

Abstract
Voltage-gated ion channels regulate the electric activity of excitable tissues, such as the heart and brain. Therefore, treatment for conditions of disturbed excitability is often based on drugs that target ion channels. In this study of a voltage-gated K channel, we propose what we believe to be a novel pharmacological mechanism for how to regulate channel activity. Charged lipophilic substances can tune channel opening, and consequently excitability, by an electrostatic interaction with the channel's voltage sensors. The direction of the effect depends on the charge of the substance. This was shown by three compounds sharing an arachidonyl backbone but bearing different charge: arachidonic acid, methyl arachidonate, and arachidonyl amine. Computer simulations of membrane excitability showed that small changes in the voltage dependence of Na and K channels have prominent impact on excitability and the tendency for repetitive firing. For instance, a shift in the voltage dependence of a K channel with −5 or +5 mV corresponds to a threefold increase or decrease in K channel density, respectively. We suggest that electrostatic tuning of ion channel activity constitutes a novel and powerful pharmacological approach with which to affect cellular excitability.
Introduction
Voltage-gated ion channels regulate the flow of different ions over the cell membrane in response to changes in the membrane potential. They are crucial for health, and dysfunctional channels can cause severe conditions such as epilepsy and heart arrhythmias (1–3). It is therefore important to understand how the activity of these channels is regulated and how abnormal activity can be pharmacologically tuned.
Most voltage-gated ion channels open in response to depolarizing potentials. The depolarization induces a movement within voltage-sensor domains located in the periphery of the ion channel molecule (4). This movement couples to the central ion-conducting pore, and leads to channel opening (5). A key event in this process is the outward movement of the positively charged voltage sensors. Traditional drugs acting on ion channels reduce channel activity by blocking the ion-conducting pore (6–8). This is an effective, but also quite dramatic, way to regulate channel activity and excitability in the heart and the brain. Furthermore, a pore block cannot increase the channel activity. Instead of plugging the pore, a potentially milder regulation of ion channels could be achieved by modifying their voltage dependence. Certain novel small-molecule openers, tested as anti-epileptic drugs, change the voltage dependence of channels important for neuronal excitability, by targeting the gate to keep the channel open (4). However, an alternative and yet unexplored pharmacological strategy would be to modulate the voltage dependence by directly targeting the voltage sensor itself; i.e., voltage-sensor pharmacology. This can be achieved either via mechanic or electric interactions.
In a previous study, we demonstrated that negatively charged free polyunsaturated fatty acids (PUFAs), such as docosahexaenoic acid (DHA), shifted the voltage dependence of the Shaker K channel in hyperpolarizing direction (9). Uncharged PUFAs (i.e., DHA methyl esters) were ineffective, consistent with an electrostatic mechanism in which negatively charged PUFAs attract the voltage sensors, thereby facilitating their outward movement and the consequent channel opening (10). Yet, there is a possibility that the negative charge of the PUFA is needed for proper binding, whereas the acyl tail affects the channel. To test the electrostatic mechanism therefore requires a PUFA-like molecule with a positive charge. If the electrostatic mechanism is correct, a positively charged PUFA would shift the voltage dependence in opposite direction to the negatively charged one, whereas the uncharged PUFA would have no effect. This is illustrated in Fig. 1, A and B, with different compounds having an arachidonyl backbone. Conversely, if the charge is only necessary for binding, either both the positively charged and uncharged PUFAs will be ineffective, or the positively charged PUFA will have the same effect as the negatively charged one. Charged lipophilic substances that electrostatically tune the movement of the voltage sensor would be what we believe to be a novel pharmacological approach that we call lipoelectric modification.
Figure 1.
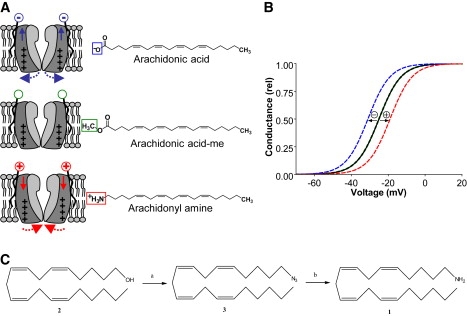
Theory behind electrostatic tuning of voltage-sensor movement. Exemplified by three compounds with an arachidonyl backbone. (A) A negatively charged compound attracts the voltage sensor and facilitates channel opening, an uncharged substance does not affect the channel, and a positively charged substance repels the voltage sensor and hinders channel opening. The structure of arachidonic acid and its two analogs are shown to the right. (B) Schematic prediction of the effect on the voltage dependence for each compound. (C) Synthesis scheme for arachidonyl amine. Arachidonyl amine (1) was synthesized from arachidonyl alcohol (2) via arachidonyl azide (3). For the conversion from arachidonyl alcohol to arachidonyl azide (a) pyridine and methanesulphonyl chloride, followed by DMF and NaN3 are used. For conversion from arachidonyl azide to arachidonyl amine (b) LiAlH4, tetrahydrofuran and diethyl ether are used.
