

Abstract
Imprinted genes are differentially expressed from the maternally and paternally inherited alleles. Accordingly, inheritance of both copies of an imprinted chromosome or region from a single parent leads to the mis-expression of the imprinted genes present in the selected region. Strains of mice with reciprocal and Robertsonian chromosomal translocations or mice with engineered chromosomal rearrangements can be used to produce progeny where both copies of a chromosomal region are inherited from one parent. In combination with systematic differential expression and methylation-based approaches, these mice can be used to identify novel imprinted genes. Advances in genome sequencing and computer-based technologies have facilitated this approach to finding imprinted genes.
There are three major sources of mouse chromosome rearrangements. First, there are mouse stocks carrying reciprocal translocations (see Glossary and Box 1), deletions and duplications, usually induced randomly by irradiation or chemical mutagenesis. Second, there are strains carrying robertsonian translocations (Box 1), which are mostly derived from wild populations [1] (http://www.mgu.har.mrc.ac.uk/stock/stock.html and http://www.jax.org/resources/documents/). These strains are valuable because they are available ‘off the shelf’ for most chromosome regions (http://www.mgu.har.mrc.ac.uk/anomaly/anomaly-intro.html). The third source derives from chromosome engineering, which is a targeted, specifically engineered approach to chromosome anomaly induction. Chromosome engineering, for instance using the Cre–loxP recombination system [2], is capable of generating specific rearrangements, including inversions, translocations, deletions and duplications of selected portions of chromosomes (Box 2).
Box 1. Generation of mouse embryos with uniparental disomy or uniparental duplication.
Fig. I.

Mice with uniparental disomy (UpDi) inherit both copies of a whole chromosome from one parent. Mice with uniparental duplication (UpDp) inherit both copies of a specific chromosome region from the same parent. To generate UpDi or UpDp embryos to study imprinting, mouse translocation stocks that include the imprinted chromosome arm or segment of interest are selected, and a suitable genetic marker gene (yellow, Fig. I) is incorporated. (a) UpDi progeny are generated by mating two mice heterozygous for the same robertsonian translocation (see Glossary). The male carries two copies of the marker gene, one copy on the metacentric Robertsonian chromosome, and the other on the normal homologue. The female carries no copies of the marker gene. (b) UpDp progeny are generated by mating two mice heterozygous for the same reciprocal translocation (see Glossary). The male carries two copies of the genetic marker (yellow), one copy on the rearranged chromosome segment and one on the normal ho mologue (blue). In both crosses, UpDi or UpDp progeny result from the complementation of rare nondisjunction gametes and can be recognized as they will carry two copies of the marker gene, both inherited from the male.
Reciprocal crosses can be used to generate marked progeny with maternal disomy or duplication. When imprinted chromosome segments are involved progeny can exhibit phenotypic abnormalities, such as neonatal lethality, growth or behavioural effects, or loss of viability. If no offspring are recovered at birth, then they should be observed in utero for abnormal phenotypes. The frequency of UpDi and UpDp zygotes and progeny will below, 1–17%, depending on the translocation used, the chromosome region being tested, and the frequency and type of nondisjunction necessary for successful complementation of unbalanced gametes. Other zygotic classes are not shown in the diagrams. Marker genes are usually chosen to give a clear phenotype, often recognizable in utero, although biochemical or DNA markers can also be used.
Reference
- a.Cattanach BM, Beechey CV. Genomic imprinting in the mouse: possible final analysis. In: Reik W, Surani A, editors. Genomic Imprinting: Frontiers in Molecular Biology. Vol. 18. IRLPress; 1997. pp. 118–145. [Google Scholar]
Box 2. Use of chromosome rearrangements to study imprinting.
Production of targeted chromosome rearrangements
Although many chromosome rearrangements are available throughout the mouse genome, their breakpoints are random. More recently, chromosome rearrangements with selected breakpoints have been induced in embryonic stem cells by chromosome engineering using the Cre–loxP recombination system [a].
