicester, UK) at RT. These probes have a spring constant of 1–2 N m−1, a resonant frequency of 55–90 kHz, and a coercive field of 500–650 Oe. Images were taken using a VEECO Multimode IIIa atomic force microscope. The scan rate was 0.1–1 Hz, depending on the scanned area, with a resolution of 256 pixels per line. Pseudo-three-dimensional (3D) AFM images were generated using WSxM software (35).
Results and Discussion
Labeling method and its verification with confocal imaging
The method for high-resolution imaging of membrane proteins by AFM and MFM is depicted in Fig. 1 A. Target proteins are labeled with biotin (step 1), which is then recognized by anti-biotin-coated microbeads containing superparamagnetic nanoparticles (step 2). The labeled cell surface is then scanned with an oscillating AFM probe coated with a thin magnetic layer (step 3). While working in the MFM mode, the AFM scans each line twice: initially in the AFM tapping mode, creating a topographic image, and then in the MFM lift mode (i.e., at a certain distance from the surface), which allows the detection of magnetic forces and creates a magnetic image of the same area. To demonstrate and characterize the major advantages of combined AFM and MFM imaging, proteins on the surface of rat aortic SMCs were nonselectively labeled with water-soluble biotin-XX SSE as described in Materials and Methods. This procedure should predominantly biotinylate the primary amines of lysine and N-termini of proteins on the extracellular surface of the cells (36). To confirm successful biotinylation of membrane proteins, biotinylated SMCs were initially labeled with the fluorescent marker FITC conjugated to anti-biotin antibodies and imaged using confocal microscopy. Fig. 1 B shows that the fluorescent signal is located peripherally (left) and overlaps with the boundaries of freshly isolated rat aortic SMCs, thus indicating that only extracellular proteins are labeled using this approach. Since subsequent AFM/MFM imaging was performed on primary cultured cells (because they have a relatively smaller height, and our AFM scanner has a height measurement limit of 2.7 μm), the same procedure was performed on rat aortic SMCs cultured for 48 h. Fig. 1 C shows fluorescent signal from cultured rat aortic SMCs labeled in the same way as the freshly isolated cells shown in Fig. 1 B, confirming successful staining of surface proteins. Note that the fluorescent signal is more diffused in cultured cells compared to freshly isolated cells due to their flat geometry (compare Fig. 1, B and C). Nevertheless, successful biotinylation of both freshly isolated (first biotinylated and then fixed) and primary cultured (first fixed and then biotinylated) rat aortic SMCs suggests that cell fixation does not significantly hinder labeling of the surface proteins.
Figure 1.
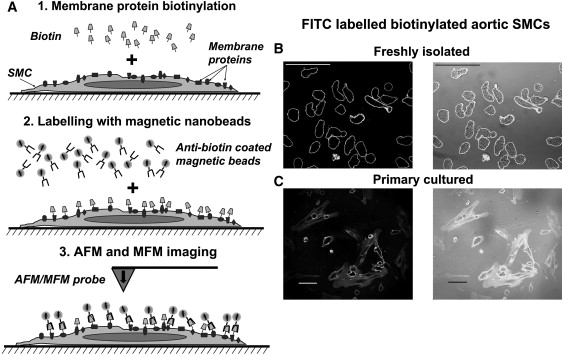
Labeling method and imaging of biotinylated intact rat aortic SMCs. (A) Schematics of the cell biotinylation and AFM/MFM imaging procedure, showing the key steps of cell membrane biotinylation with biotin XX (step 1), labeling with anti-biotin-coated superparamagnetic microbeads (step 2), and imaging with the AFM/MFM probe (step 3). (B and C) Confocal images of freshly isolated (B) and primary cultured (C) rat aortic SMCs were biotinylated as shown in A (step 1), but labeled with anti-biotin-conjugated FITC instead of magnetic microbeads (step 2). Confocal images were used to verify the experimental procedure before magnetic labeling. The left and right panels in B and C compare fluorescent and superimposed fluorescent and transmitted light images, respectively. Scale bars are 50 μm.