To test the theory of lipoelectric tuning of the voltage-sensor movement, we synthesized arachidonyl amine, which is positively charged (or uncharged depending on pH). Instead of having a negatively charged carboxyl group, it has a positively charged amine group. Thus, arachidonyl amine bears similar structure to the PUFA arachidonic acid, but is positively charged (compare structures in Fig. 1 A). As predicted from the electrostatic hypothesis, we find that the amine shifted the voltage dependence of channel opening in positive direction along the voltage axis. The negatively charged arachidonic acid shifted the voltage dependence in negative direction, whereas the uncharged arachidonic acid methyl ester was without effect. Thus, we have shown that lipophilic substances can modify channel opening, probably by interfering with the voltage sensor movement, with the direction depending on the charge of the substance. Computer simulations showed pronounced effects on excitability and repetitive firing, with small changes in the voltage dependence of K and Na channels. Voltage-sensor tuning could be a future pharmacological strategy in the design of drugs against diseases of both hypo- and hyperexcitability.
Materials and Methods
Arachidonyl amine synthesis
Arachidonyl alcohol was purchased from Nu-Chek Prep (Elysian, MN). Reactions were monitored by thin layer chromatography plates coated with silica gel 60 F254 (Merck, Darmstadt, Germany), and bands were visualized by ultraviolet and anisaldehyde. Products were purified by flash chromatography using 30–60 μM silica gel (Mallincrodt Baker, Phillipsburg, NJ). Petroleum ether used was 60/95 grade. 1H and 13C NMR were recorded on a Bruker Avance 500 spectrometer operating at 500.1 MHz and 125.8 MHz, respectively. Tetramethylsilane was used as an internal standard. The spectra were processed from the recorded FID files with Topspin 2.1 software (Bruker BioSpin, Zurich, Switzerland). Chemical shifts (δ) are reported in ppm downfield from tetramethylsilane. The following abbreviations are used: s, single; br s, broad singlet; t, triplet; m, multiplet.
The synthesis of arachidonyl amine (1 in Fig. 1 C) is schematized in Fig. 1 C. Arachidonyl alcohol (2 in Fig. 1 C) (430 mg, 1.5 mmol) was dissolved in dry pyridine (9 mL) and the solution was cooled on an ice bath. Methanesulphonyl chloride (340 mg, 3.0 mmol) was added dropwise and stirring was continued at 0°C for 3 h. The reaction mixture was poured into ice cold water and extracted with ethyl acetate. The organic phase was washed with 1 M HCl, saturated NaHCO3 and water, and finally, dried over Na2SO4. Evaporation of solvent gave 530 mg of brownish, oily product. Mesylate was dissolved in dry N,N- dimethylformamide (DMF) (20 mL) and NaN3 (480 mg, 7.4 mmol) was added in one portion. The mixture was stirred at 60°C for 30 h. DMF was evaporated and ethyl acetate was added. The solution was washed with brine and water, dried over Na2SO4 and evaporated. The crude product was purified by flash chromatography eluting with 0–5% ethyl acetate in petroleum ether. Evaporation of solvents gave 379 mg (81%) of a colorless, oily product. 1HNMR (CDCl3): δ 0.89 (t, 3J = 6.4 Hz, 3 H), 1.25–1.39 (m, 6 H), 1.42–1.49 (m, 2 H), 1.59–1.65 (m, 2 H), 2.04–2.13 (m, 4 H), 2.80–2.85 (m, 6 H), 3.27 (t, 3J = 6.9 Hz, 2 H), 5.31–5.43 (m, 8 H). The solution of arachidonyl azide (3 in Fig. 1 C) (370 mg, 1.2 mmol) in dry diethyl ether (4 mL) was added slowly into the solution of LiAlH4 (89 mg, 2.4 mmol) in dry tetrahydrofuran (4 mL). The mixture was stirred at 50°C overnight. Ethyl acetate was added, and saturated NaHCO3 was added slowly. The white precipitate was filtered and the organic phase was separated. The water phase was extracted with ethyl acetate, and combined organic layers were dried over Na2SO4 and evaporated. The crude product was purified twice by flash chromatography eluting with 0–100% methanol in dichloromethane. Evaporation of solvents gave 85 mg (30%) of brownish oil. 1H NMR (CDCl3): δ 0.89 (t, 3J = 6.6 Hz, 3 H), 1.22 (br s, 2 H), 1.26–1.49 (m, 10 H), 2.04–2.11 (m, 4 H), 2.69 (t, 3J = 6.6 Hz, 2 H), 2.81–2.85 (m, 6 H), 5.31–5.43 (m, 8 H). 13C NMR (CDCl3): δ 14.1, 22.6, 25.6 (2 C), 26.9 (2 C), 27.1, 27.2, 29.3, 31.5, 33.5, 42.2, 127.6, 127.9 (2 C), 128.1, 128.4, 128.5, 130.1, 130.5. The final product arachidonyl amine is uncharged or positively charged depending on pH. Pure aliquots, stock solutions, and the test solutions were stored in light proof containers. Pure aliquots and stock solutions were also stored under a protective atmosphere of argon.
