

Abstract
Bronchiolitis obliterans organizing pneumonia (BOOP) and acute respiratory distress syndrome (ARDS) are two clinically and histologically distinct syndromes sharing the presence of an inflammatory and fibrotic component. Apoptosis via the Fas/Fas ligand (FasL) pathway plays an important role in the development of acute lung injury and fibrosis characteristic of these and other pulmonary inflammatory and fibrotic syndromes. We evaluated the role of apoptosis via the Fas/FasL pathway in the development of pulmonary inflammation and fibrosis in reovirus 1/L-induced BOOP and ARDS. CBA/J mice were intranasally inoculated with saline, 1 × 106 (BOOP), or 1 × 107 (ARDS) PFU reovirus 1/L, and evaluated at various days postinoculation for in situ apoptosis by TUNEL analysis and Fas/FasL expression. Our results demonstrate the presence of apoptotic cells and up-regulation of Fas/FasL expression in alveolar epithelium and in infiltrating cells during the inflammatory and fibrotic stages of both reovirus 1/L-induced ARDS and BOOP. Treatment of mice with the caspase 8 inhibitor, zIETD-fmk, inhibited apoptosis, inflammation, and fibrotic lesion development in reovirus 1/L-induced BOOP and ARDS. However, CBA/KlJms-Faslpr-cg/J mice, which carry a point mutation in the Fas cytoplasmic region that abolishes the ability of Fas to transduce an apoptotic signal, do not develop pulmonary inflammation and fibrotic lesions associated with reovirus 1/L-induced BOOP, but still develop inflammation and fibrotic lesions associated with reovirus 1/L-induced ARDS. These results suggest a differential role for the Fas/FasL apoptotic pathway in the development of inflammation and fibrotic lesions associated with BOOP and ARDS.
Chronic inflammation and tissue fibrosis are leading causes of morbidity and mortality in granulomatous and interstitial lung disorders as well as in the chronic stage of acute respiratory distress syndrome (ARDS)4 (1–6). A number of distinct clinical entities are characterized by a fibrotic component, which may be distinguished by both the location of the fibrotic lesion within the lung and the presence of an interstitial pneumonia (1–6). Acute, inflammatory responses that occur in the distal air spaces of the lung (bronchioles, alveolar ducts, and alveoli) may develop into one of a limited number of nonspecific pulmonary disorders with a fibrotic component including bronchiolitis obliterans organizing pneumonia (BOOP) (3, 5, 7–12). BOOP lesions have a patchy distribution in the lung, are frequently associated with a peribronchiolar organizing pneumonia, and are associated with the presence of lipid-laden foam cells in the alveolar spaces (5, 7–12). While the structural integrity of the alveolar ducts and walls within regions of BOOP lesion development are normally preserved, the alveolar septa may be thickened with an infiltrate of inflammatory mononuclear cells (5, 7–12). ARDS is a clinical syndrome that is characterized by diffuse alveolar damage usually secondary to an intense host inflammatory response of the lung to an infectious, noninfectious, or extrapulmonary insult (6, 13–16). ARDS is a biphasic disease that progresses from an acute exudative phase, characterized by epithelial and endothelial cellular injury, neutrophilic aggregation, the formation of hyaline membranes, alveolar edema, and hemorrhage, to an organizing (chronic) phase, characterized by regeneration and healing via resolution or repair with persistent intraalveolar and interstitial fibrosis (6, 13–18). Thus, the histopathological changes observed in ARDS can be divided into the overlapping phases of exudation, regeneration, and healing, which may be distinguished by either resolution or repair leading to fibrosis (6, 13–18).
We have previously described small animal models of BOOP and ARDS elicited by respiratory infection with reovirus serotype 1, strain Lang (reovirus 1/L) (19–23). CBA/J mice inoculated by the intranasal (i.n.) route with 1 × 106 PFU reovirus 1/L develop a clinically and histopathologically severe infection with the elicitation of a nonspecific fibrotic response of the lung that is characteristic of histopathology of human BOOP lesions (19, 22, 23). These BOOP lesions, like those observed in humans, are characterized by the patchy distribution of intraluminal plugs of granulation tissue that are chiefly composed of fibro-blast-like cells and limited amounts of collagen (19). The development of BOOP lesions in this animal model is preceded by a similarly patchy distribution of peribronchiolar mononuclear cell inflammatory lesions that progress into characteristic well-developed BOOP lesions (19). In contrast, CBA/J mice inoculated with 1 × 107 PFU reovirus 1/L develop ARDS, providing a model that recapitulates both its acute exudative phase, including the formation of hyaline membranes, as well as its regenerative phase with healing by repair, leading to intraalveolar and interstitial fibrosis (20–23). As with human ARDS, histologically, our model exhibits diffuse alveolar damage, a protein-rich edema leading to the formation of hyaline membranes, hemorrhage due to vascular leakage, and disruption of the alveolar epithelium (20, 21). Additionally, diffuse infiltrates composed primarily of neutrophils (PMNs) and macrophages are predominant, which is similar to human ARDS (20–23). The chronic phase of our ARDS model demonstrates fibrosing alveolitis, which can be observed in human ARDS patients that exhibit healing by repair, leading to fibrosis (20–23). Thus, these small animal models of BOOP and ARDS provide very relevant models for deciphering common underlying cellular, biochemical, and molecular mechanisms that may alter the pulmonary environment, leading to inflammation and fibrosis.
Apoptosis, the process of programmed cell death, plays a major regulatory role in homeostasis by maintaining a balance between cell proliferation and cell death (24). In human respiratory diseases, including ARDS and other pulmonary fibrotic disorders, apoptosis may play a role in the pathogenesis of the disease process by two distinct mechanisms: (1) via delayed leukocyte apoptosis and/or (2) through enhanced endothelial and epithelial cell apoptosis (25–28). Recent studies have suggested that apoptosis of the alveolar epithelium via the Fas/Fas ligand (FasL) pathway may be an important determinant in the pathogenesis of pulmonary fibrosis in idiopathic pulmonary fibrosis (IPF), BOOP, obliterative bronchiolitis, and in acute lung injury such as ARDS (29–37). These observations support the hypothesis that apoptosis of alveolar epithelial cells potentially through the Fas/FasL pathway is involved in the pathophysiology of at least some forms of pulmonary fibrosis. We hypothesize that apoptosis through the Fas/FasL pathway plays a crucial role in inflammation and fibrosis associated with reovirus 1/L-induced BOOP and/or ARDS, two in vivo models that closely resemble the pathophysiology of their human counterparts. We demonstrate an up-regulation of both Fas and FasL as well as a significant induction of apoptosis in situ in both reovirus 1/L-induced BOOP and ARDS. Additionally, treatment of mice with the caspase-8 and caspase-6 inhibitor, N-benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl-ketone (zIETD-fmk) inhibits apoptosis, inflammation, and fibrotic lesion development in both reovirus 1/L-induced BOOP and ARDS. Finally, CBA/KlJms-Faslpr-cg/J mice, which carry a point mutation in the Fas cytoplasmic region that abolishes the ability of Fas to transduce an apoptotic signal (38), when inoculated with reovirus 1/L did not develop fibrotic lesions associated with reovirus 1/L-induced BOOP, but still developed acute inflammation and fibrosis associated with reovirus 1/L-induced ARDS. Therefore, while expression of Fas and FasL may be involved in both reovirus 1/L-induced ARDS and BOOP, a direct role for the Fas/FasL pathway is evident only in reovirus 1/L-induced BOOP.
Materials and Methods
Animals
Four- to 5-wk-old female CBA/J mice and CBA/KlJms-Faslpr-cg/J, which carry a point mutation in the Fas cytoplasmic region that abolishes the ability of Fas to transduce an apoptotic signal (38), were obtained from The Jackson Laboratory and maintained in microisolator cages under specific pathogen-free conditions in a BL-2 facility. Cages were housed in a high efficiency particulate air-filtered animal isolator clean room (Nuaire), and all animal manipulations were performed in class II biological safety cabinets. Virally primed mice were kept physically isolated from all other experimental and stock mice. All animal protocols were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee Board.
Virus
Reovirus 1/L was originally obtained from Dr. W. Joklik (Duke University School of Medicine, Durham, NC). Third-passage gradient-purified stocks were obtained by re-cloning and amplifying parental stocks on L-929 fibroblast cells (American Type Culture Collection) (19). Following the purification of new stocks, infectious viral titers were obtained by limiting dilution on L-929 monolayers (19).
Inoculation protocol
Animals were lightly anesthetized with an i.p. injection of 0.08 cc of 20% ketamine (Vetalar, 100 mg/ml; Fort Dodge Laboratories) and 2% PromAce (acepromazine maleate, 10 mg/ml; Ayerst Laboratories) before immunization. Animals were inoculated by the i.n. application of 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) of reovirus 1/L in 30 μl (15 μl in each nostril) in sterile injectable grade 0.9% NaCl (Baxter Healthcare). Control animals were inoculated with 30 μl (15 μl in each nostril) of sterile injectable grade 0.9% NaCl. After the indicated time points, animals were sacrificed with an i.p. injection of 0.2 cc sodium nembutal (50 mg/ml; Abbott Laboratories).
Caspase inhibitor administration
Mice were injected i.p. starting on day 3 postreovirus 1/L inoculation and given daily until the completion of the time course with either the pan-caspase inhibitor N-benzylcarboxy-Val-Ala-Asp-fluoromethyl-ketone (zVAD-fmk) (Kamiya Biomedicals) or the caspase-8 (FLICE) inhibitor N-benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl-ketone (zIETD-fmk) (Kamiya Biomedicals) at 5 mg/kg in 100 μl of volume. zVAD-fmk and zIETD-fmk were prepared fresh daily as a stock solution at 1 mg/ml in 10% DMSO (in PBS). Based on a series of preliminary experiments using caspase inhibitor concentrations of 1, 5, and 7.5 mg/kg, 5 mg/kg was chosen as a final dosage after evaluating inflammatory infiltration and fibrosis via H&E staining. This dosage is consistent with other published protocols evaluating caspase inhibitors in vivo (39–45). Saline-inoculated control mice treated with either zVAD-fmk or zIETD-fmk did not demonstrate any pulmonary pathology (data not shown). Reovirus 1/L-inoculated mice were also treated with 10% DMSO (in PBS) as a carrier control. As expected, these mice demonstrated significant pulmonary pathology similar to reovirus 1/L-inoculated, untreated mice (data not shown).
Bronchoalveolar lavage (BAL)
BAL was performed in situ by injecting and withdrawing a 0.5-ml aliquot of HBSS twice through an intubation needle (21 gauge). A total of 1.5 ml of HBSS was used. BAL fluid was centrifuged at 14,000 rpm for 5 min and then was frozen at −70°C until use. Cells collected by BAL were washed three times with HBSS containing 5% FCS and 0.05% azide, and suspended at 1 × 106 cells/ml for flow cytometric analysis.
Histology
Lungs were inflated in situ with 2% paraformaldehyde (Sigma-Aldrich) by intratracheal intubation, removed, and suspended in an additional 2% paraformaldehyde for 2 h at 4°C before being embedded in paraffin. H&E staining were performed on 4-μm sections. Masson's trichrome and Sirius red staining were used to visualize collagen deposition. With Masson's trichrome the nuclei stain a dark red/purple, muscle stains red, and connective tissue, including collagen, stains blue. With Sirius red, in bright-field microscopy, collagen is red on a pale yellow background. Nuclei, if stained, are black but may often be gray or brown. In normal or saline-immunized lung sections, Sirius red staining is evident only within the walls of the bronchioles and arterioles, which contain connective tissue including collagen, while the lung alveolar airspaces are not significantly stained. To score lung inflammation and fibrosis, lung samples were screened for the following three histopathological parameters: (1) deposition of extracellular matrix; (2) leukocyte infiltration (interstitial inflammation); and (3) airway obliteration due to granulation tissue formation and/or fibrosis. Each lung section was blindly evaluated and scored on a scale of 0–3 with 0 as absent (normal), 1 as mild, 2 as moderate, and 3 as severe (22, 23). Additionally, after Sirius red staining, the severity of pulmonary fibrosis was also evaluated by quantitating the amount of red stained area (connective tissue) in reovirus 1/L-induced lung samples using ImageJ sotfware analysis (46). Results are expressed as a percentage of Sirius red content in saline-inoculated, control mice. Differences between groups were examined for statistical significance using a two-tailed Student's t test. A p value of <0.05 was considered significant. Supplemental Fig. 15 demonstrates normal lung tissue stained with H&E, Masson's trichrome, and Sirius red. Images from low (×20) and high (×40) magnification on an Olympus BX40 microscope were captured with a Polaroid digital microscope camera and edited using Adobe Photoshop 5.0 software.
Hydroxyproline (HP) assay
The extent of pulmonary fibrosis was also determined by estimating total lung collagen as reflected by the measurement of HP content of the lung. Mice were sacrificed at various time points after inoculation with reovirus 1/L and the lungs were removed, lyophilized, and weighed. Total lung HP content was assayed in duplicate as previously described (20). Differences between groups were examined for statistical significance using a two-tailed Student's t test. A p value of <0.05 was considered significant.