AFM and MFM imaging of biotinylated cells labeled with superparamagnetic microbeads
AFM imaging of a small area of the cell surface (4 μm × 2 μm) of a biotinylated primary cultured SMC treated with anti-biotin-coated superparamagnetic microbeads instead of anti-biotin FITC revealed the presence of multiple bright spots corresponding to elevated heights (see Fig. 2 A; a corresponding 3D reconstruction image is shown on the right). Subsequent scanning of the same area of the cell with the MFM probe (Fig. 2 B) showed a good correlation between the AFM and MFM responses, suggesting that superparamagnetic microbeads are responsible for the majority of these spots. Of interest, both the AFM and MFM images of the biotinylated surface of intact SMCs labeled with superparamagnetic nanoparticles resemble the AFM images of immunoglobulins immobilized on the patterned monolayers (37).
Figure 2.
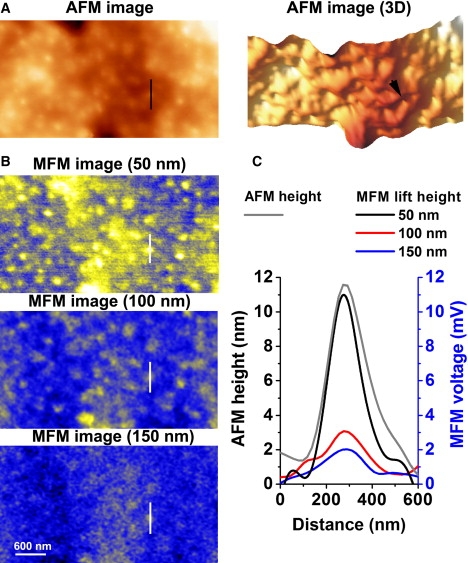
Dependence of the magnetic signal on the lift height of the MFM probe. (A) AFM image of biotinylated cultured aortic SMCs in tapping mode (left) and a 3D reconstruction (right) of a 4 × 2 μm area of the surface of a biotinylated cell. (B) MFM images of the same area at different lift heights as indicated above each panel. (C) Line analysis of the AFM and MFM responses across the selected area shown by straight lines in A and B, and by an arrowhead in the 3D reconstruction image in A.
The ability of MFM to sense the presence of magnetic particles at a distance was tested by lifting the cantilever up from 50 nm (the standard height) above the cell surface to 100 and 150 nm (Fig. 2 B). Fig. 2 C compares the MFM voltage profile of a selected particle (shown by white lines in B) at different cantilever heights imaged with MFM, and the AFM height (topographic) profile for the same particle (shown by the black line and arrowhead in Fig. 2 A). It can be clearly seen that MFM can sense the presence of magnetically labeled proteins up to 150 nm above the cell surface under our experimental conditions with no direct physical contact with labeled molecules, which is an essential step in AFM imaging. This ability of MFM to “sense” magnetically labeled proteins at a distance could potentially allow the investigation of magnetically labeled single molecules not only on the surface of the membrane, but also directly in the submembrane space after, for example, receptor internalization. As demonstrated in Fig. 2 C, a measurable magnetic response was achieved at lift heights of up to 150 nm under our experimental conditions. The maximum detection height will depend on the magnetic moment of the nanoparticles and the magnetic properties of the MFM probe (determined by the probe geometry and composition), the size of the nanoparticles, and the magnetic field applied to the specimen. All of these parameters can be precisely controlled, allowing determination of the distance from the cell surface to a magnetically labeled molecule and creating an opportunity for 3D protein mapping in the submembrane space, which is not possible with AFM alone.
Magnetic nature of MFM responses
The magnetic nature of the signal measured with MFM was confirmed in experiments with inverse magnetic polarization of the MFM probe. The schematic of this approach is demonstrated in Fig. 3 A. Fig. 3, B and C, respectively show the AFM and MFM images for the MFM probe polarized vertically down (right) and vertically up (left), whereas the magnetic microbeads were polarized up using an externally applied magnetic field (see Materials and Methods). Fig. 3 D compares cross-sectional analyses of a selected superparamagnetic bead in the AFM and MFM images for the opposite polarizations of the MFM probe. The change in sign for the magnetic but not the topographic responses with reversed polarization suggests that magnetic responses are indeed measured in the MFM mode.
Figure 3.