Using this technology, mouse strains can be established that carry duplications, deletions, inversions or translocations in selected imprinting regions. Duplications, deletions or inversions of a region can be generated if selected end points are on the same chromosome. Reciprocal translocations can be generated if end points map to nonhomologous chromosomes.
Potential uses of selected chromosome rearrangements within imprinting regions
Targeted duplications, deletions and insertions could be used to study effects of imprinted gene dosage, effects of rearrarrangement of imprinted genes within a cluster, or effects of deletion or duplication of imprinted genes on phenotype.
Reciprocal tranlocations
A targeted translocation within the distal chromosome 7 imprinting cluster has been used to study the imprinting status of imprinted genes in a new location on chromosome 11 (Fig. I). Mice with rearranged chromosomes 7 and 11 were used to study the imprinting status of the rearranged gene cluster.
Conclusion
The translocation of distal chromosome 7 imprinted genes to chromosome 11 affects the imprinting of all chromosome 7 genes telomeric to the breakpoint. The imprinting of genes proximal to the chromosome 7 breakpoint was unaffected, consistent with the hypothesis that regulatory elements lie within or proximal to Kcnq1. Similar methods could be used to rearrange other imprinting regions on other chromosomes.
Fig. I.

Distal chromosome 7 imprinting cluster. (a) Red boxes show maternally expressed genes and blue boxes show paternally expressed genes. (b) LoxP sites were inserted between Kcnq1 and Cdkn1c on the distal end of chromosome 7, and the distal end of chromosome 11 to generate a reciprocal translocation between chromosomes 7 and 11 in an embryonic stem cell. Adapted from Ref. [b].
References
- a.Mills AA, Bradley A. From mouse to man: generating megabase chromosome rearrangements. Trends Genet. 2001;17:331–339. doi: 10.1016/s0168-9525(01)02321-6. [DOI] [PubMed] [Google Scholar]
- b.Cleary MA, et al. Disruption of an imprinted gene cluster by a targeted chromosomal tranlocation in mice. Nat. Genet. 2001;29:78–82. doi: 10.1038/ng715. [DOI] [PubMed] [Google Scholar]
Imprinting and monoallelic expression
Genes that are imprinted are expressed from either the maternally inherited allele or the paternally inherited allele, but not both. Androgenetic embryos have two paternally inherited copies, and parthenogenetic embryos have two maternally inherited copies of every chromosome. Because of their monoallelic expression, imprinted genes are mis-expressed in these embryos, whereas genes that are not imprinted are expressed normally, unless their expression is dependent on upstream imprinted genes. Androgenetic and parthenogenetic embryos do not survive beyond mid-gestation, showing that it is necessary to inherit both maternal and paternal genomes for normal mouse development.
Mice with chromosomal rearrangements, in particular Robertsonian and reciprocal translocations, can be used to generate embryos with uniparental disomy (UpDi) or uniparental duplication (UpDp) of whole chromosomes or specific chromosomal regions, respectively (Box 1). Unlike the androgenetic and parthenogenetic embryos mentioned above, only individual chromosomes or chromosomal regions are uniparentally inherited in UpDi or UpDp embryos, rather than the entire chromosome complement. Mouse stocks carrying Robertsonian and reciprocal translocations have been used extensively to define chromosome regions subject to imprinting [3] (Box 3). The advantage of using reciprocal translocations to generate embryos with maternal and paternal UpDp is that specific subregions of the genome with defined imprinting phenotypes can be studied. The embryos often live longer than androgenetic and parthenogenetic embryos, allowing the effects of inheritance of no or two expressed copies of imprinted genes on specific tissues to be examined at later stages of development. Similarly, Robertsonian chromosomes generate maternal and paternal UpDi for entire chromosomes (Box 1). Age-, strain- and tissue-matched embryos with maternally inherited UpDi or UpDp of a particular chromosome or chromosomal region can be compared with embryos with paternally inherited UpDi or UpDp of the same regions. A phenotypic difference would suggest that genes in the duplicated region could be imprinted.