Flow cytometric analysis
Cells were stained for cell surface marker expression as previously described except that all cells were also stained with Cy-Chrome-conjugated rat anti-mouse-CD45 (30-F11, leukocyte common Ag, Ly-5; BD Pharmingen), and only anti-mouse-CD45+ cells were acquired for analysis (21). The following Abs were used in this analysis: CD4 (GK1.5, L3T4; R-PE-labeled; Caltag Laboratories), CD8a (53-6.7, PerCP-labeled; BD Pharmingen), CD11b/Mac-1 (M1/70, allophycocyanin-labeled; BD Pharmingen), Ly6G (RB6-8C5, Gr-1, allophycocyanin-labeled; BD Pharmingen), Fas polyclonal Ab (A20, FITC-labeled; Santa Cruz Biotechnology), and FasL (Kay10, PE-labeled; BD Pharmingen). Isotype-matched controls were run for each sample (Caltag and BD-Pharmingen). Flow cytometric analysis was performed using a dual-laser FACSCalibur flow cytometer and the CellQuest acquisition and analysis software program (BD Biosciences).
Immunohistochemistry (IHC)
IHC was performed on paraformaldehyde-fixed and paraffin-embedded lung tissue. Five- to 8-μm sequential sections were collected on poly-l-lysine-treated slides (Sigma-Aldrich). Sections were deparaffinized in xylene and dehydrated in graded alcohol. Following deparaffinization, the tissue sections were heat treated for 10 min with a Target Retrieval Solution (S1700; Dako) following the manufacturer's instructions. The slides were then immersed in 2% hydrogen peroxide to quench endogenous peroxidase for 10 min and incubated with 5% normal goat serum (Vector Laboratories) for 1 h. For IHC for Fas analysis, incubation with a rabbit anti-mouse Fas polyclonal Ab (A20, 1/100 dilution; Santa Cruz Biotechnology) for 30 min was performed followed by incubation with biotinylated anti-rabbit IgG (1/200 dilution; Santa Cruz Biotechnology) for 30 min. IHC for FasL analysis was performed using the M.O.M. Immunodetection kit PK-2200 (Vector Laboratories). Sections were incubated with M.O.M. 10% blocking reagent for 1 h to block nonspecific binding. Sections were then rinsed with PBS and incubated in M.O.M. 4% diluent, followed by incubation with mouse anti-mouse FasL mAb (Kay10, 1/50 dilution; BD Pharmingen) for 30 min. Slides were then washed and incubated with M.O.M. biotinylated anti-mouse IgG reagent (1/250 dilution; Vector Laboratories) for 10 min. For IHC for caspase-8 or caspase-3, sections were incubated overnight with either a rabbit polyclonal Ab for cleaved caspase-3 (1/200 dilution; Cell Signaling Technologies) (data not shown) or a rabbit polyclonal Ab for active/cleaved caspase-8 (1/200 dilution; Imgenex) followed by incubation with a biotinylated goat anti-rabbit IgG (1/500; Santa Cruz Biotechnology) for 1 h. Immunoreactivity was demonstrated using the ABC immunostaining system kit (ABC kit; Vector Laboratories) for Fas, caspase-8, and caspase-3 or using the avidin biotin complex immunoperoxidase system from the M.O.M. immunodetection kit PK-2200 (Vector Laboratories) for FasL analysis. Color development was completed with the 3′,3-diaminobenzidine substrate (Sigma-Aldrich), and sections were counterstained with hematoxylin. For control incubations, primary Ab was replaced by normal goat or mouse serum (Vector Laboratories). Some sections were also doubled stained with the rabbit antimouse Fas polyclonal Ab (A20) and the rabbit anti-thyroid transcription factor-1 (TTF-1) polyclonal Ab, a marker for type II epithelial cells (H190, 1/100 dilution; Santa Cruz Biotechnology). Briefly, sections were first stained with the rabbit anti-mouse Fas polyclonal Ab (A20) as described above using the 3′,3-diaminobenzidine substrate. The tissue was then blocked using an avidin/biotin blocking kit (SP-2001; Santa Cruz Biotechnology), which ensures all endogenous biotin, biotin receptors, or avidin-binding sites present in the tissue are blocked. Sections were then incubated with the anti-TTF-1 polyclonal Ab followed by incubation with biotinylated anti-rabbit IgG (1/200 dilution; Santa Cruz Biotechnology) for 30 min as described. Immunoreactivity was demonstrated using the ABC immunostaining system kit (ABC kit; Vector Laboratories) and color was completed using a Vector VIP substrate kit (SK-4600; Vector Laboratories), which produces a red/violet-colored precipitate. Sections were then counterstained with methyl green (H-3402; Vector Laboratories).
DNA Nick end labeling of tissue sections
The presence of apoptosis in lung tissue sections was assessed using the TUNEL technique (ApopTag peroxidase in situ apoptosis detection kit; Serologicals) according to the manufacturer's instructions. Briefly, slides were deparaffinized and rehydrated and the specimens were permeablized with proteinase K (20 μg/ml in PBS) for 15 min at room temperature. After inactivation of endogenous peroxidase, specimens were washed in equilibration buffer. Sections were then treated with terminal deoxynucleotidyl transferase enzyme and digoxigenin-labeled nucleotides for 60 min at 37°C. Incubation with anti-digoxigenin peroxidase Ab for 30 min at room temperature was performed and 3′,3-diaminobenzidine substrate (Sigma-Aldrich) was used for color development. Sections were counterstained with hematoxylin. In the negative control sections, the enzyme was omitted. TUNEL analysis was quantitated by counting the number of TUNEL-positive cells in eight random 225-mm2 fields from two independent experiments with three mice per time point in a blinded fashion. Differences between groups were examined for statistical significance using a two-tailed Student's t test. A p value of <0.05 was considered significant.
RNA preparation and RT-PCR analysis
Total RNA was isolated from whole lungs by guanidium denaturation utilizing TRIzol reagent (Sigma-Aldrich). For RT-PCR analysis, cDNA was prepared by reverse transcription of isolated RNA samples using the Qia-gen one-step RT-PCR kit following the manufacturer's instructions. The PCR amplifications were performed using a 20-μl reaction volume containing 4 μl of each cDNA, 6 μl of deionized water, 4 μl of 5× buffer, 4 μl of Q buffer, 0.8 ml of dNTPs, 0.8 ml of primers, and 0.4 ml of Taq polymerase. The primers used were as follows: GADPH, Forward, 5′-CAA CGA CCC CTT CAT TGA CCT C-3′, reverse, 5′-ATC CAC GAC GGA CAC ATT GG-3′; Fas, forward, 5′-TCC TTT GAT GAT TCA GGG AGT GG-3′, reverse, 5′-ATA ACA GCA CCT TGG TCA GGG C-3′; and FasL, forward, 5′-GTC AGT TTT TCC CTG TCC ATC TTG-3, reverse, 5′-TCC TAA TCC CAT TCC AAC CAG AG-3′. The conditions for amplification were as follows: GADPH, Fas, FasL, 50°C for 30 min for 1 cycle, 95°C for 15 min for 1 cycle, 94°C for 1 min for 35 cycles, 59°C for 1 min for 1 cycle, 72°C for 1 min for 1 cycle, and 72°C for 10 min for 1 cycle. PCR products were analyzed using a 1% agarose gel and stained with ethidium bromide. Band intensities on scanned gels were analyzed using the public domain National Institutes of Health Image program. Fas and FasL band intensities were compared with GAPDH controls and reported as a ratio of Fas or FasL to GAPDH. Differences between groups were examined for statistical significance using a two-tailed Student's t test. A p value of <0.05 was considered significant.
Western blot analysis
For Western blotting, lungs from saline or reovirus 1/L-treated mice were removed and homogenized using a Tissue Tearor in 1.5 ml of 25 mM Tris (pH 8.0) containing a cocktail of protease inhibitors (N-ethylmaleimide (10 mM), benzamidine (5 mM), leupeptin (50 μg/ml), pepstatin A (5 μg/ml), PMSF (2 mM)). A 1/9 volume of 20% SDS was then added to each sample and incubated at 60°C for 15 min. Total protein was determined using a modified Bradford protein assay (Sigma-Aldrich). Bio-Rad sample buffer with 5% 2-ME was added to 20 μg of total protein from lysates or 40 μg of total protein from BAL fluid and heated at 95°C for 5 min. Samples were resolved by SDS-PAGE using Ready Gels Tris-HCl 8–16% (Bio-Rad). Western blotting was then accomplished using either the rabbit anti-mouse Fas polyclonal Ab (A20; Santa Cruz Biotechnology), the mouse antimouse FasL mAb (Kay10; BD Pharmingen), or the rabbit anti-caspase-3 antiserum (H-277, 1/500 dilution; Santa Cruz Biotechnology) with the appropriate secondary Abs. The anti-caspase-3 recognizes the full-length procaspase-3 and the p17 and p20 cleaved subunits. Bands were detected using the ECL substrate kit from Pierce. The cleaved caspase-3 band intensities were compared with background controls and reported as a ratio of cleaved caspase-3 to background. Band intensities on scanned gels were analyzed using the public domain National Institutes of Health Image program. Western analysis using an anti-actin Ab (Santa Cruz Biotechnology) was also performed to demonstrate equal loading (data not shown). Differences between groups were examined for statistical significance using a two-tailed Student's t test. A p value of <0.05 was considered significant.
Results
Apoptosis as evidenced by in situ TUNEL analysis is present in both reovirus 1/L-induced BOOP and ARDS
Apoptosis plays a major regulatory role in homeostasis by maintaining a balance between cell proliferation and cell death and is implicated in the pathogenesis of human respiratory diseases, including ARDS and other pulmonary fibrotic disorders (25–28). With this in mind, we assessed the role of apoptosis in the reovirus 1/L-induced models of both ARDS and BOOP, which we developed in our laboratory. CBA/J mice were inoculated by the i.n. route with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L, sacrificed at the indicated time points, and apoptosis in situ was assessed by TUNEL analysis on paraffin-embedded lung sections. The extent of pulmonary fibrosis was also determined by estimating total lung collagen as reflected by the measurement of HP content of the lung (20–23). We have previously described the histological development of both reovirus 1/L-induced ARDS and BOOP (19–23). For reovirus 1/L-induced ARDS, at day 9 postinoculation, there is a severe pneumonia (peribronchiolar lesions with lymphocytic infiltration) with the presence of hyaline membranes, which are pathognomonic for human ARDS (20, 21). In addition to areas of mononuclear cell infiltration, edema, and hyaline membranes, the development of fibrotic lesions also occurs during the recovery phase of the infection. Between days 12 and 14 postinoculation, young, cellular fibrous polyps of collagenous tissue can be observed that develop into discrete fibroblastic polyps in the alveolar ducts (20, 21). Therefore, the peak acute inflammatory phase of the disease is represented at day 9 postinoculation while the peak fibrotic phase of the disease is represented by days 12–14 (20, 21). For reovirus 1/L-induced BOOP, at day 7 postinoculation, there is an inflammatory cellular infiltrate characterized by mononuclear cells and macrophages that demonstrates a typical focus of an active viral pneumonia (19). Fibrous plugs characteristic of BOOP fibrotic lesions are visible at day 14 postinoculation and well-formed BOOP fibrotic lesions (fibrous cellular plug present in alveolar ducts) peak at day 21 postinoculation (19). Therefore, these peak days, days 9 and 14 for reovirus 1/L-induced ARDS and days 7, 14, and 21 for reovirus 1/L-induced BOOP, were chosen as key time points to investigate the role of apoptosis in these models. For reovirus 1/L-induced ARDS, TUNEL analysis was evaluated on days 3, 5, 7, 9, 14, and 21 days postinoculation (Fig. 1A and data not shown). Peak apoptosis as measured by TUNEL expression was observed on day 9 postinoculation during the peak inflammatory response and declined by day 14 postinoculation when peak fibrotic lesions were observed as determined by HP content (Fig. 1A) (20, 21). Similarly, we compared the presence of apoptotic cells via TUNEL analysis on days 5, 7, 9 14, and 21 in the reovirus 1/L-induced BOOP (Fig. 1B and data not shown). Significant apoptosis as measured by TUNEL-positive cells was observed mainly at days 7–14, with a decrease in the number of TUNEL-positive cells observed on day 21, which corresponded to the peak fibrotic response as determined by HP content (Fig. 1B) (19, 22, 23). No apoptotic cells (<1%) were detected in control, saline-inoculated mice at any time point after inoculation (Fig. 1 and data not shown). Evidence of apoptosis in reovirus 1/L-induced BOOP and ARDS suggests that cell death may play an important role in lung tissue injury and apoptosis occurring during the regeneration phase of the diseases may explain the development and persistence of fibrosis.
FIGURE 1.