Molecular biology and expression of ion channels
Experiments were carried out on the Shaker H4 channel (accession number NM_167595.3) (11) made incapable of fast inactivation by the Δ(6–46) deletion (12). The Bluescript II KS(+) plasmid was linearized with HindIII, and cRNA synthesized using the T7 mMessage mMachine kit (Ambion, Austin, TX). To get oocytes, female Xenopus laevis was anesthetized with 1.4 g/L ethyl 3-aminobenzoate methanesulfonate salt (tricaine). A batch of oocytes was removed and the surgery wound stitched together. Animal experiments were approved by the local Animal Care and Use Committee at Linköping University. Oocytes were separated by incubation for ∼2 h in a Ca-free O-R2 solution (13) (in mM: 82.5 NaCl, 2 KCl, 5 HEPES, and 1 MgCl2; pH adjusted to 7.4 by NaOH) containing Liberase Blendzyme 3 (0.7 U/mL; Roche Diagnostics, Bromma, Sweden). Healthy matured oocytes were singled out and incubated at 11°C in a modified Barth's solution (MBS; in mM: 88 NaCl, 1 KCl, 2.4 NaHCO3, 15 HEPES, 0.33 Ca(NO3)2, 0.41 CaCl2, and 0.82 MgSO4; pH adjusted to 7.6 by NaOH) supplemented with penicillin (25 U/mL), streptomycin (25 μg/mL), and sodium pyruvate (2.5 mM) overnight before injection. Fifty nL of cRNA (50 pg) was injected into each oocyte using a Nanoject injector (Drummond Scientific, Broomall, PA). Injected oocytes and uninjected oocytes for control experiments were maintained at 11°C in MBS containing pyruvate and antibiotics until subject to electrophysiological experiments. All chemicals are from Sigma-Aldrich (Stockholm, Sweden) if not stated otherwise.
Electrophysiology
Currents were measured with the two-electrode voltage-clamp technique (CA-1B amplifier; Dagan, Minneapolis, MN) 3–6 days after injection. The amplifier's leak and capacitance compensation were used and the currents were low-pass filtered at 5 kHz. All experiments were done at room temperature (20–23°C). The holding potential was set to −80 mV and steady-state currents achieved by stepping to potentials between −80 and +50 mV in 5 mV increments. The control solution contained (in mM): 88 NaCl, 1 KCl, 15 HEPES, 0.4 CaCl2, and 0.8 MgCl2. pH was adjusted to 7.4 with NaOH yielding a final sodium concentration of ∼100 mM. Pure arachidonyl amine or arachidonic acid methyl ester (methyl arachidonate; Sigma-Aldrich) were dissolved to 100 mM in 99.5% ethanol and stored at −20°C until use. Right before experiments stock solutions were diluted in control solution to the desired concentration. The effective test-compound concentration was assumed to be 70% of the nominal concentration due to fatty acid binding to the chamber (previously quantified in Börjesson et al. (10)). All concentrations given in this work are the effective test-compound concentrations. It should be noted that the quantification was done for a fatty acid and not arachidonyl amine. However, considering the resemblance in structure we believe the amount of chamber binding to be roughly similar. Pure control solution was added to the bath using a gravity-driven perfusion system. All test solutions were added manually with a glass Pasteur pipette to avoid binding to the perfusion system (the added volume was several times larger than the bath solution volume). Indomethacin (Sigma-Aldrich) was used to prevent possible conversion by cyclooxygenase as previously described for arachidonic acid (10).
Analysis
The steady-state current, I, at each test potential was measured at the end of the stimulation pulse (60–90 ms after the onset of the pulse). However, at voltages above 0 mV, arachidonyl amine induced an inactivation of the current and reduced the steady-state current (Fig. 2 B). To eliminate this current-reducing effect we fitted an exponential curve to the inactivating current and extrapolated this curve back to time for onset of the voltage pulse. This value was assumed to be equal to the maximum current in the absence of inactivation. Because it takes some time for the channel to open, this procedure slightly overestimates the inactivation-induced current reduction. Using the K channel model described below it can be shown that the compensation should be 30–35% less than what is suggested by the extrapolation method. However, this overestimation did not affect the measurement of the voltage dependence. The K conductance, G, was calculated as G(V) = I(V)/(V − VK), where V is the absolute membrane potential and VK the reversal potential for the K ion (−80 mV). Shifts of the G(V) curve were quantified by sliding the control curve along the voltage axis to overlap the test compound curve. In cases with altered curve slope the control curve was slid until the curves overlapped at the 10% level of the control curve. Average values are expressed as mean ± SE. Statistical analyses on G(V) shifts were done with two-tailed one sample t-test with the mean value compared to a hypothetical value of 0. For comparison of shifts at different pH two-tailed unpaired t-test was used; p < 0.05 is considered as significant.
Figure 2.