Box 3. The Harwell mouse imprinting map.
The Harwell imprinting map (http://www.mgu.har.mrc.ac.uk/imprinting/imprinting.html) illustrates regions of the mouse genome associated with phenotypic anomalies that could be attributable to imprinting (imprinting effects). Genetic breeding experiments using mice with chromosome translocations show that imprinting effects are localized to particular chromosome regions, implying that genes within these regions must function differently according to their parental origin. Furthermore, uniparental inheritance of imprinting regions could lead to anomalous phenotypes from before birth to adulthood. Virtually the whole mouse autosomal genome has been scanned for imprinting effects, and imprinting regions have been found on chromosomes 2, 6,7, 9, 11, 12, 17 and 18. The first imprinted genes were discovered in the early 1990s, and more than 50 have now been reported, most of these locating to the ‘imprinting regions’ of the genome.
Fig. I.

Map of chromosomes 2, 6, 7 and 9. Yellow regions have abnormal imprinting phenotypes with maternal (Mat) or paternal (Pat) duplication. Imprinted genes in blue are paternally expressed. Imprinted genes in red are maternally expressed. Imprinted genes within clusters are not necessarily in correct order Asterisks show paternally expressed small nucleolar RNAs.
References
- a.Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature. 1985;315:496–498. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- b.Cattanach BM, Beechey CV. Genomic imprinting in the mouse: possible final analysis. In: Reik W, Surani A, editors. Genomic Imprinting: Frontiers in Molecular Biology. Vol. 18. IRL Press; 1997. pp. 118–145. [Google Scholar]
Engineered chromosome translocations could be used in a similar way to establish translocation stocks for generating UpDp offspring of preselected chromosome regions with the advantage that smaller or overlapping chromosome segments could be studied to allow a finer dissection of imprinting phenotypes. Engineered deletions and duplications of individual genes in the candidate regions could be used to study the effects of imprinted gene dosage on phenotypes. Selected translocations and inversion chromosomes can also be used to rearrange imprinted regions of the genome and study the phenotypic consequences (Box 2). For example, a reciprocal translocation chromosome has been engineered with one breakpoint within a cluster of imprinted genes on mouse chromosome 7 and the other on chromosome 11 (Box 2) [4]. Genes remaining on mouse chromosome 7 maintain correct imprinted expression. Imprinted genes translocated to mouse chromosome 11 show a loss of imprinting, consistent with the view of coordinate control of clusters of imprinted genes.
Extent of imprinting
Translocation mice have been used to define at least 11 regions of the mouse genome that have imprinting phenotypes ranging from early embryonic lethality to postnatal effects on growth and development. The number of imprinted genes currently defined is ~60 (http://www.mgu.har.mrc.ac.uk/imprinting/imprinting.html). Estimates of the total number of imprinted genes in the mouse are very difficult to make, one estimate based on the genomic regions of the mouse for which UpDi or UpDp results in deleterious phenotypes, suggest there could be ~100 imprinted genes [5]. This agrees with earlier estimates of 100–200 imprinted genes, based on the number of imprinted genomic regions and human disease loci [6] and on a differential methylation screen [7]. Although the great majority of reported imprinted genes locate to these defined imprinting regions, a minority map to regions of the genome that show no detectable phenotype when inherited uniparentally (http://www.mgu.har.mrc.ac.uk/imprinting/imprinting.html). There are regions of the genome that elicit imprinted phenotypes not yet accounted for by known imprinted genes; for example, proximal chromosome 2 (C.V. Beechey and B.M.Cattanach, unpublished). Together, these factors suggest that there are additional imprinted genes in the mouse genome that have yet to be defined. Novel imprinted genes will further characterize the phenotypes (which frequently relate to growth and development) observed in maternal and paternal UpDi and UpDp mice.