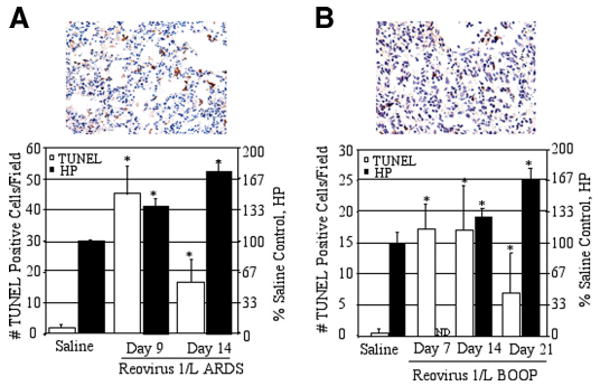
Apoptosis is evident in reovirus 1/L-induced ARDS and BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and sacrificed at the indicated time points. Apoptosis in situ was assessed using the TUNEL technique on paraffin-embedded lung sections. The extent of pulmonary fibrosis was also determined by estimating total lung collagen as reflected by HP content of the lung. A, Reovirus 1/L-induced ARDS; representative in situ TUNEL staining on day 9 postreovirus 1/L-induced ARDS (top). Histograms are TUNEL-positive cells (open) and HP content (filled) in saline-inoculated control (day 9) and on days 9 and 14 after reovirus 1/L-induced ARDS (bottom). B, Reovirus 1/L-induced BOOP; representative in situ TUNEL staining on day 14 after reovirus 1/L-induced BOOP (top). Histograms are TUNEL-positive cells (open) and HP content (filled) in saline-inoculated control (day 7) and on days 7, 14, and 21 after reovirus 1/L-induced BOOP (bottom). TUNEL data represent mean ± SD of eight 225-mm2 fields from two independent experiments with three mice per time point. HP data represent the mean ± SD of four mice per time point. Results are expressed as a percentage of HP content in saline-inoculated, control mice. ND, not determined. *, p < 0.05 compared with saline-inoculated, control mice.
Fas and FasL are expressed during reovirus 1/L-induced BOOP and ARDS
Apoptosis mediated by the Fas/FasL pathway has been suggested to play an important role in the development of acute lung injury and fibrosis (29–37). To determine whether the Fas/FasL pathway was activated in either reovirus 1/L-induced ARDS or BOOP, CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and sacrificed at the indicated key time points. Lungs were paraffin embedded and stained with an Ab to either Fas or FasL. Immunohistochemical staining of lung tissue at various time points after reovirus 1/L inoculation demonstrated significant Fas expression in alveolar epithelial cells and infiltrating cells on days 7, 9, 12, and 14 postreovirus 1/L inoculation in reovirus 1/L-induced ARDS (Fig. 2A, top left and data not shown). Positive immunostaining for FasL was detected in the alveolar epithelium and infiltrating cells on days 9 and 14 postinfection in reovirus 1/L-induced ARDS (Fig. 2A, bottom left and data not shown). Similarly, Fas and FasL expression via IHC was observed in reovirus 1/L-induced BOOP with peak Fas and FasL expression being observed between days 14 and 21 after reovirus 1/L inoculation (data not shown). In control, saline-inoculated animals, constitutive, positive immunostaining for Fas was detected only in bronchiolar epithelial cells, not in alveolar epithelial cells, while FasL immunostaining was not observed in any cells at any time point (data not shown). To determine whether alveolar type II epithelial cells expressed the Fas Ag, two-color IHC for Fas and TTF-1 was performed on lung tissue sections obtained from reovirus 1/L-induced ARDS on day 9 postinoculation (Fig. 2A, top right and bottom right). TTF-1 binds to regulatory elements located in the promoters of a number of transcriptional targets in the lung (47, 48). In the postnatal lung, TTF-1 is most abundant in type II epithelial cells in the alveolus, where it regulates surfactant protein synthesis (47, 48). ARDS-induced lung sections stained positively with the anti-Fas Ab (brown precipitate) and TTF-1 Ab, a marker for alveolar epithelial type II cells (red/purple precipitate) (Fig. 2A, top right and bottom right), demonstrating the presence of the Fas Ag on alveolar type II epithelial cells. The TTF-1 Ag stains predominantly within the nucleus of positive cells. Fig. 2A (top right) demonstrates costaining of epithelial cells for Fas Ag in a predominantly involved section of the lung, while Fig. 2A (bottom right) demonstrates costaining of epithelial cells in a predominantly uninvolved, adjacent section of the lung. An enlarged image of an alveolar epithelial cell stained positively for both TTF-1 (predominant purple staining nucleus) and Fas (predominant brown staining cytoplasm) is shown in the indicated boxed region (Fig. 2A, bottom right). These results support the conclusion that alveolar epithelial cells up-regulate Fas Ag after reovirus 1/L-induced ARDS.
FIGURE 2.
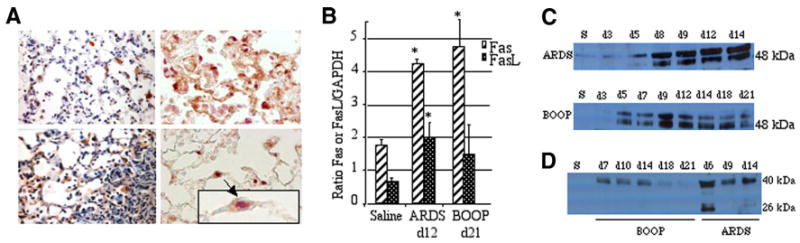
Both Fas and FasL are expressed in reovirus 1/L-induced ARDS and BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and sacrificed at the indicated time points. A, Lungs of reovirus 1/L-induced ARDS were paraffin embedded and stained with an Ab to either Fas (top left) or FasL (bottom left) on day 9. IHC for Fas and FasL is representative of four experiments with two mice per time point. Lung sections from reovirus 1/L-induced ARDS were also dual stained with an Ab to Fas (brown-precipitate) and TTF-1, a marker for alveolar epithelial type II cells (purple precipitate) (top right and bottom right) on day 9. IHC for Fas and TTF-1 is representative of four individual mice. An enlarged image of an alveolar epithelial cell (arrow) stained positively for both TTF-1 (predominant purple staining nucleus) and Fas (predominant brown staining cytoplasm) is shown in the indicated boxed region. B, RNA was prepared from whole lung tissue of either saline or reovirus 1/L-inoculated mice. Relative expression of Fas or FasL on day 12 reovirus 1/L-induced ARDS or on day 21 reovirus 1/L-induced BOOP was determined by comparing the ratio of Fas or FasL mRNA with the housekeeping gene, GAPDH. Histograms represent densitometric data from the mean ± SD autoradiogram signals from three mice per time point. *, p < 0.05 compared with saline-inoculated, control mice. C, Western analysis from whole lung lysates for protein expression of Fas overtime in reovirus 1/L-induced ARDS and BOOP; representative of two independent experiments (S, saline day 9). D, Western analysis from BAL fluid for protein expression of FasL overtime in reovirus 1/L-induced ARDS and BOOP. Representative of two independent experiments (S, saline day 9).
The induction of both Fas and FasL was also evaluated at the mRNA and protein level from whole cell extracts from total lungs. RNA was prepared from whole lung tissue from either saline- or reovirus 1/L-inoculated mice and analyzed for the expression of both Fas and FasL mRNA on day 12 postinoculation in reovirus 1/L-induced ARDS and on day 21 postinoculation in reovirus 1/L-induced BOOP via RT-PCR (Fig. 2B). An significant increase in the ratio of Fas to the housekeeping gene, GAPDH, in both reovirus 1/L-induced ARDS and reovirus 1/L-induced BOOP as compared with control, saline-inoculated mice was demonstrated (Fig. 2B). Additionally, a significant increase in FasL expression was also demonstrated in reovirus 1/L-induced ARDS as compared with control, saline-inoculated mice (Fig. 2B). Expression of Fas and FasL proteins was assessed by Western blot analysis on whole cell extracts from total lungs or BAL fluid from saline- or reovirus 1/L-inoculated mice over a full time course. Expression of Fas (48 kDa; Fig. 2C) and FasL proteins (40 kDa membrane bound, 26 kDa cleaved; Fig. 2D) were observed in whole lung lysates (Fig. 2C) and in BAL fluid (Fig. 2D) over time in reovirus 1/L-induced ARDS and BOOP.
Fas and FasL expressions on the infiltrating cells in reovirus 1/L-induced BOOP and ARDS were also determined via flow cytometry. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L, and cells recovered by BAL were analyzed for coexpression of cell surface phenotype markers and cell surface expression of Fas and FasL. Peak inflammation for reovirus 1/L-induced ARDS occurred on day 9 postinoculation, while peak inflammation for reovirus 1/L-induced BOOP occurred on day 7 postinoculation (19–21). Cells recovered from ARDS-induced mice on day 9 postinoculation (Fig. 3A) were first stained with CD4, CD8, Mac1 (macrophages and other cells), or GR1 (PMNs and macrophages), and then positive cells were gated and analyzed for the expression of either Fas or FasL. Of the CD4+ cells, 52% expressed Fas Ag, while 39% expressed FasL. Of the CD8+ cells, 37% expressed Fas, while 41% expressed FasL. Of the Mac1+ cells (macrophages; PMNs, some B cells), 45% expressed Fas, while 55% expressed FasL. Of the GR1+ cells (PMNs and macrophages), 30% expressed Fas, while 37% expressed FasL. Cells recovered from BOOP-induced mice on day 7 postinoculation (Fig. 3B) were first stained with CD4, CD8, or Mac1, and then positive cells were gated and analyzed for the expression of either Fas or FasL. Of the CD4+ cells, 44% expressed Fas, while 37% expressed FasL. Of the CD8+ cells, 34% expressed Fas, while 54% expressed FasL. Of the Mac1+ cells, 71% expressed Fas, while 70% expressed FasL. These results demonstrate a clear up-regulation of the expression of both Fas and FasL on the infiltrating cells in reovirus 1/L-induced ARDS and BOOP. Taken together, these data strongly suggest that the Fas/FasL apoptotic pathway is activated in both reovirus 1/L-induced ARDS and BOOP.
FIGURE 3.
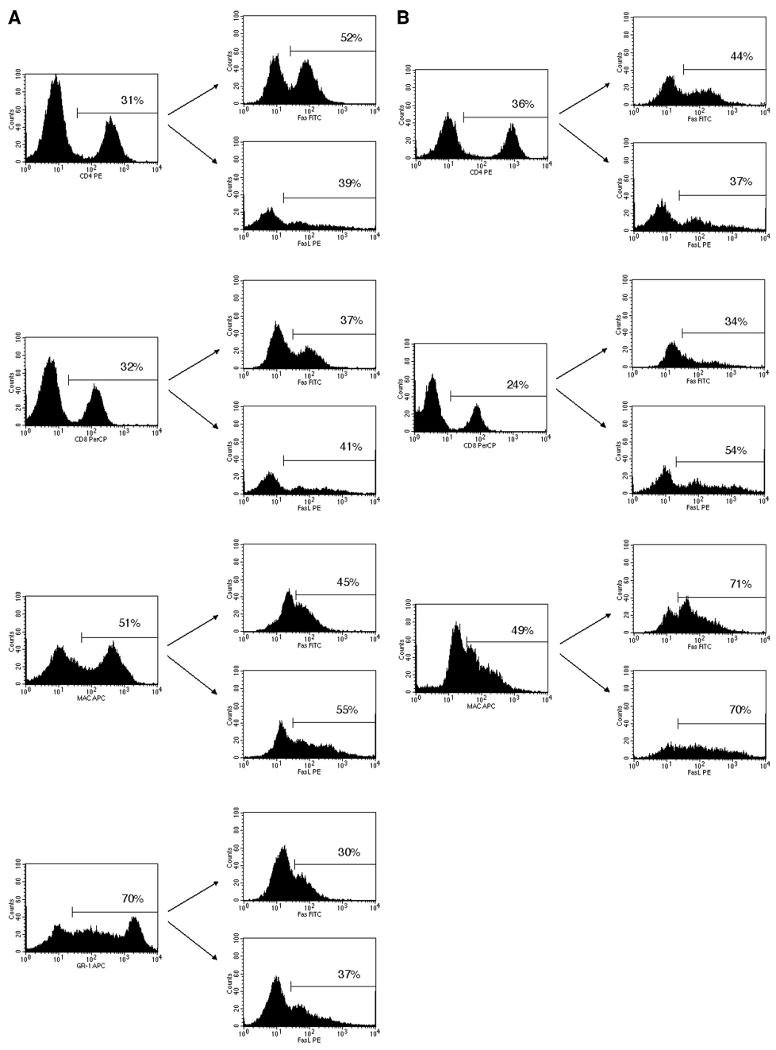
Fas and FasL are expressed on the surface of infiltrating cells in reovirus 1/L-induced ARDS and BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and cells recovered by BAL were analyzed for the coexpression of cell surface phenotype markers with surface expression of Fas and FasL. Infiltrating cells were first stained with either anti-CD4, anti-CD8, anti-Mac1, or anti-GR1. The gated positive cells were then analyzed for the expression of either Fas or FasL. A, Cells recovered from ARDS-induced mice on day 9 postinoculation. B, Cells recovered from BOOP-induced mice on day 7 postinoculation. The percentage of the total cells expressing either anti-CD4, anti-CD8, anti-Mac1, or anti-GR1 is shown in the histograms on the left. The percentage of the gated population expressing either Fas or FasL is shown in the histograms on the right. The results shown represent one of the three independent experiments demonstrating similar results.