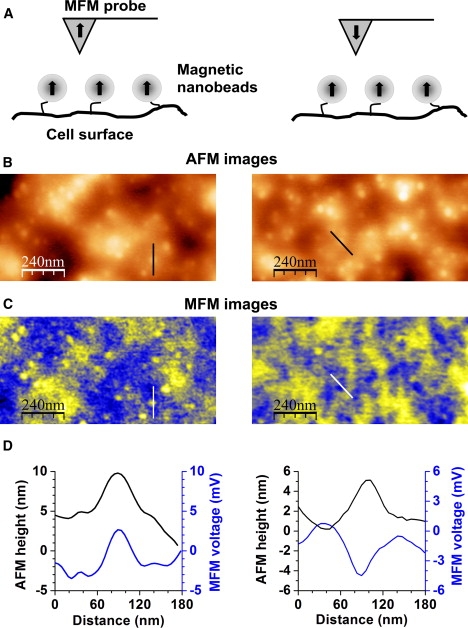
Magnetic nature of MFM responses. (A) Schematic of the experiment with the MFM probe polarized vertically up (left picture) and vertically down (right picture). (B) AFM images of two areas of a primary cultured biotinylated SMC measured with the MFM tip polarized “up” (left column) and “down” (right column). (C) Corresponding MFM images for the two selected cell areas. Note that for magnetic repolarization, the MFM probe had to be removed from the instrument; therefore, the images on the left and right do not exactly represent the same area of the cell. (D) Comparison of the topographic (AFM) and magnetic (MFM) profiles (respectively shown by straight lines in B and C) for a single superparamagnetic microbead for different polarizations of the MFM tip. Black and blue lines depict variations in the AFM height and the MFM voltage, respectively.
Mapping of individual ET receptors in intact SMCs
The methodological approach described above was then adapted to investigate the distribution of ET receptors on the surface of SMCs, as schematically depicted in Fig. 4 A. ET receptors (which are ubiquitously expressed in the vasculature) are activated by the agonist ET-1, leading to vessel constriction (25). The ET-1 peptide is composed of 21 amino acids and includes one lysine residue at position 9 (24); thus it possesses two primary amines, on lysine-9 and cysteine-1 at the N-terminus. These can be biotinylated (step 1; see Materials and Methods for details) before applying ET-1 on SMCs (step 2). Receptor-bound biotinylated ET-1 (bET-1) will be recognized by anti-biotin-coated superparamagnetic microbeads (step 3) and then visualized with the AFM/MFM probe (step 4). To independently verify this methodological approach and confirm that bET-1 was able to recognize ET receptors on the cell surface, cells were labeled with 100 nM of bET-1 and then imaged by confocal microscopy. Cultured SMCs were fixed with 3% PFA (to prevent possible internalization of activated receptors) before bET-1 treatment and then labeled with anti-biotin conjugated FITC. Confocal images of a representative group of cells thus treated are shown in Fig. 4 B, demonstrating successful labeling of cells. Of interest, both diffused and bright-spot fluorescent signals were observed, suggesting uneven distribution and perhaps clustering of ET receptors in SMCs. The bright spots (Figs. 4 and 5 C) are likely due to a specific interaction between bET-1 and ET receptors, because the pretreatment of cells with an excess of nonbiotinylated ET-1 (200 nM) before cell incubation with bET-1 inhibited the FITC fluorescent signal (Fig. 5 B), which did not differ from that measured in control untreated cells (Fig. 5 A).
Figure 4.
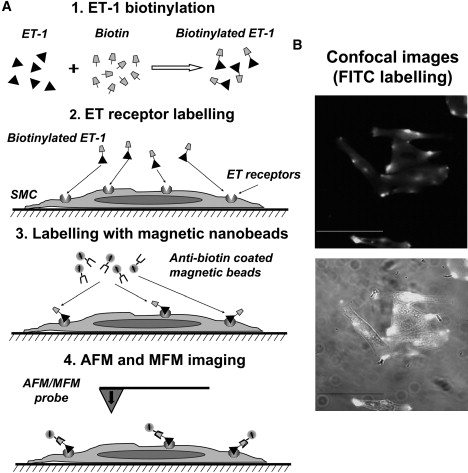
Method for visualizing ET receptors in biotinylated cultured SMCs, and AFM/MFM imaging verified by confocal microscopy. (A) Schematics of the experimental procedure. The key steps include biotinylation of ET-1 (step 1), labeling of the surface ET receptors with bET-1 (step 2), labeling of bET-1 bound to the receptor with anti-biotin-coated superparamagnetic microbeads (step 3), and imaging of the labeled receptors with AFM/MFM (step 4). (B) Confocal imaging of cultured cells treated with 100 nM bET-1-and then labeled with anti-biotin FITC-conjugated antibodies instead of anti-biotin-coated superparamagnetic microbeads. The bottom panel shows superimposed fluorescent and transmitted light images. Scale bars are 50 μm.