Function of imprinting
The majority of imprinted genes for which function has been assigned control embryonic or postnatal growth [8]. There could, however, be an ascertainment bias given that many have been identified because they contribute to a growth phenotype. Furthermore, mouse embryo chimeras formed from fusions between androgenetic and normal embryos are oversized, whereas chimeras formed from normal and gynogenetic embryos (with two maternal genomes) tend to be growth retarded [9–11]. Imprinted genes involved in embryonic growth have also been detected indirectly; for example, Mash2 was identified as the result of gene targeting [12]. However, one can anticipate that uncharacterized imprinted genes could have functions unrelated to embryonic growth.
Detecting imprinted genes using monoallelic expression
Embryos or mice with UpDi and UpDp only identify chromosomal regions that might contain imprinted genes. To identify the individual genes, there are several methods currently in use (reviewed in Ref. [13]). Here, we discuss some approaches based on expression, methylation and genetics, and two new strategies for imprinted gene identification. The validity of imprinted gene expression is achieved using allele-specific assays, and we discuss these later.
Subtractive hybridization
Subtractive hybridization is a method for comparing mRNA present in one population and not in another. Subtractive cDNA hybridization between normal and parthenogenetic embryos was used to identify Peg3 [14], a paternally expressed gene on proximal chromosome 7, Peg1/Mest [15], a paternally expressed gene that maps to chromosome 6, and Peg5 or Neuronatin, a paternally expressed gene on sub-distal chromosome 2 [16]. The advantage of using parthenogenotes is that the whole genome is surveyed at once. The disadvantages are that the embryos die early, meaning that genes expressed later in development are not surveyed and tissue-specific comparisons are not feasible. Mouse embryonic fibroblast lines established from androgenetic and parthenogenetic embryos overcome this problem of limited material and have been exploited to identify the paternally expressed genes Zac1 and Sgce from a subtractive screen [17].
Differential display
Differential display is a PCR-based method that uses arbitrary primers paired with a polyA-anchored primer for identifying expression differences between pools of RNA, typically from different conditions (i.e. physiological or anatomical) using the same genetic background. A variation of differential display called allelic message display (AMD) compares identical cells or tissues from two polymorphic parental mouse strains and their corresponding reciprocal F1 hybrids. AMD produces short cDNAs derived from the 3′ end of mRNAs that are rich in strain-specific polymorphisms. Biallelically expressed polymorphic transcripts are displayed in the parental strains and in both reciprocal hybrids, whereas monoallelic transcripts appear in one but not both reciprocal F1 hybrids. This method surveys the whole genome, but it is limited by the sequence combinations of the primers. The gene Impact was identified this way, and was the first imprinted gene detected on mouse chromosome 18 [18].
Another allelic differential display strategy was developed using placental mRNA derived from two species of North American deermice, Peromyscus maniculatus and Peromyscus polionotus. Deermice are more closely related to hamsters than to Mus, but laboratory stocks of both wild-type and genetically variant Peromyscus are available. These mice display a high degree of polymorphism, making them useful for screens that depend on allelic variation [5]. The paternally expressed Dlk1 gene was identified and closely linked to the imprinted Gtl2 gene on mouse chromosome 12. At the same time, a different approach using chromosome translocations made similar findings. Mice with maternal and paternal UpDi for chromosome 12 had been used to characterize a set of parental-origin-specific developmental defects [19]. The maternal expression of Gtl2 was confirmed, and additional candidate imprinted genes responsible for the defects were sought using database searches for genes and expressed sequence tags (ESTs) in the vicinity of Gtl2. Dlk was identified 80 kb upstream of Gtl2, and the paternal expression of this gene was validated using RNA from embryos with maternal and paternal UpDi for chromosome 12 [20]. Although it might seem redundant to detect imprinted genes with different screens, maximizing the scope of gene detection exploits different criteria. Thus, selection bias resulting from one particular method can be overcome.