Both inflammation and fibrosis are diminished in zIETD-fmk-treated reovirus 1/L-induced ARDS and BOOP
To demonstrate a direct role for apoptosis is the pathophysiology of reovirus 1/L-induced ARDS or BOOP, the effects of the pan-caspase inhibitor zVAD-fmk or the initiator caspase inhibitor zIETD-fmk (caspase-8 inhibitor) was determined in reovirus 1/L-induced ARDS and BOOP. Mice were inoculated i.n. with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and were left untreated or treated with 5 mg/kg of either zVAD-fmk or zIETD-fmk daily beginning on day 3 postinoculation until sacrifice. Paraffin-embedded lung sections were then stained with either H&E or Masson's trichrome to determine the extent of inflammation and fibrosis at key time points. To score lung inflammation and fibrosis, lung samples were blindly evaluated for three histopathological parameters as described in Materials and Methods and scored on a scale of 0–3, with 0 as absent (normal), 1 as mild, 2 as moderate, and 3 as severe (22, 23). In reovirus 1/L-induced ARDS, inflammation was evaluated on day 9 (Fig. 4A) and fibrosis was evaluated on day 14 (Fig. 4B). As can be observed histologically, treatment of reovirus 1/L-induced ARDS with the initiator caspase inhibitor zIETD-fmk (caspase-8 inhibitor) significantly attenuated both the inflammatory (Fig. 4A; untreated, 2.9 ± 0.2 vs zIETD-fmk-treated, 1.78 ± 0.3; p < 0.05) and fibrotic (Fig. 4B; untreated, 2.8 ± 0.4 vs zIETD-fmk-treated, 1.87 ± 0.2; p < 0.05) phase of ARDS, while treatment with the pan-caspase inhibitor zVAD-fmk was less effective (Fig. 4A (zVAD-fmk treated; 2.3 ± 0.6) vs 4B (zVAD-fmk treated; 2.2 ± 0.2)). Similarly, in reovirus 1/L-induced BOOP, treatment with zIETD-fmk was more effective in inhibiting inflammation (not shown) and fibrotic lesions formation on day 21 (Fig. 4C; untreated, 2.3 ± 0.2 vs zIETD-fmk-treated, 1.3 ± 0.7; p < 0.05) than treatment with zVAD-fmk (Fig. 4C; untreated vs zVAD-fmk treated (1.8 ± 0.4)). Supplemental Fig. 1 demonstrates normal lung tissue stained with H&E and Mas-son's trichrome.
FIGURE 4.
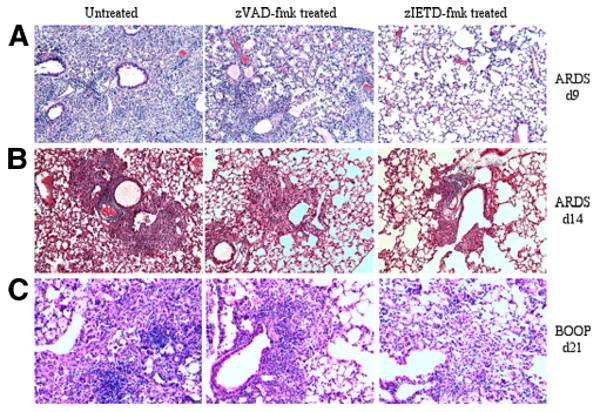
Both inflammation and fibrosis are diminished in zIETD-fmk-treated reovirus 1/L-induced ARDS or BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and were left untreated or treated with 5 mg/kg of either zVAD-fmk or zIETD-fmk daily beginning on day 3 postinoculation. A, Reovirus 1/L-induced ARDS at day 9 (H&E). B, Reovirus 1/L-induced ARDS at day 14 (Masson's trichrome). C, Reovirus 1/L-induced BOOP at day 21 (H&E). Objective magnification, ×20. Results are representative of four independent experiments evaluating four mice per time point.
To support the histological evaluation, total lung collagen to evaluate the extent of pulmonary fibrosis was evaluated by the biochemical measurement of the HP content of the lungs on day 14 for reovirus 1/L-induced ARDS and day 21 from reovirus 1/L- induced BOOP. Values were expressed as the percentage of that obtained in control mice. As shown in Fig. 5A, a 1.5-fold (BOOP) to 2.4-fold (ARDS) increase in HP accumulation in the lungs was observed after infection with reovirus 1/L as compared with saline-inoculated controls. In contrast, treatment of reovirus 1/L-induced ARDS or BOOP with zIETD-fmk resulted in a significant decrease in HP content as compared with untreated animals (Fig. 5A). An ∼1.5-fold increase in HP content was observed in zIETD-fmk treated, reovirus 1/L-induced ARDS as compared with a 2.4-fold increase in HP content in untreated reovirus 1/L-induced ARDS. In reovirus 1/L-induced BOOP, HP content was reduced to almost control, uninfected levels in zIETD-fmk treated animals (Fig. 5A). While zIETD-fmk significantly reduced HP content in reovirus 1/L-induced ARDS and BOOP, HP content was not significantly affected by treatment of either reovirus 1/L-induced ARDS or BOOP with zVAD-fmk (Fig. 5A). Therefore, our results demonstrate both histologically and biochemically that the administration of zIETD-fmk was effective at inhibiting inflammation and fibrosis associated in reovirus 1/L-induced ARDS and BOOP.
FIGURE 5.
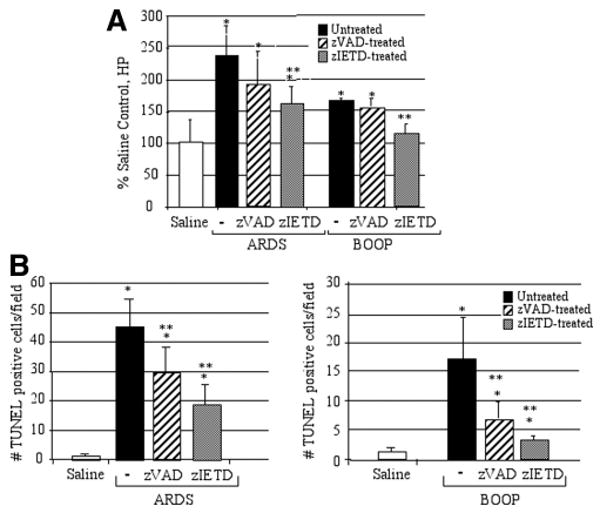
Collagen deposition and apoptosis are diminished in zIETD-fmk treated reovirus 1/L-induced ARDS or BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and were left untreated or treated with 5 mg/kg of either zVAD-fmk or zIETD-fmk daily beginning on day 3 postinoculation until sacrifice. A, The extent of pulmonary fibrosis was determined by estimating total lung collagen as reflected by the measurement of the HP content of the lung. Results are expressed as a percentage of HP content in saline-inoculated, control mice. Results shown are for day 14 ARDS and day 21 BOOP. Each data point represents the mean ± SD of six mice. *, p < 0.05 compared with saline-inoculated, control mice (day 14); **, p < 0.05 compared with reovirus 1/L-inoculated mice. B, Apoptosis in situ was assessed using TUNEL labeling on paraffin-embedded lung sections in saline (day 9 ARDS, day 14 BOOP), reovirus 1/L-induced ARDS (day 9) and reovirus 1/L-induced BOOP (day 14). TUNEL data represent mean ± SD of eight 225-mm2 fields from two independent experiments with three mice per time point. *, p < 0.05 compared with saline-inoculated, control mice; **, p < 0.05 compared with reovirus 1/L-inoculated mice.
Apoptosis as evidenced by in situ TUNEL analysis is diminished in zIETD-fmk-treated reovirus 1/L-induced ARDS and BOOP
In addition to the histological and biochemical analysis of zVAD-fmk- and zIETD-fmk-treated animals, apoptosis in situ was also evaluated via TUNEL analysis on paraffin-embedded lung sections. Peak TUNEL activity was evaluated on day 9 for reovirus 1/L-induced ARDS (Fig. 5B) and day 14 for reovirus 1/L-induced BOOP (Fig. 5B). Treatment of both reovirus 1/L-induced ARDS (Fig. 5B) and BOOP (Fig. 5B) in vivo with zIETD-fmk significantly reduced the number of TUNEL-positive cells as compared untreated animals. An ∼60% reduction in the number of TUNEL-positive cells was observed in zIETD-fmk-treated reovirus 1/L-induced ARDS (Fig. 5B) and a 75% reduction in the number of TUNEL-positive cells in reovirus 1/L-induced BOOP (Fig. 5B). While treatment of reovirus 1/L-induced ARDS or BOOP with zVAD-fmk also resulted in a reduction of TUNEL-positive cells, this reduction was less than that observed after zIETD-fmk treatment (30% in reovirus 1/L-induced ARDS (Fig. 5B) and 56% in reovirus 1/L-induced BOOP (Fig. 5B)).
Finally, we performed immunostaining for caspase-8 in untreated and zIETD-fmk-treated mice (Fig. 6A). In both zIETD-fmk-treated BOOP and ARDS on day 14 postinoculation, immunostaining for caspase-8 was significantly diminished as compared with untreated BOOP or ARDS (Fig. 6A). In control, saline-inoculated animals, positive immunostaining using the active/cleaved caspase-8 Ab was randomly detected in alveolar epithelial cells at a very low level (data not shown). Furthermore, Western analysis from whole lung lysates for protein expression of cleaved caspase-3 was determined in reovirus 1/L-induced ARDS on day 9 postinoculation and in reovirus 1/L-induced BOOP on day 14 postinoculation after treatment with either zVAD-fmk or zIETD-fmk (Fig. 6B). In reovirus 1/L-induced BOOP or ARDS, cleaved caspase-3 was significantly induced as compared with saline inoculated mice (Fig. 6B). Treatment of either reovirus 1/L-induced BOOP or ARDS with zIETD-fmk significantly reduced the induction of cleaved caspase-3 as compared with untreated reovirus 1/L-induced lungs. However, while treatment of either reovirus 1/L-induced BOOP or ARDS with zVAD-fmk reduced the expression of cleaved caspase-3, this reduction was not statistically significant. Immunostaining for cleaved caspase-3 in either zVAD-fmk- or zIETD-fmk-treated reovirus 1/L-induced BOOP and ARDS demonstrated similar results as were observed via Western analysis (data not shown). Collectively, these results confirm that treatment of mice in vivo with zIETD-fmk significantly inhibited both inflammation and fibrosis in both reovirus 1/L-induced BOOP and ARDS.
FIGURE 6.
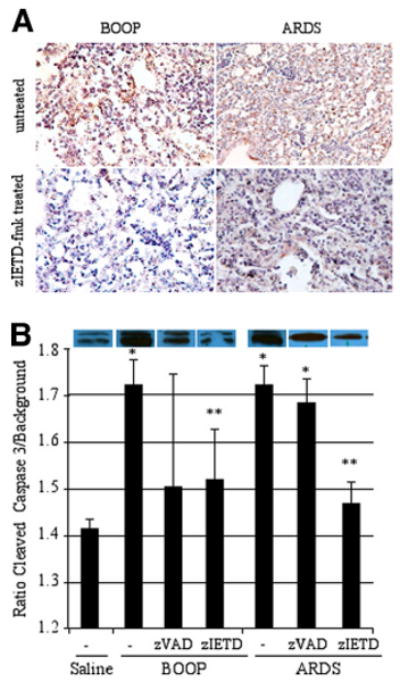
The presence of both caspase-8 and caspase-3 are diminished in zIETD-fmk reovirus 1/L-induced ARDS or BOOP. CBA/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and were left untreated or treated with 5 mg/kg of either zVAD-fmk or zIETD-fmk daily beginning on day 3 postinoculation until sacrifice. A, Lungs of reovirus 1/L-induced BOOP and ARDS on day 14 postinoculation were paraffin embedded and stained with a rabbit polyclonal Ab for active/cleaved caspase-8 in either untreated (top) or zIETD-fmk-treated (bottom) reovirus 1/L-iduced BOOP or ARDS. IHC for active/cleaved caspase-8 is representative of two experiments with two mice per time point. B, Western analysis from whole lung lysates for protein expression of cleaved caspase-3 was determined in reovirus 1/L-induced ARDS on day 9 postinoculation and in reovirus 1/L-induced BOOP on day 14 postinoculation. Relative expression of cleaved caspase-3 was determined by comparing the ratio of the cleaved caspase-3 band to background. Histograms represent densitometric data from the mean ± SD autoradiogram signals from three mice for the saline, ARDS-induced, and BOOP-induced mice and from at least four mice in the zVAD-fmk- and zIETD-fmk-treated groups. *, p < 0.05 compared with saline-inoculated, control mice; **, p < 0.05 compared with reovirus 1/L-inoculated mice.