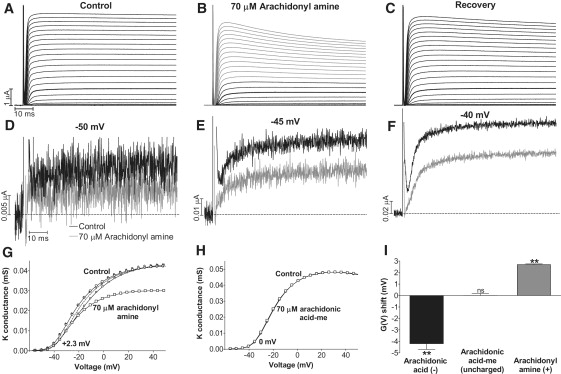
Effects of arachidonyl amine on the Shaker K channel. (A–C) Currents associated with voltage-clamp steps between −80 and +50 mV in 5 mV increments. Holding voltage is −80 mV. Arachidonyl amine (70 μM) reduced the steady-state current at the most negative voltages (compare black traces in B with A) and induced inactivation at more positive voltages (gray traces in B). Recovery after wash with control solution. Thick trace in A–C marks the current at −25 mV. (D–F) Currents at −50, −45, and −40 mV with (gray) and without (black) 70 μM arachidonyl amine. (G) Arachidonyl amine (70 μM) shifted the G(V) curve in positive direction along the voltage axis. Control (○), 70 μM arachidonyl amine (□), inactivation-corrected (see Materials and Methods) effect of arachidonyl (small □), inactivation-corrected recovery (small ○). Dashed curve is control curve shifted +2.3 mV. (H) Arachidonic acid-me (70 μM) did not shift the G(V) curve. Control (○), 70 μM arachidonic acid-me (□). (I) Summary of the induced G(V) shifts for different forms of arachidonyl compounds (70 μM). n = 5–8. Error bars = SE.
Computer simulations of excitability
To explore the effects on neuronal excitability of voltage-sensor modifying substances we used a simple and well-characterized excitable membrane, the node of Ranvier of the frog myelinated axon (14) with minor modifications (9). The system is based on one type of Na channel, one type of delayed rectifier K channel, and a leakage channel. The following rate constants were used for the computations:
V is the absolute membrane voltage, ΔVx is the modification of the voltage dependence of each gate (m, h, n). The rate constants are given in ms−1. The initial state (at V = −70 mV in all simulations) of each gate is given by
The development of each state is calculated as
where dt is a sufficiently short time step of 1 μs. The conductances G are calculated as
The maximum conductances are GNa,max = 2.5 μS, GK,max = 0.4 μS, and Gleak = 0.03 μS.
The four calculated currents are:
IC is the capacitive current and IS is the continuous stimulus current injected to elicit repetitive firing in the nodal system. The reversal potential for the currents are VNa = 50 mV, VK = −90 mV, and Vleak = −71 mV. The change in membrane voltage is calculated as
where CM is the membrane capacitance set to 2 μF/cm2 (i.e., 2 pF/node of 100 μm2). A holding potential of −70 mV is used in the beginning of all simulations. Repetitive firing is defined as firing after >100 ms of continuous stimulation.
Results
Arachidonyl amine on uninjected X. laevis oocytes
X. laevis oocytes is a common and reliable model system both for expression of ion channels and for exploration of ion channels using electrophysiological techniques. To ensure that the arachidonyl amine does not affect endogenous Xenopus oocyte channels, it was tested on uninjected oocytes. Neither 21 nor 70 μM arachidonyl amine induced any endogenous currents, and did not affect the resting membrane potential and membrane permeability of Xenopus oocytes. At 210 μM, the arachidonyl amine was toxic, by inducing massive leakage in the oocytes. Thus, concentrations >70 μM could not be reliably used for oocyte experiments. However, prominent PUFA effects on the Shaker K channel have been reported at concentrations between 3 and 70 μM (10).
The effect of arachidonyl amine on the Shaker K channel
Having established that moderate concentrations of arachidonyl amine did not affect uninjected Xenopus oocytes, the arachidonyl amine was used to test the hypothesis of lipoelectric voltage-sensor tuning. Arachidonyl amine (70 μM) had a clear effect on the Shaker K current (Fig. 2, A and B). At voltages more negative than −10 mV, the arachidonyl amine reduced the steady-state current, with no obvious effect on the time course (Fig. 2 B, black traces). Fig. 2, D–F, shows magnified traces at three different voltages. The reduction is 49% at −50 mV, 41% at −45, and 33% at −40. At voltages more positive than −10 mV, the arachidonyl amine induced an inactivation (Fig. 2 B; gray traces). The arachidonyl amine effect was quickly reversed after wash with control solution, except for a small inactivation component remaining at the most positive voltages (Fig. 2 C). When the steady-state conductance (at the end of the pulse) versus voltage, G(V), was plotted, two effects were observed: The arachidonyl amine i), shifted the G(V) curve in the positive direction along the voltage axis; and ii), reduced the maximum conductance (Fig. 2 G, large squares). However, if the effect on the inactivation was compensated for (see Materials and Methods), the reduction was completely eliminated (Fig. 2 G, small squares). The mean shift in voltage dependence, measured at the foot of the curve, by 70 μM arachidonyl amine was +2.7 ± 0.6 mV (n = 8; p = 0.003). This should be compared with arachidonic acid, which shifted the G(V) curve with −4.2 ± 0.5 mV (from Börjesson et al. (10)), and with the uncharged methyl ester of the arachidonic acid (arachidonic acid-me), which did not affect the voltage dependence at all (Fig. 2 H; mean shift 0.0 ± 0.2 mV; n = 6; p = 1.0). A comparison of the induced G(V) shifts by 70 μM of the negatively charged, the uncharged, and the positively charged forms of arachidonic acid at pH 7.4 is shown in Fig. 2 I. These data clearly support our lipoelectric hypothesis.