Serial analysis of gene expression
Serial analysis of gene expression (SAGE) is a method for generating expression profiles of the mRNAs in a given cell type [21] and can be adapted for imprinted gene detection. SAGE is not dependent on the prior availability of transcript information, and it can detect relatively low levels of transcripts. The method entails generating cDNA from mRNA, which is reduced to short stretches of expressed sequence (SAGE TAGs). The method assures that any given mRNA will give rise to only one specific TAG. Once generated, pools of tags are ligated into long concatemers, which are cloned and sequenced. The TAGs are extracted from the sequences and deposited into a database. The number of times a specific tag is present in the database corresponds to the abundance of its parental transcript in the starting mRNA. Profiles from different samples are then compared to find differences in gene expression. SAGE has been adapted for expression profiling from samples with limited numbers of cells [22]; this is especially useful for comparing isolated tissues from UpDp embryos.
Microarrays
Imprinting is an illustration of how the merging of classic genetic resources and dna microarray technology can result in rapid progress in gene identification. Microarrays can be used together with chromosome rearrangements to compare gene transcripts in maternal versus paternal UpDp and UpDi tissues. A secondary computer-based screen excludes sequences that do not map to the chromosomal region under study. Known imprinted genes from loci with biparental inheritance present on the arrays act as internal controls. Although microarrays are limited in that they survey only sequences that have been deposited in the UniGene database (http:www.ncbi.nlm.nih.gov/UniGene), and so are included in the array (unlike subtractive hybridization and differential display, which in theory survey all transcripts) they screen thousands of genes at once. (Less than 50% of the mouse clones are currently mapped in UniGene and a bioinformatics tool for combining map position data from the MIT STS marker set with the UniGene dataset and ultimately with gene clusters on microarrays would vastly improve computer-based mapping.)
A radiation-induced chromosome rearrangement in the mouse that generates embryos with UpDp for proximal mouse chromosomes 7 and 11 has been tested [23]. In a limited survey of a few organs of this mouse, a new imprinted transcript (paternally expressed brain-specific transcript) was identified [23]. Microarrays from parthenogenetic versus normal embryos or androgenetic embryos have identified Dlk1 [24] and three novel imprinted genes, Asbn4, Ata3 and Dcn [25]. This is a successful high-throughput screen, but it is limited to genes expressed in the early parthenogenetic and androgenetic embryos before they die.
Microarrays generate false positives, including downstream targets of imprinted genes or genes that are mis-regulated as a result of the chromosome rearrangement itself. Other false positives arise as a result of dissection contamination in tissue-specific analyses of embryos; this is especially apparent when sub-dividing complex tissues such as the brain. This can be overcome using embryonic fibroblast lines [17] and embryonic stem cells with UpDi of selected chromosomes (L. Lefebvre et al., unpublished), although not all imprinted genes are expressed in embryonic stem cells, and it is not clear whether they all maintain monoallelic expression in cell lines.
Antisense and noncoding RNAs
Some imprinted genes are associated with antisense or noncoding transcripts. However, several antisense transcripts, such as Igf2as and Zfp127as, are not listed within the UniGene mouse dataset, so do not feature on commercial microarrays. A database of antisense transcripts (http://www.hgmp.mrc.ac.uk/Research/Antisense) [26] has been established and could be a useful tool for identifying new imprinted genes. The imprinted murine noncoding Ipw transcript, is also not listed in UniGene [23]. Including these antisense and noncoding sequences in the next generation of Affymetrix mouse chips would be useful. Development of custom arrays of specific chromosomes to combine with chromosome translocations and tissue-specific chips for organ-specific analysis would also aid novel gene identification.
Single nucleotide polymorphisms
The rapid increase in the number of single nucleotide polymorphisms (SNPs) from mapped transcribed sequences means that an adaptation of a polymorphism-based approach can be used [27] with intercrosses between different mouse strains. Mice that have polymorphisms can be used to distinguish the parental origin of a transcribed allele of a gene in F1 hybrid mice. temperature-modulated heteroduplex analysis (also known as DHPLC) can distinguish between heteroduplexes (products from both alleles of a gene) and homoduplexes (product of one allele). Therefore, monoallelic transcripts can be distinguished from biallelic transcripts (after conversion to double-stranded cDNAs). SNPs in biallelicly expressed genes would show heterozygosity (both alleles would be expressing), whereas monoallelic expression of a gene would show homozygosity at the SNP (E. Coghill, pers. commun.). One limitation would be that imprinted genes without SNPs would not be assayed, but the rapid increase in the number of SNPs in the public domain make this a feasible new approach.