Figure 5.
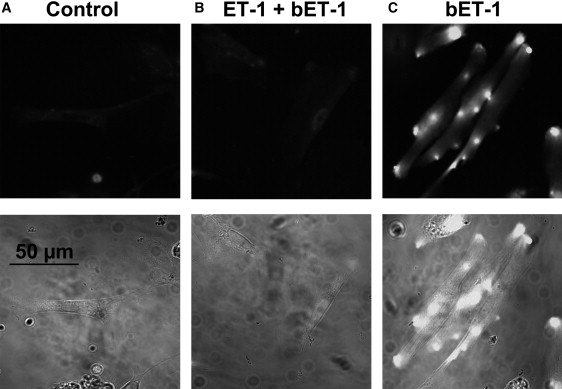
Specificity of bET-1 labeling. (A–C) Confocal images of primary cultured aortic SMCs: nontreated (control) (A), pretreated with 200 nM of nonmodified ET-1 and then treated with bET-1 (100 nM) (B), and treated with bET-1 (100 nM) (C).
Cells labeled with bET-1 were then treated with anti-biotin-coated superparamagnetic microbeads and subjected to AFM and MFM imaging. Fig. 6 A shows an AFM image of the surface of a cultured SMC at low resolution. To visualize individual proteins, three different surface areas of the cell (shown by squares labeled 1–3 in Fig. 6 A) were imaged with AFM at high spatial resolution (Fig. 6 B). Analysis of these AFM images revealed distinctive white spots on the surface of the SMCs, which can be clearly seen as topographic peaks in the 3D reconstruction (bottom panels in Fig. 6 B). Of interest, high-resolution AFM imaging revealed that these topographic features are distributed on the cell surface both as individual peaks and as complexes of various sizes. Subsequent MFM imaging of the same areas (Fig. 6 C) revealed a nearly perfect match between the AFM and MFM responses, suggesting that the topographic peaks in the AFM images represent ET receptors selectively labeled with superparamagnetic microbeads. It is noteworthy that the line cross-section analysis shown in Fig. 6 D (panel 3i), which compares the AFM height and MFM voltage profiles, clearly demonstrates three distinctive amplitude peaks with AFM and only two with MFM, indicating the presence of only two ET receptors. Therefore, the use of MFM in addition to AFM imaging offers an additional opportunity to distinguish between specifically magnetically labeled receptors and other nonspecific topographic features present on the cell surface. This advantage of MFM/AFM imaging is further illustrated by control experiments in which nonbiotinylated cells were treated with superparamagnetic microbeads (Fig. S2). The distinctive topographic features seen in the AFM image (Fig. S2 A) are absent in the MFM image of the same cell area (Fig. S2 B; also see the line analysis in Fig. S3 C). These data also confirm the absence of nonspecific interactions of anti-biotin-coated microbeads with the cell surface of nonbiotinylated control cells, and thus independently support the results obtained in control experiments with FITC described above and in Fig. 5.
Figure 6.