pH dependence of amine effect
If the effect of the amine on the channel's voltage dependence is caused by an electrostatic mechanism, then the potency of the amine should depend on pH. Specifically, the amine should be more effective at low pH, because a larger fraction of amine is protonated and thus positively charged. The pH dependence of the arachidonyl amine effect followed this prediction. Whereas 21 μM arachidonyl amine at pH 7.4 did not affect the channel's voltage dependence, it induced a significant effect at pH 6.5. Fig. 3, A–C, show large current reductions of the amine at pH 6.5 at voltages between −20 and −10 mV. (Note that more positive voltage steps are needed to open the channel at pH 6.5 than at pH 7.4, as expected from protonable surface charges (15).) Fig. 3 D shows that the G(V) curve is clearly shifted in the positive direction along the voltage axis, with no reduction of the maximum conductance after compensation for the induced inactivation (small squares). The mean shift was +4.5 ± 0.4 mV (n = 5; p = 0.0003). Although 70 μM arachidonyl amine had a clear effect at pH 7.4 (Fig. 2), it was abolished at pH 9.0 (Fig. 3 E; mean shift −0.1 ± 0.1 mV; n = 3; p = 0.4). We also explored the effect of 70 μM arachidonyl amine at pH 6.5, but found that this induced massive leakage of the oocyte, similar to that for 210 μM at pH 7.4 described above. Thus, it seems that the cellular membrane is disrupted if the number of positively charged arachidonyl amine molecules exceeds a certain level. Shift data from the pH experiments are summarized in Fig. 3 F.
Figure 3.
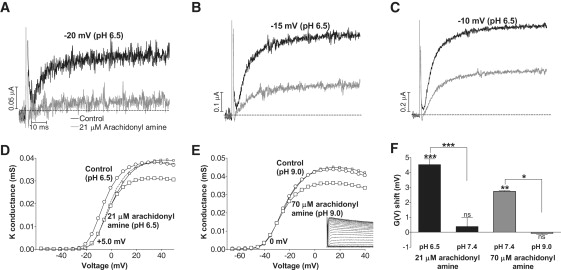
pH dependence of the arachidonyl amine effect. (A-C) Arachidonyl amine (21 μM) induced dramatic current reduction at the most negative voltages at pH 6.5. Control (black), amine (gray). (D) The G(V) shift was more pronounced at pH 6.5 than at pH 7.4. Control (○), 21 μM arachidonyl amine (□), 21 μM arachidonyl amine compensated for inactivation (small □). Dashed line is control curve shifted +5.0 mV. (E) The G(V) shift was abolished at pH 9.0 whereas the inactivation remained (see inset). Control (○), 70 μM arachidonyl amine (□), 70 μM arachidonyl amine compensated for inactivation (small □). (F) Summary of the pH dependence of arachidonyl amine effects. Error bars = SE.
However, despite the lack of effect on the channel's voltage dependence at pH 9.0, the arachidonyl amine inactivated the channel at positive voltages (Fig. 3 E, inset). This suggests that the effect on the channel's voltage dependence and the effect on the inactivation are caused by two different mechanisms. Thus, we find it likely that the arachidonyl amine, independent of its charge, binds to the channel and speeds up slow inactivation, possibly by destabilizing the turret region of the channel (16,17). From this position, depending on its charge, it affects the voltage sensor electrostatically.
Computer modeling of excitability
What impact do these relatively small shifts in the voltage dependence of a Kv channel have on cellular excitability? It is known that intravenously injected PUFAs prevent ventricular fibrillation in dog (18), but because multiple ion channels are active it is difficult if not impossible to determine, in an excitable cell, which channel(s) is the primary target for PUFAs, especially because PUFAs also affect other types of voltage-gated ion channels (19,20). To obtain information regarding the role of PUFA effects on different ion channels, we carried out computer simulations on an excitable model membrane: the myelinated frog axon (14). This simple simulation system contains one type of Na channel, one type of delayed-rectifier K channel and one type of leakage channel.
Fig. 4 A shows the effect of two different persistent stimulus currents (IS; 0.6 nA in the upper row and 1.0 nA in the lower row), in five different conditions. The middle column shows control cases. For the smaller stimulus current only a single action potential was elicited and there was no repetitive firing. Shifting the voltage dependence of the K current by −5 mV did not affect the excitability, whereas a shift by +5 mV induced repetitive firing. The larger stimulus current induced repetitive firing in the control case. Here, −5 mV abolished repetitive firing whereas +5 mV had no effect, other than a slight increase in firing frequency. Similar effects were obtained if the number but not the voltage dependence of the K channel is changed by multiplying or dividing by 3 respectively.
Figure 4.