Detecting imprinted genes with other approaches
Methylation-based gene finding
The mechanisms that regulate imprinting are not completely understood, but include allele-specific DNA methylation that is established in the gametes and can act to suppress or activate genes [28]. Differential methylation of the alleles of a gene allows methylation representational difference analysis (Me-RDA) to be used to detect novel imprinted genes (Fig. 1). Several imprinted genes have been identified using Me-RDA on mice with maternal and paternal UpDps for distal chromosome 2, including Nesp and Gnasxl [29,30] and Nespas [31]. This approach is not dependent on prior transcript information. However, because Me-RDA is a subtractive hybridization technique that relies on reassociation in complex DNA representations, some sequences reassociate less favourably and can become lost in the process.
Fig. 1.
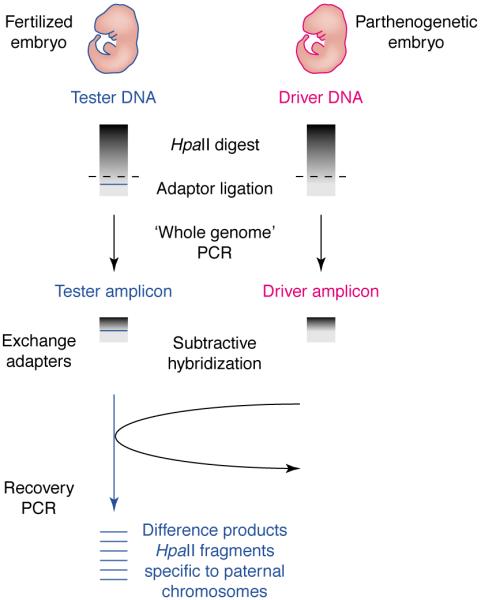
Methylation RDA (Me-RDA) can be used to compare gene expression between normal (fertilized) and parthenogenetic embryos. Genomic DNA is digested with HpaII and adapters are ligated before PCR. Rounds of subtractive hybridization remove DNA species in common between the embryos and enrich for differentially methylated products, which are recovered by PCR.
Genetic methods
Imprinted gene mutations have been identified in mice as a result of screening mutants for imprinted phenotypes [32]. The Mnt mutation was identified as a severe growth retardation phenotype only transmitted through males, and the Oed-Sml mutation was identified from two different mutant phenotypes that are produced according to the parent transmitting the mutation. Consistent with their imprinting inheritances, both Mnt and Oed-Sml map within known imprinting regions of mouse chromosomes: Mnt near Igf2 on chromosome 7, and Oed-Sml close to Gnas on distal 2. Further strategies to identify imprinted genes and phenotypes in a similar way are ongoing using the ethylnitrosourea (ENU) mutation screen at the MRC Mammalian Genetics Unit (Harwell, UK) (J. Peters, pers. commun.).
Validation of imprinting
Expression profiles alone are not sufficient to confirm imprinting, and it is essential that candidate genes be characterized further by allele-specific assays to confirm their monoallelic expression. Polymorphism-based assays can be used with interspecific F1 hybrids; for example, from a cross of C57BL/6J laboratory mouse with a different subspecies such as M. m. castaneus to produce BXC (C57BL/6J mother; Mus musculus castaneus father) hybrids and vice versa. Assays to detect polymorphisms present in the transcripts include RNase protection or reverse transcription PCR (RT–PCR) followed by restriction fragment length polymorphism (RFLP) analysis (i.e. cutting with a restriction enzyme whose recognition site is present in only one of the alleles and gel analysis of the products). An RNase protection assay on interspecies hybrid RNA was used in the classic experiments assaying the H19 gene for monoallelic expression [33]. RT–PCR followed by RFLP analysis has been widely used to confirm monoallelic expression, for example Peg3 [14] and Peg1/Mest [15]. Allele-specific assays using RT–PCR or northern analysis on embryos or tissues with specific chromosome UpDi or UpDp do not depend on polymorphism detection as the gene is either expressed or not. Dlk and Gtl2 [20], Nesp and Gnasxl [29] were assayed this way. Human imprinted genes can be assayed for imprinting status using transcribed SNPs to distinguish the parental expressing alleles.