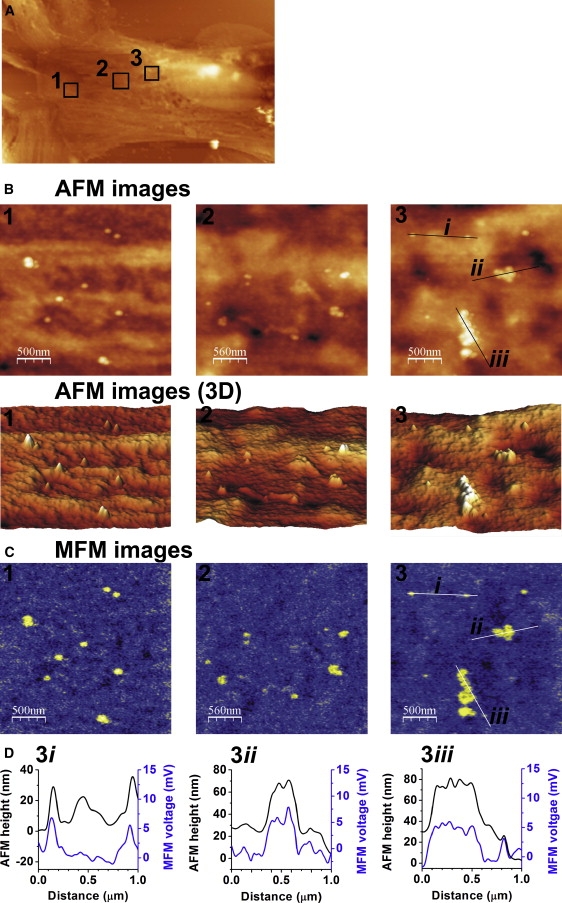
AFM/MFM imaging of ET receptors on the surface of aortic SMCs. (A) AFM image (50 × 30 μm) of the surface area of a cultured SMC. (B) AFM images of three selected areas at high spatial resolution (2.5 × 2.5 μm) shown by squares 1–3 in A (upper panels) and their corresponding 3D reconstruction (bottom panels). (C) Corresponding MFM images of the three areas shown in B. Note a correlation between amplitude heights measured with AFM and magnetic responses measured with MFM, confirming the specificity of the receptor labeling. (D) Line section analysis of the AFM topographic (black lines) and MFM magnetic (blue lines) responses for two individual ET receptors (i) and two receptor complexes (ii and iii) marked by straight lines in the third selected area (right columns in B and C).
Furthermore, these results suggest that native ET receptors can exist as large receptor complexes (conglomerates) in intact aortic SMCs. The AFM topographic and MFM magnetic profiles for two separate receptors and the two receptor conglomerates (straight lines denoted as i, ii, and iii in the third square in the AFM image in B and the MFM image in C) are compared in Fig. 6 D. This analysis demonstrates a complex organization of the receptor conglomerate, which could be composed of three to five individual ET receptors (e.g., panel 3iii in Fig. 6 D). It is unlikely that such conglomerates are due to multiple magnetic particles bound to bET-1, as only two potential biotinylated sites (cystein-1 and lysine-9 residues) per the agonist are available. A computational molecular analysis of the interaction between ET-1 and the ET receptor suggests that cystein-1 at the N-terminus and lysine-9 of ET-1 preserve a substantial accessible area after the agonist docks into the receptor (38), and therefore either of the biotinylated amino acid residues might theoretically interact with an anti-biotin-coated microbead or an FITC molecule. On the other hand, numerous functional studies suggest that the N-terminus is essential for selective interaction with ETA receptors (27). Also, the removal of cysteine-1 at the N-terminus renders ET-1 inactive in the intact rat aorta, where the ETA receptor subtype is dominant (39). These observations may indicate that cysteine-1 at the N-terminus of ET-1 may not be easily accessible for molecular interaction. Furthermore, selective biotinylation of ET-1 on lysine-9 did not alter its functional activity against the ETA receptors (40), thus supporting our functional studies with bET-1 shown in Fig. S1. Finally, taking into account that in our labeling procedure the biotinylated agonist first binds to the receptor and then interacts with superparamagnetic microbeads, and considering that the two biotinylated sites in the ET-1 molecule are likely to be in close proximity to each other (due to the presence of two disulfide bonds), it is reasonable to speculate that only one biotinylated site per ET-1 molecule would be available for the interaction with a superparamagnetic microbead or an FITC molecule, perhaps at lysine-9. However, further focused structural studies will be necessary to address the question as to which biotinylated amino acid in ET-1 is mainly responsible for the interaction with superparamagnetic beads.