Both inflammation and fibrosis are attenuated in reovirus 1/L-induced BOOP but not in reovirus 1/L-induced ARDS in CBA/KlJms-Faslpr-cg/J mice
To demonstrate a direct role for the Fas/FasL apoptotic pathway in either reovirus 1/L-induced ARDS or BOOP, CBA/KlJms-Faslpr-cg/J mice, which carry a point mutation in the Fas cytoplasmic region that abolishes the ability of Fas to transduce an apoptotic signal (38), were inoculated i.n. with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L. Animals were evaluated at key time points on days 9 and 14 postinoculation for reovirus 1/L-induced ARDS and on days 7 and 21 for reovirus 1/L-induced BOOP. Paraffin-embedded lung sections were stained with H&E, Masson's trichrome, or Sirius red (Fig. 7). Interestingly, inoculation of CBA/KlJms-Faslpr-cg/J mice with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L resulted in decreased mortality. Typically, 20% mortality is observed for reovirus 1/L-induced BOOP (19). However, no mortality was observed during 21 days in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP). In reovirus 1/L-induced ARDS, 45–60% mortality was observed between days 9 and 14 postinoculation with 1 × 107 PFU reovirus 1/L (20, 21). However, in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU (ARDS) reovirus 1/L, only 15% of the animals died by day 14 postinoculation. All remaining animals were sacrificed on day 14 postinoculation, so that mortality after day 14 could not be assessed. In reovirus 1/L-induced ARDS, inflammation was evaluated on day 9 and fibrosis was evaluated on day 14 (Fig. 7A). As can be observed histologically, inflammation typically observed on day 9, after reovirus 1/L-induced ARDS, was attenuated in CBA/KlJms-Faslpr-cg/J mice vs CBA/J mice (Fig. 7A; CBA/J, 2.9 ± 0.2 vs CBA/KlJms-Faslpr-cg/J, 1.8 ± 0.4; p < 0.05). However, by day 14 postinoculation, CBA/KlJms-Faslpr-cg/J mice developed similar histological features (both fibrosis and inflammation) as compared with CBA/J mice (Fig. 7A; CBA/J, 2.8 ± 0.4 vs CBA/KlJms-Faslpr-cg/J, 2.8 ± 0.5) as determined by either H&E or Masson's trichrome staining. In reovirus 1/L-induced BOOP, inflammation was evaluated on day 7 and fibrosis was evaluated on day 21 (Fig. 7B; CBA/J vs CBA/KlJms-Faslpr-cg/J). Unlike CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU (ARDS) reovirus 1/L, CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP) demonstrated an attenuation of both inflammation (Fig. 7B; 2.1 ± 0.3 vs 1.6 ± 0.4; p < 0.05) and fibrosis (Fig. 7B; 2.6 ± 0.4 vs 1.5 ± 0.4; p < 0.05).
FIGURE 7.
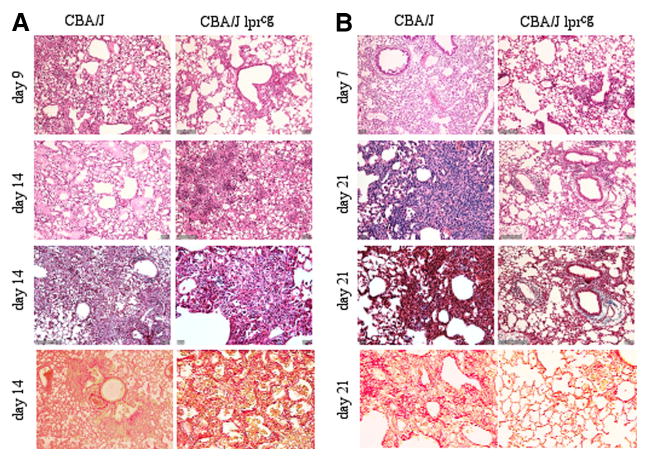
Both inflammation and fibrosis are diminished in reovirus 1/L-induced BOOP in CBA/KlJms-Faslpr-cg/J mice but not in reovirus 1/L-induced ARDS in CBA/KlJms-Faslpr-cg/J mice. CBA/J or CBA/KlJms-Faslpr-cg/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L. Paraffin-embedded lung sections were stained with H&E (first and second panels), Masson's trichrome (third panel), or Sirius red (fourth or bottom panels). A, Reovirus 1/L-induced ARDS in CBA/J or CBA/KlJms-Faslpr-cg/J mice; B, Reovirus 1/L-induced BOOP in CBA/J or CBA/KlJms-Faslpr-cg/J mice; Objective magnification, ×20. Representative of two experiments with two mice per time point.
To more clearly demonstrate the presence of fibrotic lesions histologically in either CBA/J or CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU (ARDS) or 1 × 106 PFU reovirus 1/L (BOOP), tissue sections were stained with Sirius red on day 14 for reovirus 1/L-induced ARDS and on day 21 for reovirus 1/L-induced BOOP (Fig. 7, bottom panels). With Sirius red, in bright-field microscopy, collagen is stained red on a pale yellow background. In normal or saline-immunized lung sections, Sirius red staining is evident only within the walls of the bronchioles and arterioles, which contain connective tissue including collagen, while the lung alveolar airspaces are not stained (not shown). As can be observed, significant staining for collagen via Sirius red is evident in either CBA/J or CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU (ARDS) (Fig. 7A, bottom panel; 2.8 ± 0.8 vs 2.7 ± 0.3). However, while significant staining for collagen is observed in CBA/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP), Sirius red staining is dramatically reduced in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP) (Fig. 7B, bottom panel; 2.5 ± 0.5 vs 1.5 ± 0.5, p < 0.05). The severity of pulmonary fibrosis was also evaluated by quantitating the amount of Sirius red (connective tissue) staining in reovirus 1/L-induced lung samples (day 14 for reovirus 1/L-induced ARDS and day 21 for reovirus 1/L-induced BOOP) using ImageJ sotfware analysis (Fig. 8A) (46). Results are expressed as a percentage of Sirius red staining in saline-inoculated, control mice (Fig. 8A). As shown in Fig. 8A, significant Sirius red staining was observed in both CBA/J mice and CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU reovirus 1/L (ARDS) as compared with normal saline-inoculated control mice. In CBA/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP), as expected, a significant increase in Sirius red staining over saline-inoculated mice was observed (Fig. 8A). However, while a significant increase in Sirius red staining was also observed in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP) as compared with saline-inoculated mice, this increase in staining was significantly decreased as compared with CBA/J mice inoculated with 1 × 106 PFU reovirus 1/L (BOOP) (Fig. 8A). Supplemental Fig. 1 demonstrates normal lung tissue stained with H&E, Masson's trichrome, and Sirius red.
FIGURE 8.
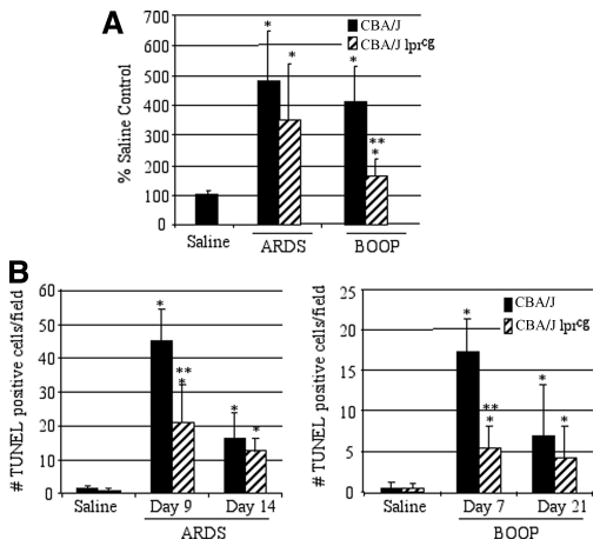
Apoptosis is reduced in reovirus 1/L-inoculated CBA/K1Jms-Faslpr-cg/J mice. CBA/J or CBA/KlJms-Faslpr-cg/J mice were i.n. inoculated with either 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L and sacrificed at the indicated time points. A, Sirius red staining in both CBA/J or CBA/KlJms-Faslpr-cg/J mice was quantitated in reovirus 1/L-induced ARDS (day 14) and in reovirus 1/L-induced BOOP (day 21) using ImageJ software. Results are expressed as the percentage of Sirius red content in saline-inoculated, control mice. Sirius red data represent mean ± SD of two experiments with two mice per time point. *, p < 0.05 compared with saline-inoculated, control mice. **, p < 0.05 compared with reovirus 1/L-inoculated mice. B, Apoptosis in situ was assessed using TUNEL labeling on paraffin-embedded lung sections. TUNEL-positive cells in saline (day 9 ARDS; day 7 BOOP) and in reovirus 1/L-induced ARDS on days 9 and 14 or in reovirus 1/L-induced BOOP on days 7 and 21 are shown. TUNEL data represent mean ± SD of eight 225-mm2 fields from two experiments with two mice per time point. *, p < 0.05 compared with saline-inoculated, control mice; **, p < 0.05 compared with reovirus 1/L-inoculated mice.
Apoptosis in situ was also evaluated via TUNEL analysis in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 106 PFU (BOOP) or 1 × 107 PFU (ARDS) reovirus 1/L (Fig. 8B). Apoptosis was significantly decreased on day 9 in CBA/KlJms-Faslpr-cg/J mice as compared with CBA/J mice inoculated with 1 × 107 PFU (ARDS) reovirus 1/L (Fig. 8B). Similarly, apoptosis was significantly decreased on day 7 in CBA/KlJms-Faslpr-cg/J mice as compared with CBA/J mice inoculated with 1 × 106 PFU (BOOP) reovirus 1/L (Fig. 8B). However, the numbers of apoptotic cells in CBA/KlJms-Faslpr-cg/J mice inoculated with 1 × 107 PFU (ARDS) reovirus 1/L were greater than those observed in either CBA/KlJms-Faslpr-cg/J mice or CBA/J mice inoculated with 1 × 106 PFU (BOOP) (Fig. 8B). Taken together, these results suggest that while expression of Fas and FasL may be involved in both reovirus 1/L-induced ARDS and BOOP, a direct role for the Fas/FasL pathway is evident only in reovirus 1/L-induced BOOP.
Discussion
BOOP and ARDS are two clinical and histologically distinct syndromes sharing the presence of both an inflammatory infiltrate and a fibrotic component (6–17). We have previously demonstrated that CBA/J mice infected with 1 × 106 PFU reovirus 1/L develop a clinically and histopathologically severe infection with the elicitation of a nonspecific fibrotic response of the lung (BOOP) (19, 22, 23). In contrast, CBA/J mice infected with 1 × 107 PFU reovirus 1/L develop ARDS and provide a model that recapitulates both its acute exudative phase, including the formation of hyaline membranes, as well as its regenerative phase, with healing by repair, leading to intraalveolar and interstitial fibrosis (20–22). Both of these models accurately recapitulate the pathophysiology of BOOP or ARDS that is observed in human patients, and thus they provide very relevant models for deciphering common underlying cellular, biochemical, and molecular mechanisms that may alter the pulmonary environment, leading to inflammation and fibrosis. Apoptosis especially via the Fas/FasL ligand pathway has been suggested to play an important role in the development of acute lung injury and fibrosis characteristic of these and other pulmonary fibrotic syndromes (25–37). Therefore, we evaluated the role of apoptosis via the Fas/FasL pathway in the development of inflammation and pulmonary fibrosis in reovirus 1/L-induced BOOP and ARDS. Our results demonstrated the presence of apoptotic cells and Fas/FasL expression in both the alveolar epithelium and in the infiltrating cells during the fibrotic stage of ARDS (days 12–14) and BOOP (days 14–21). While treatment of mice with the pan-caspase inhibitor zVAD-fmk led to a limited reduction in fibrosis, the caspase-8/6-specific inhibitor zIETD-fmk inhibited apoptosis and fibrotic lesion development in both reovirus 1/L-induced BOOP and ARDS. However, CBA/KlJms-Faslpr-cg/J mice, which carry a point mutation in the Fas cytoplasmic region that abolishes the ability of Fas to transduce an apoptotic signal (38), did not develop fibrotic lesions associated with reovirus 1/L-induced BOOP, but still developed fibrosis associated with reovirus 1/L-induced ARDS. Therefore, while the Fas/FasL apoptotic pathway may be involved in both reovirus 1/L-induced ARDS and BOOP, a direct role for the Fas/FasL pathway is evident only in reovirus 1/L-induced BOOP. Other alternative or redundant pathways in addition to the Fas/FasL pathway that also lead to apoptosis may be involved in the histopathological development of ARDS. Taken together, these results suggest a role for specific caspase inhibitors in the prevention of apoptosis, leading to a decrease in fibrosis, a strategy that may contribute to the prevention and treatment of pulmonary fibrotic diseases.