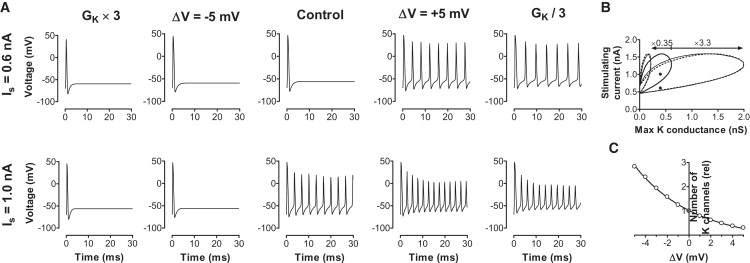
Impact of altered voltage dependence or density of K channels on cellular excitability. Tested by computer simulations on the myelinated frog axon. (A) A persistent stimulus current of 0.6 nA (upper row) elicited one action potential in the control case. A +5 mV shift in the K channels' voltage dependence or a reduced number of K channels induced repetitive firing. A stimulus current of 1.0 nA (lower row) induced repetitive firing in the control case. A −5 mV shift in the K channels' voltage dependence or an increased number of K channels abolished repetitive firing. (B) The thick continuous line encircles the area in which repetitive firing occurs. A shift of −5 mV reduced the area of repetitive firing (small thin circle) to 0.35 of its original value (small dotted circle). A shift of +5 mV increased the area of repetitive firing (large thin circle) with a factor of 3.3 (large dotted circle). (C) The equivalent change in K channel density for different G(V) shifts. The continuous line is A = exp(−ΔV/s), where s = 4.7 mV.
To explore these effects quantitatively, we calculated which combinations of K channel densities and stimulus currents give repetitive firing for a fixed Na channel density (2.5 μS). Fig. 4 B shows the area where repetitive firing occurs (encircled with a thick continuous line). Even in the absence of K channels, repetitive firing was possible. If the K conductance exceeded ∼0.6 μS, repetitive firing was not possible, independent of the size of the stimulus current used. A stimulus current below ∼0.5 nA depolarized the cell too little to induce repetitive firing, and stimulus currents above ∼1.5 nA depolarized the cell too much, thus preventing repetitive firing. If the voltage dependence of the K channel was shifted −5 mV, the area of repetitive firing (delineated with a thin continuous line) was contracted to ∼1/3 of its original size. The dotted line is control area multiplied with 0.35 with respect to the x axis. Thus a −5 mV shift in the K channel voltage dependence corresponds to an almost threefold increase in K channel density. Conversely, if the voltage dependence of the K channel was shifted +5 mV, the area of repetitive firing (delineated with a thin continuous line) was expanded to ∼3 times its original size. The dotted line is control area multiplied with 3.3 with respect to the x axis. Thus a +5 mV shift in the K channel voltage dependence corresponds to a threefold reduction in K channel density. The cases from Fig. 4 A are shown as black dots in Fig. 4 B. The corresponding change in K channel density is shown in Fig. 4 C. A doubling in density occurs every 3.2 mV. It should be noted that the effects described here are independent of the Na channel density (4). Thus a change in the K channel's voltage dependence is a simple, fast, and reversible way to mimic large changes in K channel density, and consequently to affect excitability.
The effects described in this and previous (9,10) investigations may not necessarily be isolated to K channels. Effects of PUFAs on Na channel activation and Na channel inactivation has been described (19,21). What impact do effects on other channels than K channels have on excitability? To make an initial estimation, we calculated the minimum stimulus current needed to induce repetitive firing. We first tested the role of K channels (n parameter) for repetitive firing (Fig. 5 A). Shifting the K channel activation in negative direction along the voltage axis increased the threshold, and at ∼−2 mV shift the repetitive firing was abolished (seen as a vertical line). Reciprocally, a shift in positive direction decreased the threshold. We next explored the role of Na channel activation (m parameter) and Na channel inactivation (h parameter), in isolation and in combination, for repetitive firing (Fig. 5 B). Isolated effects on either of the two Na channel parameters (activation or inactivation) abolished repetitive firing, if the shift exceeded 1 mV in positive or negative direction, respectively. Shifting the Na channel activation and inactivation, together with +2 mV, also abolished repetitive firing. This indicates that the excitable system of the frog node of Ranvier explored by Frankenhauser and Huxley (14) is remarkably close to the border of repetitive firing with respect to several gating parameters.
Figure 5.
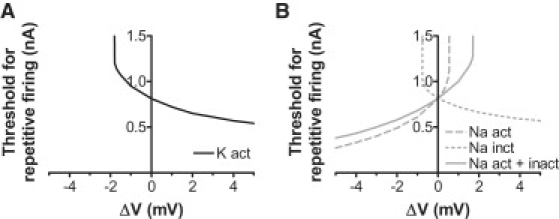
Importance of the voltage dependencies of K and Na channels for repetitive firing tested by computer simulations of the myelinated frog axon. (A) A shift of the voltage dependence of the K channel activation in positive direction along the voltage axis decreased the threshold for repetitive firing. A shift in negative direction increased the threshold, and a shift of −2 mV abolished repetitive firing (seen as a vertical line). (B) A shift in the voltage dependence of the Na channel inactivation had similar effects as for K channel activation; a shift in positive direction reduced the threshold for repetitive firing and a shift in negative direction increased the threshold; −1 mV abolished repetitive firing. A shift of Na channel activation had the opposite effect; +1 mV abolished repetitive firing. The continuous line shows the effect of a combined shift of Na channel activation and inactivation.