The existence of receptor conglomerates on the surface of SMCs observed in high-resolution AFM/MFM images could explain the existence of the bright spots of fluorescence observed on the surface of FITC-labeled SMCs (Fig. 6 B). Although intracellular conglomerates of ET receptors were previously demonstrated in vascular endothelial cells by means of gold particle immunostaining and electron microscopy (26), this is the first direct demonstration (to our knowledge) of ET receptor clusters on the surface of native cells. It is noteworthy that previous reports demonstrated colocalization of ET receptors with caveolin in COS cells (where ETA receptors were overexpressed) (41) and association of endogenous ETA and ETB receptors with lipid rafts in primary cultured rat peritubular SMCs (42). However, in our preparation, the bright spots of FITC fluorescence (which may be associated with ET receptor clusters) were not clearly associated with the fluorescence caveolin-1 Cy3 conjugate when examined under confocal microscope (Fig. S3). Also, the size of the largest cluster visualized by AFM/MFM imaging is greater than the typical size of caveolae (50–100 nm). These results suggest that ET receptor clusters in rat aortic SMCs may be organized in a different manner and may not be necessarily associated with caveolae, although colocalization of individual receptors or small receptor groups with caveolae cannot be excluded.
In conclusion, we have successfully applied the novel methodological approach that combines AFM and MFM to study specific macromolecules on the surface of intact cells with nanoscale resolution. MFM imaging depends on long-range (noncontact) interactions between the MFM probe and the target protein, in contrast to AFM imaging, which relies on measurement of the force of short-range (contact) interactions between target proteins and a functionalized (ligand-coated) AFM tip. The combined use of AFM and MFM for molecular mapping therefore offers a range of advantages, such as 1), the potential ability to map individual molecules in the submembrane space; 2), a higher level of accuracy in determining the distribution of specific molecules; 3), less physical distortion of molecules on the cell surface, due to decreased direct interactions between target molecules and the MFM tip (although AFM topographic scanning is a necessary initial step in the MFM imaging, it is performed in the tapping mode with a nonfunctionalized MFM probe); and 4), an increased lifetime of MFM tips compared to functionalized (ligand-coated) AFM tips, allowing a greater number of scans of the cell surface.
MFM imaging provides high lateral resolution for protein mapping, comparable to that of AFM (Fig. 6 D). In our case, it was limited to 50 nm (i.e., the size of commercially acquired superparamagnetic microbeads). However, Schreiber and colleagues (34) demonstrated that 15 nm superparamagnetic nanoparticles could be detected by MFM at RT. Recently, Kaiser et al. (43) demonstrated that subnanometer-scale magnetic imaging is achievable with the use of an atomically sharp magnetic tip. Therefore, one can expect that the use of smaller magnetic nanoparticles and a sharper MFM probe in future studies will significantly improve the lateral resolution of this method.
Although we performed our experiments on fixed cells to characterize and verify this methodological approach, MFM imaging could also be adopted to study protein dynamics in live cells, since AFM was previously used to study the surface topography of living cells in solution (44,45). This offers a potential advantage of AFM/MFM imaging over electron microscopy, which delivers a comparable resolution but cannot be used in live cells.
Supporting Material
Verification of functional bioavailability of ET-1 and references, and three figures, are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01628-2.
Supporting Material
Acknowledgments
We thank Adrian Rogers for technical help and support with the confocal imaging.
This work was supported by the Engineering and Physical Sciences Research Council (grant EP/E014925).
References
- 1.Giepmans B.N., Adams S.R., Tsien R.Y. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J., Campbell R.E., Tsien R.Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 3.Bruchez M.P. Turning all the lights on: quantum dots in cellular assays. Curr. Opin. Chem. Biol. 2005;9:533–537. doi: 10.1016/j.cbpa.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Lewis A., Taha H., Ammann E. Near-field optics: from subwavelength illumination to nanometric shadowing. Nat. Biotechnol. 2003;21:1378–1386. doi: 10.1038/nbt898. [DOI] [PubMed] [Google Scholar]