While apoptosis of the infiltrating cells and proliferating resident cells participate in the resolution of acute inflammation, failure to clear unwanted cells by apoptosis may prolong the inflammatory response (24–28). This prolonged and persistent inflammation may then lead to excessive apoptosis of resident epithelial cells, leading to the development of fibrosis (24–28). Recent studies have suggested that apoptosis of the alveolar epithelium via the Fas/FasL pathway may be an important determinant in the pathogenesis of pulmonary fibrosis in diseases such as IPF and in acute lung injury such as ARDS (29–34, 49, 50). In patients with acute lung injury, Fas and FasL were coexpressed on alveolar epithelium cells, on infiltrating inflammatory cells, and on sloughed epithelial cells in the alveolar spaces of patients who died (25–28, 51, 52). In patients with IPF, the expression of Fas and Fas signaling molecules including FAS-associated death domain (FADD), a signal transducer downstream of Fas, as well as caspase-1 and caspase-3, were up-regulated in bronchiolar and alveolar epithelial cells, and FasL was expressed in the infiltrating cells (30, 53–55). Additionally, sFas and sFasL were found in the BAL fluid of patients with ARDS, IPF, and collagen vascular disease (30, 32, 36, 51, 52, 56). Significantly higher levels of soluble Fas and FasL were observed in ARDS patients with worse clinical outcomes and in those who died (32), suggesting that sFasL and sFas released in the airspaces of patients with acute lung injury leads to activation of the Fas/FasL pathway, which contributes to the severe epithelial damage that occurs in ARDS (32, 51). In support of this hypothesis, the concentration of sFasL found in the BAL fluid of ARDS patients was sufficient to induce Fas-dependent apoptosis of primary cultures of human distal lung epithelial cells (32, 57), suggesting that sFasL is released in vivo during human disease as a “biologically active, death-inducing effector molecule capable of inducing apoptosis in Fas-susceptible target cells of the lungs” (32). In contrast, sFasL has also been found to exert an antiapoptotic effect by competing with membrane-bound FasL for binding to Fas (58). Patients with BOOP expressed both sFas and sFasL (36, 37), suggesting that perhaps elevated levels of sFas may abrogate the cytotoxicity of sFasL in BOOP patients who respond better to therapy (36). These observations have led to the suggestion that an additional cofactor or other molecule found in the BAL fluid may be required for the effect of sFasL (36).
Apoptosis and Fas/FasL pathway expression have also been described in animal models of pulmonary fibrosis, including bleomycin-induced fibrosis (29–31, 59–61). While up-regulation of Fas and FasL and excessive apoptosis of bronchiolar and alveolar epithelial cells were demonstrated after bleomycin instillation, the role of the Fas/FasL pathway remains controversial. While it has been reported that administration of the soluble form of Fas or anti-FasL Ab prevented the development of bleomycin-induced fibrosis (29, 30), suggesting a role for Fas/FasL pathway in fibrotic lesion development, the results of bleomycin-induced fibrosis in Fas- or FasL-deficient mice remain equivocal. In one study, both C3H-lpr/lpr (lpr) (Fas deficient) and C3H-gld/gld (gld) (FasL deficient) mice were resistant toward bleomycin-induced fibrosis, suggesting an essential role for the Fas/FasL pathway in the development of pulmonary fibrosis (29, 30). However, another study demonstrated that lpr and gld mice were just as susceptible to bleomycin-induced fibrosis as were wild-type mice (60), suggesting that the Fas/FasL pathway is not a prerequisite for the development of bleomycin-induced fibrosis. While a clear reason for this discrepancy was not given, it was suggested that the differences in the strain of mice used (C3H vs C57BL/6) as well as the timing of the analysis of apoptosis could be responsible for this discrepancy (29, 30, 60). In models of acute lung injury including Ig deposition, successive exposure to hemorrhage shock and cecal ligation and puncture (CLP), septic shock, and cadmium-induced lung injury, Fas was up-regulated in alveolar and inflammatory cells, FasL-positive inflammatory cells were present in the airspaces, intratracheal administration of an anti-Fas blocking Ab attenuated lung injury, and measures of lung injury parameters were found to be reduced in both lpr and gld mice (33, 34, 62– 64). Therefore, strategies to inhibit Fas-mediated epithelial apoptosis may preserve epithelial function in patients who develop acute lung injury. A recent report investigated the role of apoptotic cell suicide in lung tissues of mice with CLP-induced polymicrobial sepsis (65). These authors demonstrated that synthetic double-stranded small interfering RNA (siRNA) targeting FADD introduced into the tissues of the whole animal suppressed apoptosis induction in septic lungs, prevented acute lung injury development, and dramatically improved the survival of CLP mice (65). A similar critical role of epithelial cell apoptosis via activation of the Fas/FasL pathway has been also described in a model of hyperoxia-induced acute lung injury in Pneumocystis murina-infected mice (66). These observations support the hypothesis that apoptosis of alveolar epithelial cells potentially through the Fas/FasL pathway is involved in the pathophysiology of acute lung injury and pulmonary fibrosis.
To directly investigate the role of the Fas/FasL pathway in the models of reovirus 1/L-induced BOOP and ARDS developed in our laboratory (19–23), we used the CBA/KlJms-Faslpr-cg/J strain of mice, which express a spontaneous mutation, lprcg (38). Unlike the lpr strain, lprcg mice express full-length Fas mRNA as abundantly as do wild-type mice, but due to a point mutation in the Fas cytoplasmic region, Fas is unable to transduce an apoptotic signal (38). In the reovirus 1/L model system, we have clearly demonstrated that in both reovirus 1/L-induced ARDS and BOOP, (1) apoptosis of the alveolar epithelium is evident via TUNEL analysis; (2) Fas and FasL are up-regulated in situ in alveolar epithelium and in cells of the inflammatory infiltrate; (3) sFasL is found in the BAL fluid; (4) treatment of mice with zIETD-fmk (caspase-8 inhibitor) is more effective at attenuating inflammation and fibrosis than is treatment with the pan-caspase inhibitor zVAD-fmk; and (5) Fas-deficient animals are resistant to reovirus 1/L-induced BOOP but are still susceptible to reovirus 1/L-induced ARDS. These data would support a direct role for the Fas/FasL pathway in the development of fibrotic lesions associated with reovirus 1/L-induced BOOP but not reovirus 1/L-induced ARDS. These data are consistent with our finding that unlike reovirus 1/L-induced ARDS, reovirus 1/L-induced BOOP is dependent on the presence of T cells (22, 23), suggesting that the predominant infiltration of T cells that express FasL in reovirus 1/L-induced BOOP is responsible for damage to Fas-expressing alveolar epithelial cells, leading to fibrosis. To support this conclusion, we have previously demonstrated in reovirus 1/L-induced BOOP that depletion of either CD4+ or CD8+ T cells before reovirus 1/L infection inhibited fibrotic lesion development (23), and neonatally thymectomized CBA/J mice, which lack mature peripheral T cells, when infected with 1 × 106 PFU reovirus 1/L (BOOP) do not develop intraalveolar fibrosis associated with BOOP (22). In contrast, thymectomized CBA/J mice when infected with 1 × 107 PFU reovirus 1/L (ARDS) still develop all of the histological features of ARDS, including fibrosis (22). Therefore, excessive Fas/FasL interaction mediated through the interaction of infiltrating T cells with alveolar epithelial cells may directly lead to pulmonary fibrosis in reovirus 1/L-induced BOOP. This is supported by other studies demonstrating that intratracheal administration of Fas-activating Ab mimicking Fas/FasL cross-linking caused alveolar epithelial apoptosis and pulmonary fibrosis, which was diminished in lpr mice (67, 68), and selective inactivation of Fas in T cells caused massive leukocyte infiltration in the lungs together with increased inflammatory cytokine production and pulmonary fibrosis resembling idiopathic pulmonary fibrosis in humans (69). Additionally, chimeric mice lacking Fas in either myeloid cells (macrophages) or nonmyeloid cells (epithelial cells) demonstrated that only animals expressing Fas in nonmyeloid cells showed significant increases in lung injury parameters, suggesting that Fas-mediated lung injury requires the expression of Fas on nonmyeloid cells of the lung (70, 71). Since reovirus 1/L can infect a variety of cell types, including epithelial cells, it was essential to determine whether epithelial cell apoptosis induced by reovirus 1/L infection contributed to the pathophysiology of reovirus 1/L-induced BOOP or ARDS. In these experiments, we did not observe significant TUNEL activity at the height of reovirus 1/L replication in vivo (days 1–3) (Fig. 1, data not shown), suggesting that epithelial cell apoptosis induced by reovirus 1/L is limited (19, 20). These data are consistent with published data on apoptosis induced by reovirus 1/L infection. Reovirus 1/L infection occurs following receptor-mediated endocytosis after the virions bind to cell surface molecules, including junctional adhesion molecule 1 (JAM1), and to other uncharacterized cell surface carbohydrate moieties, but not to sialic acid (72). Reovirus strains differ in their capacity to induce apoptosis, and the reovirus T3 prototype strain Dearing (T3D) induces apoptosis much more efficiently both in vivo and in vitro than does reovirus 1/L, since virion binding to both junctional adhesion molecule 1 and sialic acid is essential for optimal expression of apoptosis in infected cells that does not occur with reovirus 1/L (72). Additionally, it has been demonstrated that the ability of reovirus strains to infect and propagate in tissue culture cells does not correlate with its capacity to induce apoptosis (72). These observations suggest that direct apoptosis of alveolar epithelial cells due to infection with reovirus 1/L is minimal. Therefore, most apoptosis observed in both reovirus 1/L-induced ARDS and BOOP is mediated through the interaction of FasL-expressing infiltrating cells with Fas-expressing alveolar epithelial cells or through other apoptotic pathways.
While the Fas/FasL pathway is up-regulated in reovirus 1/L-induced ARDS, it may represent only one potential mechanism leading to alveolar epithelial cell damage and eventual fibrosis in reovirus 1/L-induced ARDS. This hypothesis is consistent with our observation that CBA/KlJms-Faslpr-cg/J mice, which lack a functional Fas protein, when inoculated with 1 × 107 PFU reovirus 1/L still develop ARDS. We also demonstrate that the caspase-8 inhibitor zIETD-fmk, but not the caspase-3 inhibitor zVAD-fmk, significantly attenuated inflammation and fibrosis in both reovirus 1/L-induced ARDS and BOOP. This was evident through both a reduction in histopathology and in lung HP content as a measure of fibrosis. However, note that the caspase-8 inhibitor zIETD-fmk was more effective at reducing fibrosis in BOOP than in ARDS. While zIETD-fmk significantly reduced fibrosis as measured by HP content as compared with untreated reovirus 1/L-induced ARDS, the HP content in zIETD-fmk reovirus 1/L-induced ARDS was still significantly different than in control, saline-inoculated lungs. This observation is also consistent with a greater reduction in apoptotic cells in reovirus 1/L-induced BOOP after treatment with zIETD-fmk as compared with reovirus 1/L-induced ARDS. In fact, the numbers of TUNEL-positive cells in zIETD-fmk-treated reovirus 1/L-induced ARDS are greater than the numbers of TUNEL-positive cells in untreated reovirus 1/L-induced BOOP. These data, in concert with the differential response in CBA/KlJms-Faslpr-cg/J to reovirus 1/L-induced BOOP vs ARDS, suggest an essential role for the Fas/FasL pathway in reovirus 1/L-induced BOOP but not in reovirus 1/L-induced ARDS.
Multiple reports have demonstrated that administration of the angiotensin-converting enzyme inhibitor captopril, which inhibits Fas-induced apoptosis in vitro, or the caspase inhibitor zVAD-fmk inhibited apoptosis and prevented fibrotic lesion development in bleomycin-treated animals (42, 43, 59, 73) or in sepsis-induced acute lung injury (41). However, it has been suggested that a high degree of caspase-3 inhibition may be necessary to completely block apoptotic cell death in a CLP-induced sepsis model system (74). Therefore, high and persistent levels of caspase inhibition may be needed clinically, and zVAD-fmk may be a low-potency caspase-3 inhibitor (74). While treatment of reovirus 1/L-induced ARDS and BOOP with zVAD-fmk inhibited caspase activation, as demonstrated by a decrease in the amount of cleaved caspase-3 in treated animals, we did not observe a significant reduction in inflammation or fibrosis in either model. Perhaps the need in our model for persistent and complete caspase blockage was not achieved and that the use of a more potent caspase-3 inhibiter such as M867 may prove effective (74). In a model of liver injury, IETD-CHO, a caspase-8 inhibitor, effectively prevented hepatocellular apoptosis, hemorrhage, and liver failure after Fas receptor activation by inhibiting not only caspase-8 activation but cytochrome c release by inhibiting the activation of caspase-3 and caspase-9 (39, 40). These authors suggested that Fas may rely solely on caspase-8 activation and the mitochondria to activate caspase-3, which can process more procaspase-8 and thus propagate the amplification of the apoptotic signal (39, 40). Additionally, it was recently reported that silencing of Fas but not caspase-8 in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhage shock and sepsis, an indirect pathway to acute lung injury, suggesting a pathophysiological role for Fas activation in nonpulmonary shock-induced acute lung injury (75). These data are not contradictory to what we present herein on reovirus 1/L-induced ARDS. In our model system i.n. inoculation of reovirus 1/L is a direct model of acute lung injury as opposed to an indirect lung injury such as that caused by extrapulmonary insult included in some models of sepsis (75), and therefore the role of the Fas/FasL pathway may not be as significant in reovirus 1/L-induced ARDS. Therefore, other alternative or redundant pathways that also lead to apoptosis may be involved in the histopathological development of ARDS in addition to the Fas/FasL pathway.