Discussion
We have shown that lipid compounds such as PUFAs affect an ion channel's voltage dependence via an electrostatic mechanism. A negatively charged PUFA, such as arachidonic acid, shifts the voltage dependence in hyperpolarizing direction along the voltage axis. The crucial experiment in this study was that the synthesized positively charged arachidonyl amine shifted the voltage dependence in positive direction along the voltage axis. The neutral methyl ester of arachidonic acid had no effect on the channel's voltage dependence. Thus, the voltage dependence of Kv channels can be tuned electrostatically by lipophilic substances, the direction of the effect depending on the charge of the compound. We propose that this be referred to as a lipoelectric modulation. Although the electrostatic mechanism explored in this study applies to free PUFAs, it is probably also applicable for a variety of other lipophilic substances with a charged group or moiety.
Lipids, both in the form of phospholipids in the membrane and as free fatty acids, are interesting and potent channel regulators (4). An interaction between the lipid bilayer and the voltage sensor in Kv channels was suggested some time ago (22), and a close contact between negative phospholipid headgroups and positive charges in the voltage sensors has later been seen, both in the Kv1.2/2.1 chimera crystal structure (23) and in molecular dynamics studies (24–26). When experimentally removing the headgroups, the voltage dependence and channel function is greatly disturbed (27–30). Therefore, negatively charged headgroups may function as counter charges for the gating charges (27). Also for PUFAs the charged headgroup is central for the effect, as shown here and in other investigations (10). A certain level of basal physiological lipoelectric modulation is to be expected, because PUFAs exist at μM concentrations in serum and, likely, in cerebrospinal fluid (31). At these concentrations PUFAs have clear effects not only on Kv channels but also on a variety of other voltage-gated ion channels (20), some of which may be tuned electrostatically. The effect of increased PUFAs on Kv channels is also suggested to be an important component of the ketogenic diet used for epilepsy treatment (9). Positively charged PUFAs are, to our knowledge, not naturally occurring in the human body. In this study they are used merely to show the principle of electrostatic tuning. The clear effect on the voltage dependence makes the PUFA amine an interesting model substance for future synthesis of positively charged but more specific drugs. The amine effect also raises the question of whether or not other positively charged substances act through lipoelectric modulation. For instance, amphiphilic cysteine-knot toxins, such as hanatoxin, partition into the membrane, from where they interact closely with the voltage-sensor domain and inhibit channel activation (32). Because hanatoxin possesses several positive residues at a reasonable depth on the channel-interacting side it has been speculated that part of its action is to electrostatically prevent the voltage-sensor movement (4).
Changing the voltage dependence of a channel is a powerful way to affect cellular excitability and the tendency for repetitive firing, as demonstrated by computer simulations in this study. Shifting the voltage dependence of a K channel with only a few mV is shown to effectively prevent or induce repetitive firing, depending on the direction of the shift. We have also shown that small changes in the voltage dependence of the activation and/or inactivation of a Na channel has great impact on repetitive firing. For PUFAs, this is highly relevant because they affect activation and to a larger extent the voltage dependence of inactivation of Na channels (19,20). The dominant hyperpolarizing shift of Na channel inactivation would act in a similar manner as the K channel effect reported, here and previously, i.e., to reduce cellular excitability. The simulations on Na channels also demonstrate that pharmacological lipoelectric modulation of Na channel voltage dependence, in either direction, would have significant effects on excitability. Changes in excitability can also be estimated by determining the minimum current needed to elicit an action potential (i.e., rheobase) or the minimum slope of an activating current ramp needed to elicit an action potential (that gives information about accommodation of the nerve). In the frog myelinated axon the rheobase depends almost solely on the voltage dependence of the Na channel activation (m gate), whereas accommodation in the ramp experiments depends to a large extent on the voltage dependence of the Na channel inactivation (h gate) (33,34). Thus, excitability of nerve cells is critically dependent on the channels' voltage dependencies, suggesting that pharmacological compounds with extremely small effects on the voltage sensing machineries of the channel could be potent drugs against diseases of both hypo- and hyperexcitability.
Another notable finding here is that arachidonyl amine induces massive leakage of the oocyte, at a concentration of 210 μM at neutral pH or at 70 μM at pH 6.5. Thus, it appears that the cellular membrane is distorted or damaged by a certain dose of positively charged arachidonyl amine molecules. Arachidonyl amine is not the only amine reported to be toxic. When mapping the cytotoxicity of different polyamines, the positive charge(s) on polyamines were identified to be important for toxicity (35,36). Both the number of charges and their geometrical arrangement are determining toxicity factors (35). The polyamines first induce membrane leakage, followed by decreased metabolic activity, possibly through interactions with membrane lipids. Perhaps this mechanism is shared by arachidonyl amine. Thus, this suggests that lipoelectric pharmacological compounds have to be channel specific, not only to induce specific effects, but also to reduce the cellular toxicity.
In this study, a PUFA-like amine was used to test the hypothesis of electrostatic tuning of an ion channel's voltage sensitivity. We suggest that the PUFA-like amines are useful tools to differentiate between electrostatic and nonelectrostatic effects of PUFAs.
Acknowledgments
We thank Peter Larsson (University of Miami) and Stefan Thor (Linköping University) for helpful comments on the manuscript.
This work was supported by the Swedish Research Council (grant No. 5630 and No. 5914), the Swedish Heart-Lung Foundation, the Swedish Brain Foundation, Linköping University, ALF-grants from the County Council of Östergötland, King Gustaf V and Queen Victoria's Freemason Foundation, Queen Silvia's Anniversary Foundation, and Stina and Birger Johansson's Foundation.