- 5.Möller C., Allen M., Müller D.J. Tapping-mode atomic force microscopy produces faithful high-resolution images of protein surfaces. Biophys. J. 1999;77:1150–1158. doi: 10.1016/S0006-3495(99)76966-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frederix P.L., Akiyama T., Engel A. Atomic force bio-analytics. Curr. Opin. Chem. Biol. 2003;7:641–647. doi: 10.1016/j.cbpa.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Stoffler D., Goldie K.N., Aebi U. Calcium-mediated structural changes of native nuclear pore complexes monitored by time-lapse atomic force microscopy. J. Mol. Biol. 1999;287:741–752. doi: 10.1006/jmbi.1999.2637. [DOI] [PubMed] [Google Scholar]
- 8.Erickson E.S., Mooren O.L., Dunn R.C. Activation of ryanodine receptors in the nuclear envelope alters the conformation of the nuclear pore complex. Biophys. Chem. 2004;112:1–7. doi: 10.1016/j.bpc.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Franco-Obregón A., Wang H.W., Clapham D.E. Distinct ion channel classes are expressed on the outer nuclear envelope of T- and B-lymphocyte cell lines. Biophys. J. 2000;79:202–214. doi: 10.1016/S0006-3495(00)76284-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller D.J., Hand G.M., Sosinsky G.E. Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J. 2002;21:3598–3607. doi: 10.1093/emboj/cdf365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrera N.P., Betts J., Edwardson J.M. Atomic force microscopy reveals the stoichiometry and subunit arrangement of the α4β3δ GABA(A) receptor. Mol. Pharmacol. 2008;73:960–967. doi: 10.1124/mol.107.042481. [DOI] [PubMed] [Google Scholar]
- 12.Barrera N.P., Herbert P., Edwardson J.M. Atomic force microscopy reveals the stoichiometry and subunit arrangement of 5-HT3 receptors. Proc. Natl. Acad. Sci. USA. 2005;102:12595–12600. doi: 10.1073/pnas.0503253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrera N.P., Shaifta Y., Edwardson J.M. AFM imaging reveals the tetrameric structure of the TRPC1 channel. Biochem. Biophys. Res. Commun. 2007;358:1086–1090. doi: 10.1016/j.bbrc.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 14.Henderson R.M., Schneider S., Oberleithner H. Imaging ROMK1 inwardly rectifying ATP-sensitive K+ channel protein using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1996;93:8756–8760. doi: 10.1073/pnas.93.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Huizen R., Czajkowsky D.M., Li M. Images of oligomeric Kv β2, a modulatory subunit of potassium channels. FEBS Lett. 1999;457:107–111. doi: 10.1016/s0014-5793(99)01021-2. [DOI] [PubMed] [Google Scholar]
- 16.Suhara W., Kobayashi M., Mikoshiba K. Visualization of inositol 1,4,5-trisphosphate receptor by atomic force microscopy. Neurosci. Lett. 2006;391:102–107. [Google Scholar]
- 17.Barrera N.P., Ormond S.J., Edwardson J.M. Atomic force microscopy imaging demonstrates that P2X2 receptors are trimers but that P2X6 receptor subunits do not oligomerize. J. Biol. Chem. 2005;280:10759–10765. doi: 10.1074/jbc.M412265200. [DOI] [PubMed] [Google Scholar]
- 18.Lange T., Jungmann P., Schillers H. Reduced number of CFTR molecules in erythrocyte plasma membrane of cystic fibrosis patients. Mol. Membr. Biol. 2006;23:317–323. doi: 10.1080/09687860600738304. [DOI] [PubMed] [Google Scholar]
- 19.Stroh C., Wang H., Hinterdorfer P. Single-molecule recognition imaging microscopy. Proc. Natl. Acad. Sci. USA. 2004;101:12503–12507. doi: 10.1073/pnas.0403538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stroh C.M., Ebner A., Hinterdorfer P. Simultaneous topography and recognition imaging using force microscopy. Biophys. J. 2004;87:1981–1990. doi: 10.1529/biophysj.104.043331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinterdorfer P., Dufrêne Y.F. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods. 2006;3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- 22.Arakaki A., Hideshima S., Osaka T. Detection of biomolecular interaction between biotin and streptavidin on a self-assembled monolayer using magnetic nanoparticles. Biotechnol. Bioeng. 2004;88:543–546. doi: 10.1002/bit.20262. [DOI] [PubMed] [Google Scholar]