Alternative pathways, which lead to apoptosis induction or an imbalance between apoptosis-inducible and inhibitory genes, have also been suggested to be activated in bleomycin-induced fibrosis and other models of acute lung injury (31, 76–78). These include the induction of proapoptotic cytokines such as TNF-α and TGFβ (62, 63, 77, 78). TGFβ1 and TNF-α act as enhancers of Fas-mediated apoptosis of lung epithelial cells (79–82). We have also demonstrated that TGFβ1 is up-regulated in both reovirus 1/L-induced ARDS and BOOP and that neonatally thymectomized mice that develop ARDS after reovirus 1/L inoculation express significant levels of TGFβ1 and TNF-α (21–23), suggesting that apoptotic pathways regulated by TGF/β and TNF-α may be activated in reovirus 1/L-induced ARDS. The proapoptotic Bcl-2 family member Bid was also shown to be required for the development of pulmonary fibrosis in bleomycin where mice lacking Bid exhibited significantly less pulmonary fibrosis despite similar levels of TGFβ in BAL fluid (76). The authors suggested that TGFβ overexpression in the lung leads to epithelial cell apoptosis and tissue fibrosis (77) through a Bax-dependent Bid-activated pathway involving matrix metalloproteinase-12 (78). Furthermore, Bak, a member of the Bcl-2 family, has recently been identified as a proapoptotic factor in the TNF-α-induced apoptotic pathway (TRAIL) in caspase 3-deficient cells, demonstrating a caspase 3-independent function of Bak in the TNF-α-induced apoptotic pathway (83). These data are consistent with our observation that while CBA/KlJms-Faslpr-cg/J mice, which lack a functional Fas protein, when inoculated with 1 × 107 PFU reovirus 1/L develop ARDS, a significant attenuation of fibrosis is observed in reovirus 1/L-induced ARDS after treatment with the caspase-8 inhibitor zIETD-fmk. These results suggest that pathways other than the Fas/FasL pathway, which can activate caspase-8 including TRAIL apoptotic pathways or other pathways of FADD activation, such as activation of the TNF receptor I (p55), might also be responsible for the differential results observed in reovirus 1/L-induced ARDS in zVAD-fmk-treated mice vs CBA/KlJms-Faslpr-cg/J mice. In support of this assumption, in ARDS patients, TRAIL levels in the BAL fluid were correlated with clinical severity and the presence of neutrophils, which are also the major infiltrating cells in reovirus 1/L-induced ARDS (20, 84). Finally, the death of epithelial cells and other cells in the lungs in acute lung injury can occur not only by regulated (apoptosis) but also in conjunction with non-regulated mechanisms of cell death, including cell necrosis (25, 85–87). In fact, the broad-spectrum caspase inhibitor zVAD-fmk has also been demonstrated to modulate the major types of cell death, where the addition of zVAD-fmk blocks apoptotic cell death, sensitizes cells to necrotic cell death, and induces autophagic cell death (88). Therefore, the inability of zVAD-fmk to inhibit the development of either reovirus 1/L-induced ARDS or BOOP may be due to the activation or enhancement of these other pathways of cell death. We are currently exploring these and other additional apoptotic pathways in the multifactorial disease process associated with reovirus 1/L-induced ARDS.
In conclusion, our data strongly support the hypothesis that apoptosis of alveolar epithelial cells potentially through the Fas/FasL pathway is involved in the pathophysiology of pulmonary fibrosis associated with reovirus 1/L-induced BOOP. Apoptosis occurring during the regeneration phase of the diseases may help explain the development and persistence of fibrosis in these model systems. While blockage of the Fas/FasL apoptotic pathway may be a useful therapeutic in the treatment of reovirus 1/L-induced BOOP, it may be less important in reovirus 1/L-induced ARDS, where the involvement of multiple apoptotic pathways may be significant. These data suggest that while the Fas/FasL pathway may play a role in eliminating injured epithelial cells that need to be replaced by epithelial renewal, the severity of lung injury may determine whether epithelial renewal is overshadowed by a fibrotic response. Therefore, the underlying insult leading to the pathophysiology of acute lung injury and fibrosis will be important in determining the pulmonary fibrotic response and its response to therapeutic interventions.
Supplementary Material
Acknowledgments
We thank Margaret Romano for her assistance with the preparation of histological sections and Kalyn Brown for her assistance with the RT-PCR analysis.
Footnotes
This work was supported by U.S. Public Health Service Grant AI R01 40175 (to L.L.) and a grant from Eli Lilly & Co.
Abbreviations used in this paper: ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; BOOP, bronchiolitis obliterans organizing pneumonia; CLP, cecal ligation and puncture; FADD, Fas-associated death domain; FasL, Fas ligand; HP, hydroxyproline; IHC, immunohistochemistry; i.n., intranasal; IPF, idiopathic pulmonary fibrosis; reovirus 1/L, reovirus serotype 1, strain Lang; TTF-1, thyroid transcription factor-1; zIETD-fmk, N-benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethyl-ketone; zVAD-fmk, N-benzylcarboxy-Val-Ala-Asp-fluoromethyl-ketone.
The online version of this article contains supplemental material.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Cooper JA., Jr Pulmonary fibrosis: pathways are slowly coming into light. Am J Resp Cell Mol Biol. 2000;22:520–523. doi: 10.1165/ajrcmb.22.5.f185. [DOI] [PubMed] [Google Scholar]
- 2.Kuwano K, Hagimoto N, Hara N. Molecular mechanisms of pulmonary fibrosis and current treatment. Curr Mol Med. 2001;1:551–573. doi: 10.2174/1566524013363401. [DOI] [PubMed] [Google Scholar]
- 3.Katzenstein AL, Mukhopadhyay S, Myers JL. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol. 2008;39:1275–1294. doi: 10.1016/j.humpath.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Afshar K, Sharma OP. Interstitial lung disease: trials and tribulations. Curr Opin Pulm Med. 2008;14:427–433. doi: 10.1097/MCP.0b013e3283043e58. [DOI] [PubMed] [Google Scholar]
- 5.Drakopanagiotakis F, Polychronopoulos V, Judson MA. Organizing pneumonia. Am J Med Sci. 2008;335:34–39. doi: 10.1097/MAJ.0b013e31815d829d. [DOI] [PubMed] [Google Scholar]
- 6.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 7.Epler GR, Colby TV, McLoud TC, Carrington CB, Gaensler EA. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985;312:152–158. doi: 10.1056/NEJM198501173120304. [DOI] [PubMed] [Google Scholar]
- 8.Epler GR. Bronchiolitis obliterans organizing pneumonia: definition and clinical features. Chest. 1992;102(Suppl):2S–6S. doi: 10.1378/chest.102.1_supplement.2s. [DOI] [PubMed] [Google Scholar]
- 9.Colby TV. Pathologic aspects of bronchiolitis obliterans organizing pneumonia. Chest. 1992;102(Suppl):38S–43S. doi: 10.1378/chest.102.1_supplement.38s. [DOI] [PubMed] [Google Scholar]
- 10.Epler GR. Bronchiolitis obliterans organizing pneumonia. Arch Int Med. 2001;161:158–164. doi: 10.1001/archinte.161.2.158. [DOI] [PubMed] [Google Scholar]
- 11.Schlesinger C, Koss MN. The organizing pneumonias: an update and review. Curr Opin Pulm Med. 2005;11:422–430. doi: 10.1097/01.mcp.0000175521.41729.07. [DOI] [PubMed] [Google Scholar]
- 12.Cordier JF. Cryptogenic organising pneumonia. Eur Respir J. 2006;28:422–446. doi: 10.1183/09031936.06.00013505. [DOI] [PubMed] [Google Scholar]
- 13.Corrin B. Diffuse alveolar damage, the pathological basis of adult respiratory distress syndrome. In: Corrin B, editor. Pathology of the Lung. Churchill Livingstone; London: 2000. pp. 121–143. [Google Scholar]
- 14.Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med. 2004;41:460–470. doi: 10.7326/0003-4819-141-6-200409210-00012. [DOI] [PubMed] [Google Scholar]
- 15.Castro CY. ARDS and diffuse alveolar damage: a pathologist's perspective. Semin Thorac Cardiovasc Surg. 2006;18:13–19. doi: 10.1053/j.semtcvs.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 17.Ware LB. Clinical year in review III: asthma, lung transplantation, cystic fibrosis, acute respiratory distress syndrome. Proc Am Thorac Soc. 2007;4:489–493. doi: 10.1513/pats.200707-102TT. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda Y, Ishizaki M, Masuda Y, Kimura G, Kawanami O, Masugi Y. The role of intra-alveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol. 1987;126:171–181. [PMC free article] [PubMed] [Google Scholar]
- 19.Bellum SC, Dove D, Harley RA, Greene WB, Judson MA, London L, London SD. Respiratory reovirus 1/L induction of intraluminal fibrosis: a model for the study of bronchiolitis obliterans organizing pneumonia. Am J Pathol. 1997;150:2243–2254. [PMC free article] [PubMed] [Google Scholar]
- 20.London L, Majeski EI, Pantilia MK, Harley RA, London SD. Respiratory reovirus 1/L induction of diffuse alveolar damage: a model of acute respiratory distress syndrome. Exp Mol Pathol. 2002;72:24–36. doi: 10.1006/exmp.2001.2414. [DOI] [PubMed] [Google Scholar]
- 21.London L, Majeski EI, Altman-Hamamdzic S, Enockson C, Pantilia MK, Harley RA, London SD. Respiratory reovirus 1/L induction of diffuse alveolar damage: pulmonary fibrosis is not modulated by corticosteroids in acute respiratory distress syndrome in mice. Clin Immunol. 2002;103:284–295. doi: 10.1006/clim.2002.5214. [DOI] [PubMed] [Google Scholar]
- 22.Majeski EI, Harley RA, Bellum S, London SD, London L. Differential role for T cells in the development of fibrotic lesions associated with reovirus 1/L-induced bronchiolitis obliterans organizing pneumonia versus acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2003;28:208–217. doi: 10.1165/rcmb.4891. [DOI] [PubMed] [Google Scholar]
- 23.Majeski EI, Paintlia MK, Lopez AD, Harley RA, London SD, London L. Respiratory reovirus 1/L induction of intraluminal fibrosis, a model of bronchiolitis obliterans organizing pneumonia, is dependent on T lymphocytes. Am J Pathol. 2003;63:1467–1479. doi: 10.1016/S0002-9440(10)63504-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, Harrington EO, Rounds S. Apoptosis and lung injury. Keio J Med. 2005;54:184–189. doi: 10.2302/kjm.54.184. [DOI] [PubMed] [Google Scholar]
- 26.Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc. 2005;2:214–220. doi: 10.1513/pats.200504-031AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Souza PM, Lindsay MA. Apoptosis as a therapeutic target for the treatment of lung disease. Curr Opin Pharmacol. 2005;5:232–137. doi: 10.1016/j.coph.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Shu R, Filippatos G, Uhal BD. Apoptosis in lung injury and remodeling. J Appl Physiol. 2004;97:1535–1542. doi: 10.1152/japplphysiol.00519.2004. [DOI] [PubMed] [Google Scholar]
- 29.Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, Suda T, Kunitake R, Maeyama T, Miyazaki H, Hara N. Essential roles for the Fas-Fas ligand pathways in the development of pulmonary fibrosis. J Clin Invest. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kunitake R, Kaneko Y, Hara N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell Mol Biol. 1999;20:53–60. doi: 10.1165/ajrcmb.20.1.2941. [DOI] [PubMed] [Google Scholar]
- 31.Kuwano K, Hagimoto N, Tanaka T, Kawasaki M, Kunitake R, Miyazaki H, Kaneko Y, Matsuba T, Maeyama T, Hara N. Expression of apoptosis regulatory genes in epithelial cells in pulmonary fibrosis in mice. J Pathol. 2000;190:221–229. doi: 10.1002/(SICI)1096-9896(200002)190:2<221::AID-PATH495>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 32.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury. J Immunol. 1999;163:2217–2225. [PubMed] [Google Scholar]
- 33.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, Nakajima H. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med. 2001;163:762–769. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- 34.Wang HC, Shun CT, Hsu SM, Kuo SH, Luh KT, Yang PC. Fas/Fas ligand pathway is involved in the resolution of type II pneumocyte hyperplasia after acute lung injury: evidence from a rat model. Crit Care Med. 2002;30:1528–1534. doi: 10.1097/00003246-200207000-00022. [DOI] [PubMed] [Google Scholar]