References
- 1.Jentsch T.J. Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 2.Jespersen T., Grunnet M., Olesen S.P. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 3.Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- 4.Börjesson S.I., Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem. Biophys. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- 5.Long S.B., Campbell E.B., Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 6.Zhou M., Morais-Cabral J.H., MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 7.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ragsdale D.S., McPhee J.C., Catterall W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 9.Xu X.P., Erichsen D., Elinder F. Polyunsaturated fatty acids and cerebrospinal fluid from children on the ketogenic diet open a voltage-gated K channel: a putative mechanism of antiseizure action. Epilepsy Res. 2008;80:57–66. doi: 10.1016/j.eplepsyres.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Börjesson S.I., Hammarström S., Elinder F. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 2008;95:2242–2253. doi: 10.1529/biophysj.108.130757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamb A., Tseng-Crank J., Tanouye M.A. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1988;1:421–430. doi: 10.1016/0896-6273(88)90192-4. [DOI] [PubMed] [Google Scholar]
- 12.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 13.Wallace R.A., Jared D.W., Sega M.W. Protein incorporation by isolated amphibian oocytes. 3. Optimum incubation conditions. J. Exp. Zool. 1973;184:321–333. doi: 10.1002/jez.1401840305. [DOI] [PubMed] [Google Scholar]
- 14.Frankenhaeuser B., Huxley A.F. The action potential in the myelinated nerve fiber of Xenopus laevis as computed on the basis of voltage clamp data. J. Physiol. 1964;171:302–315. doi: 10.1113/jphysiol.1964.sp007378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hille B., Woodhull A.M., Shapiro B.I. Negative surface charge near sodium channels of nerve: divalent ions, monovalent ions, and pH. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1975;270:301–318. doi: 10.1098/rstb.1975.0011. [DOI] [PubMed] [Google Scholar]
- 16.Larsson H.P., Elinder F. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 2000;27:573–583. doi: 10.1016/s0896-6273(00)00067-2. [DOI] [PubMed] [Google Scholar]
- 17.Kurata H.T., Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Billman G.E., Kang J.X., Leaf A. Prevention of sudden cardiac death by dietary pure omega-3 polyunsaturated fatty acids in dogs. Circulation. 1999;99:2452–2457. doi: 10.1161/01.cir.99.18.2452. [DOI] [PubMed] [Google Scholar]
- 19.Xiao Y.F., Sigg D.C., Leaf A. The antiarrhythmic effect of n-3 polyunsaturated fatty acids: modulation of cardiac ion channels as a potential mechanism. J. Membr. Biol. 2005;206:141–154. doi: 10.1007/s00232-005-0786-z. [DOI] [PubMed] [Google Scholar]
- 20.Boland L.M., Drzewiecki M.M. Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem. Biophys. 2008;52:59–84. doi: 10.1007/s12013-008-9027-2. [DOI] [PubMed] [Google Scholar]
- 21.Leifert W.R., McMurchie E.J., Saint D.A. Inhibition of cardiac sodium currents in adult rat myocytes by n-3 polyunsaturated fatty acids. J. Physiol. 1999;520:671–679. doi: 10.1111/j.1469-7793.1999.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elinder F., Arhem P., Larsson H.P. Localization of the extracellular end of the voltage sensor S4 in a potassium channel. Biophys. J. 2001;80:1802–1809. doi: 10.1016/S0006-3495(01)76150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 24.Jogini V., Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 2007;93:3070–3082. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freites J.A., Tobias D.J., White S.H. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bjelkmar P., Niemelä P.S., Lindahl E. Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLOS Comput. Biol. 2009;5:e1000289. doi: 10.1371/journal.pcbi.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt D., Jiang Q.X., MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y., Ramu Y., Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–829. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramu Y., Xu Y., Lu Z. Enzymatic activation of voltage-gated potassium channels. Nature. 2006;442:696–699. doi: 10.1038/nature04880. [DOI] [PubMed] [Google Scholar]
- 30.Milescu M., Bosmans F., Swartz K.J. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraser D.D., Whiting S., Cunnane S.C. Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology. 2003;60:1026–1029. doi: 10.1212/01.wnl.0000049974.74242.c6. [DOI] [PubMed] [Google Scholar]
- 32.Swartz K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon. 2007;49:213–230. doi: 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frankenhaeuser B., Vallbo A.B. Accommodation in myelinated nerve fibers of Xenopus laevis as computed on the basis of voltage clamp data. Acta Physiol. Scand. 1965;63:1–20. doi: 10.1111/j.1748-1716.1965.tb04037.x. [DOI] [PubMed] [Google Scholar]
- 34.Vallbo A.B. Accommodation related to inactivation of the sodium permeability in single myelinated nerve fibers from Xenopus laevis. Acta Physiol. Scand. 1964;61:429–444. [PubMed] [Google Scholar]
- 35.Fischer D., Li Y., Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 36.Aravindan L., Bicknell K.A., Williams A.C. Effect of acyl chain length on transfection efficiency and toxicity of polyethylenimine. Int. J. Pharm. 2009;378:201–210. doi: 10.1016/j.ijpharm.2009.05.052. [DOI] [PubMed] [Google Scholar]