- 23.Amemiya Y., Tanaka T., Matsunaga T. Novel detection system for biomolecules using nano-sized bacterial magnetic particles and magnetic force microscopy. J. Biotechnol. 2005;120:308–314. doi: 10.1016/j.jbiotec.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 24.Masaki T. Historical review: endothelin. Trends Pharmacol. Sci. 2004;25:219–224. doi: 10.1016/j.tips.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Rubanyi G.M., Polokoff M.A. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol. Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- 26.Loesch A. Localisation of endothelin-1 and its receptors in vascular tissue as seen at the electron microscopic level. Curr. Vasc. Pharmacol. 2005;3:381–392. doi: 10.2174/157016105774329381. [DOI] [PubMed] [Google Scholar]
- 27.Huggins J.P., Pelton J.T., Miller R.C. The structure and specificity of endothelin receptors: their importance in physiology and medicine. Pharmacol. Ther. 1993;59:55–123. doi: 10.1016/0163-7258(93)90041-b. [DOI] [PubMed] [Google Scholar]
- 28.Tammaro P., Smith A.L., Smirnov S.V. Pharmacological evidence for a key role of voltage-gated K+ channels in the function of rat aortic smooth muscle cells. Br. J. Pharmacol. 2004;143:303–317. doi: 10.1038/sj.bjp.0705957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beck R., Bertolino S., Smirnov S.V. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ. Res. 1998;83:923–931. doi: 10.1161/01.res.83.9.923. [DOI] [PubMed] [Google Scholar]
- 30.Qian Y., Galli A., Blakely R.D. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J. Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roubert P., Gillard V., Braquet P. Down-regulation of endothelin binding sites in rat vascular smooth muscle cells. Am. J. Hypertens. 1990;3:310–312. doi: 10.1093/ajh/3.4.310. [DOI] [PubMed] [Google Scholar]
- 32.Marsault R., Feolde E., Frelin C. Receptor externalization determines sustained contractile responses to endothelin-1 in the rat aorta. Am. J. Physiol. 1993;264:C687–C693. doi: 10.1152/ajpcell.1993.264.3.C687. [DOI] [PubMed] [Google Scholar]
- 33.Bremnes T., Paasche J.D., Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J. Biol. Chem. 2000;275:17596–17604. doi: 10.1074/jbc.M000142200. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber S., Savla M., Agarwal G. Magnetic force microscopy of superparamagnetic nanoparticles. Small. 2008;4:270–278. doi: 10.1002/smll.200700116. [DOI] [PubMed] [Google Scholar]
- 35.Horcas I., Fernández R., Baro A.M. WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007;78:013705. doi: 10.1063/1.2432410. [DOI] [PubMed] [Google Scholar]
- 36.Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 1992;3:2–13. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 37.Wadu-Mesthrige K., Amro N.A., Liu G. Fabrication of nanometer-sized protein patterns using atomic force microscopy and selective immobilization. Biophys. J. 2001;80:1891–1899. doi: 10.1016/s0006-3495(01)76158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orry A.J., Wallace B.A. Modeling and docking the endothelin G-protein-coupled receptor. Biophys. J. 2000;79:3083–3094. doi: 10.1016/S0006-3495(00)76543-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forget M.A., Lebel N., Fournier A. Biological and molecular analyses of structurally reduced analogues of endothelin-1. Mol. Pharmacol. 1996;49:1071–1079. [PubMed] [Google Scholar]
- 40.Magazine H.I., Andersen T.T., Malik A.B. Evaluation of endothelin receptor populations using endothelin-1 biotinylated at lysine-9 sidechain. Biochem. Biophys. Res. Commun. 1991;181:1245–1250. doi: 10.1016/0006-291x(91)92072-r. [DOI] [PubMed] [Google Scholar]
- 41.Chun M., Liyanage U.K., Lodish H.F. Signal transduction of a G protein-coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc. Natl. Acad. Sci. USA. 1994;91:11728–11732. doi: 10.1073/pnas.91.24.11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gambara G., Billington R.A., Filippini A. NAADP-induced Ca(2+) signaling in response to endothelin is via the receptor subtype B and requires the integrity of lipid rafts/caveolae. J. Cell. Physiol. 2008;216:396–404. doi: 10.1002/jcp.21407. [DOI] [PubMed] [Google Scholar]
- 43.Kaiser U., Schwarz A., Wiesendanger R. Magnetic exchange force microscopy with atomic resolution. Nature. 2007;446:522–525. doi: 10.1038/nature05617. [DOI] [PubMed] [Google Scholar]
- 44.Pesen D., Hoh J.H. Micromechanical architecture of the endothelial cell cortex. Biophys. J. 2005;88:670–679. doi: 10.1529/biophysj.104.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radmacher M., Tillamnn R.W., Gaub H.E. From molecules to cells: imaging soft samples with the atomic force microscope. Science. 1992;257:1900–1905. doi: 10.1126/science.1411505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.