- 35.Hansen PR, Holm AM, Svendsen UG, Olsen PS, Andersen CB. Apoptosis and formation of peroxynitrite in the lungs of patients with obliterans bronchiolitis. J Heart Lung Transplant. 2000;19:160–166. doi: 10.1016/s1053-2498(99)00115-1. [DOI] [PubMed] [Google Scholar]
- 36.Kuwano K, Kawasaki M, Maeyama T, Hagimoto N, Nakamura N, Shirakawa K, Hara N. Soluble form of Fas and Fas ligand in BAL fluid from patients with pulmonary fibrosis and bronchiolitis obliterans organizing pneumonia. Chest. 2000;118:451–458. doi: 10.1378/chest.118.2.451. [DOI] [PubMed] [Google Scholar]
- 37.Lappi-Blanco E, Soini Y, Paakko P. Apoptotic activity is increased in the newly formed fibromyxoid connective tissue in bronchiolitis obliterans organizing pneumonia. Lung. 1999;177:367–376. doi: 10.1007/pl00007654. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzawa A, Moriyama T, Kaneko T, Tanaka M, Kimura M, Ikeda H, Katagiri T. A new allele of the lpr locus. lprcg that complements the gld gene in induction of lymphadenopathy in the mouse. J Exp Med. 1990;171:519–531. doi: 10.1084/jem.171.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a post-mitochondrial processing of caspase-8. Toxicol Sci. 2000;58:109–117. doi: 10.1093/toxsci/58.1.109. [DOI] [PubMed] [Google Scholar]
- 40.Bajt ML, Vonderfecht SL, Jaeschke H. Differential protection with inhibitors of caspase-8 and caspase-3 in murine models of tumor necrosis factor and Fas receptor-mediated hepatocellular apoptosis. Toxicol Appl Pharmacol. 2001;175:243–252. doi: 10.1006/taap.2001.9242. [DOI] [PubMed] [Google Scholar]
- 41.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol. 2000;157:597–603. doi: 10.1016/S0002-9440(10)64570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal UD. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am J Physiol. 2000;279:L143–L151. doi: 10.1152/ajplung.2000.279.1.L143. [DOI] [PubMed] [Google Scholar]
- 43.Kuwano K, Kunitake R, Maeyama T, Hagimoto N, Kawasaki M, Matsuba T, Yoshimi M, Inoshima I, Yoshida K, Hara N. Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am J Physiol. 2001;280:L316–L325. doi: 10.1152/ajplung.2001.280.2.L316. [DOI] [PubMed] [Google Scholar]
- 44.Hotchkiss RS, Chang KC, Swanson PE, Tinsley KW, Hui JJ, Klender P, Xanthoudakis S, Roy S, Black C, Grimm E, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1:496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- 45.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malkusch W, Rehn B, Bruch J. Advantages of Sirius red staining for quantitative morphometric collagen measurements in lungs. Exp Lung Res. 1995;21:67–77. doi: 10.3109/01902149509031745. [DOI] [PubMed] [Google Scholar]
- 47.Bohinski RJ, DiLauro R, Whitsett JA. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol Cell Biol. 1994;14:5671–5681. doi: 10.1128/mcb.14.9.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.deFelice M, Silberschmidt D, DiLauro R, Xu Y, Wert SE, Weaver TE, Bachurski CJ, Clark JC, Whitsett JA. TTF-1 phosphorylation is required for peripheral lung morphogenesis, perinatal survival, and tissue-specific gene expression. J Biol Chem. 2003;278:35574–35583. doi: 10.1074/jbc.M304885200. [DOI] [PubMed] [Google Scholar]
- 49.Antoniou KM, Pataka A, Bouros D, Siafakas NM. Pathogenetic pathways and novel pharmacotherapeutic targets in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2007;20:453–461. doi: 10.1016/j.pupt.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Uhal BD. Epithelial apoptosis in the initiation of lung fibrosis. Eur Respir J. 2003;44:7S–9S. doi: 10.1183/09031936.03.00000303. [DOI] [PubMed] [Google Scholar]
- 51.Mecklenburgh K, Murray J, Brazil T, Ward C, Rossi AG, Chilvers ER. Role of neutrophil apoptosis in the resolution of pulmonary inflammation. Monaldi Arch Chest Dis. 1999;54:345–349. [PubMed] [Google Scholar]
- 52.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:237–243. doi: 10.1164/ajrccm.161.1.9810007. [DOI] [PubMed] [Google Scholar]
- 53.Maeyama M, Kuwano K, Kawasaki M, Kunitaki R, Hagimoto N, Matsuba T, Yoshimi M, Inoshima I, Yoshida K, Hara N. Upregulation of Fassignaling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur Respir J. 2001;17:180–189. doi: 10.1183/09031936.01.17201800. [DOI] [PubMed] [Google Scholar]
- 54.Barbas-Filho JV, Ferreira MA, Sesso A, Kairalla RA, Carvalho CRRR, Capelozzi VL. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis and usual interstitial pneumonia. J Clin Pathol. 2001;54:132–138. doi: 10.1136/jcp.54.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. 2002;161:1783–1796. doi: 10.1016/S0002-9440(10)64455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamura M, Matute-Bello G, Liles WC, Hayashi S, Kajikawa O, Lin SM, Frevert CW, Martin TR. Differential response of human lung epithelial cells to fas-induced apoptosis. Am J Pathol. 2004;164:1949–1958. doi: 10.1016/S0002-9440(10)63755-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, Tschopp J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–1213. doi: 10.1084/jem.187.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand in mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol. 1997;16:91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 60.Aoshiba K, Yasui S, Tamaoki J, Nagai A. The Fas/Fas-ligand system is not required for bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2000;162:695–700. doi: 10.1164/ajrccm.162.2.9907012. [DOI] [PubMed] [Google Scholar]
- 61.Golan-Gerstl R, Wallach-Dayan SB, Amir G, Breuer R. Epithelial cell apoptosis by fas ligand-positive myofibroblasts in lung fibrosis. Am J Respir Cell Mol Biol. 2007;36:270–275. doi: 10.1165/rcmb.2006-0133OC. [DOI] [PubMed] [Google Scholar]
- 62.Neff TA, Guo RF, Neff SB, Sarma JV, Speyer CL, Gao H, Bernacki KD, Huber-Lang M, McGuire S, Hoesel LM, et al. Relationship of acute lung inflammatory injury to Fas/FasL system. Am J Pathol. 2005;166:685–694. doi: 10.1016/S0002-9440(10)62290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perl M, Chung CS, Perl U, Lomas-Neira J, de Paepe M, Cioffi WG, Ayala A. Fas-induced pulmonary apoptosis and inflammation during indirect acute lung injury. Am J Respir Crit Care Med. 2007;176:591–601. doi: 10.1164/rccm.200611-1743OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwon KY, Jang JH, Choi WI, Ramachandran S, Cho CH, Cagle PT. Expression of apoptotic nuclei by ultrastructural terminal deoxyribonucleotidyl transferase mediated dUTP nick end labeling and detection of FasL, caspases and PARP protein molecules in cadmium induced acute alveolar cell injury. Toxicology. 2006;218:197–204. doi: 10.1016/j.tox.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 65.Matsuda N, Yamamoto S, Takano K, Kageyama S, Kurobe Y, Yoshihara Y, Takano Y, Hattori Y. Silencing of Fas-associated death domain protects mice from septic lung inflammation and apoptosis. Am J Respir Crit Care Med. 2009;179:806–815. doi: 10.1164/rccm.200804-534OC. [DOI] [PubMed] [Google Scholar]
- 66.Beck JM, Preston AM, Wilcoxen SE, Morris SB, Sturrock A, Paine R., 3rd Critical roles of inflammation and apoptosis in improved survival in a model of hyperoxia-induced acute lung injury in Pneumocystis murina-infected mice. Infect Immun. 2009;77:1053–1060. doi: 10.1128/IAI.00967-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hagimoto N, Kuwano K, Miyazaki H, Kunitake R, Fujita M, Kawasaki M, Kaneko Y, Hara N. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am J Respir Cell Mol Biol. 1997;17:272–278. doi: 10.1165/ajrcmb.17.3.2893. [DOI] [PubMed] [Google Scholar]
- 68.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am J Pathol. 2001;158:153–161. doi: 10.1016/S0002-9440(10)63953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hao Z, Hampel B, Yagita H, Rajewsky K. T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J Exp Med. 2004;199:1355–1365. doi: 10.1084/jem.20032196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matute-Bello G, Lee JS, Liles WC, Frevert CW, Mongovin S, Wong V, Ballman K, Sutlief S, Martin TR. Fas-mediated acute lung injury requires fas expression on nonmyeloid cells of the lung. J Immunol. 2005;175:4069–4075. doi: 10.4049/jimmunol.175.6.4069. [DOI] [PubMed] [Google Scholar]
- 71.Matute-Bello G, Wurfel NM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol. 2007;37:210–221. doi: 10.1165/rcmb.2006-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clark P, Debiasi RL, Goody R, Hoyt CC, Richardson-Burns S, Tyler KL. Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol. 2005;18:89–115. doi: 10.1089/vim.2005.18.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uhal BD, Gidea C, Bargout R, Bifero A, Ibarra-Sunga O, Papp M, Flynn K, Filippatos G. Captopril inhibits apoptosis in human lung epithelial cells: a potential antifibrotic mechanism. Am J Physiol. 1998;275:L1013–1017. doi: 10.1152/ajplung.1998.275.5.L1013. [DOI] [PubMed] [Google Scholar]
- 74.Methot N, Huang J, Coulombe N, Vaillancourt JP, Rasper D, Tam J, Han Y, Colucci J, Zamboni R, Xanthoudakis S, et al. Differential efficacy of caspase inhibitors on apoptosis markers during sepsis in rats and implication for fractional inhibition requirements for therapeutics. J Exp Med. 2004;199:199–207. doi: 10.1084/jem.20031791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perl M, Chung CS, Lomas-Neira J, Rachel TM, Biffl WL, Cioffi WG, Ayala A. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol. 2005;167:1545–1559. doi: 10.1016/S0002-9440(10)61240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Budinger GR, Mutlu GM, Eisenbart J, Fuller AC, Bellmeyer AA, Baker CM, Wilson M, Ridge K, Barrett TA, Lee VY, Chandel NS. Proapoptotic Bid is required for pulmonary fibrosis. Proc Natl Acad Sci USA. 2006;103:4604–4609. doi: 10.1073/pnas.0507604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee CG, Kang HR, Homer RJ, Chupp G, Elias JA. Transgenic modeling of transforming growth factor-β1: role of apoptosis in fibrosis and alveolar remodeling. Proc Am Thorac Soc. 2006;3:418–423. doi: 10.1513/pats.200602-017AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)-β1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem. 2007;282:7723–7732. doi: 10.1074/jbc.M610764200. [DOI] [PubMed] [Google Scholar]
- 79.Frankel SK, Cosgrove GP, Cha SI, Cool CD, Wynes MW, Edelman BL, Brown KK, Riches DW. TNF-α sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am J Respir Cell Mol Biol. 2006;34:293–304. doi: 10.1165/rcmb.2005-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- 81.Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Lauren GJ, Thompson PJ, Knight DA. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003;29:490–498. doi: 10.1165/rcmb.2002-0262OC. [DOI] [PubMed] [Google Scholar]
- 82.Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, Maeyama T, Hara N. TGF-β1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol. 2002;168:6470–6478. doi: 10.4049/jimmunol.168.12.6470. [DOI] [PubMed] [Google Scholar]
- 83.Eigo S, Hiroaki K, Kazunari T. Identification of a caspase 3-independent role of pro-apoptotic factor Bak in TNF-α-induced apoptosis. FEBS Lett. 2002;528:63–99. doi: 10.1016/s0014-5793(02)03193-9. [DOI] [PubMed] [Google Scholar]
- 84.Lee KS, Choi YH, Kim YS, Baik SH, Oh YJ, Sheen SS, Park JH, Hwang SC, Park KJ. Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir Med. 2008;102:464–469. doi: 10.1016/j.rmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 85.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol. 2007;4:419–429. [PubMed] [Google Scholar]
- 86.Perl M, Lomas-Neira J, Chung C, Ayala A. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury: a unifying hypothesis?: what we have learned from small interfering RNAs. Mol Med. 2008;14:465–475. doi: 10.2119/2008-00011.Perl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? Am J Physiol. 2008;294:L632–L641. doi: 10.1152/ajplung.00262.2007. [DOI] [PubMed] [Google Scholar]
- 88.Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;358:44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.