Bioinformatics
The utility of bioinformatics to questions of imprinting has been illustrated in studies of Beckwith–Wiedemann syndrome (BWS) in several ways. BWS is a human disease with parent-of-origin-associated prenatal overgrowth and cancer pre-disposition, located on human chromosome 11p15.5. MTR1, a candidate gene for involvement in BWS, was identified through a screen for evolutionarily conserved sequences between human and Caenorhabditis elegans. Genomic sequence from a P1-derived artificial chromosome (PAC) encompassing the BWS region was translated in all reading frames and compared with the C. elegans protein database at the Sanger Centre to reveal a putative C. elegans protein belonging to a family of transient receptor (trp) proteins. BLAST searches against the human EST databases found similarity to the human TRP family. The novel, evolutionarily conserved, paternally expressed gene termed MTR1 (for MLSN1- and TRP-related gene 1) is also found in the mouse (Mtr1) and maps to distal chromosome 7, an imprinted region that shares conserved linkage with human chromosome 11p15.5 [34].
Bioinformatics can also facilitate understanding of the mechanisms of imprinted gene expression. In human and mouse, most imprinted genes are arranged in chromosomal clusters and share a high degree of conserved linkage (gene order) between the two species. This linked organization suggests coordinated mechanisms controlling imprinting. Identification of conserved elements responsible for the control of imprinted gene expression is therefore important, and comparisons between mouse and human sequence in imprinted regions of the genome can detect putative control regions. Several studies have used comparative genome sequence analyses for identification of conserved functional elements, and with the sequence of the human genome now available and rapid progress with the mouse genome sequence, this is an increasingly powerful tool. The BWS region on 11p15.5 has been studied systematically by comparative sequence analysis, because understanding the molecular basis of imprinted gene control will help elucidate the underlying defects that cause disease. Human and mouse sequence comparisons of the region on mouse distal chromosome 7 (human 11p15.5) using software to predict putative genes and analyze sequence motifs have revealed a number interesting features [35]. As might be expected, gene order is conserved between the human and mouse, as are transcriptional orientation and gene structure throughout the region. CpG islands are often associated with genes [36], and the densities of CpG islands in the BWS region and the comparable mouse region have been compared with other regions rich in genes that are not imprinted. This comparison showed a higher density of CpG islands in the BWS region [35,37] as well as clusters of conserved non-exonic, non-CpG island sequences [37]. Similarly, a genome sequence comparison between human, mouse and sheep in the Dlk1-Gtl2 imprinted region revealed associations of imprinted gene clusters with high densities of CpG islands [38]. The BWS region does, however, show some differences in organization between mouse and human, including an insertion of two large repetitive clusters into the human region and an additional gene [35].
The maternally expressed KvLQT gene is disrupted in some cases of BWS, and sequence comparisons between the mouse and human gene shows a region with characteristics of an imprinting control element. RT–PCR of the human and mouse loci identified an evolutionarily conserved maternally methylated CpG island in an intron of KvLQT1 and an associated paternally expressed antisense transcript [39]. Defining imprinting control regions such as this is an important aspect of understanding the mechanisms of imprinted gene expression.
Imprinting is largely conserved between mouse and human, most imprinted genes are imprinted in both species, notable exceptions are Igf2r and Impact. The reasons the two species have differences in imprinting of specific genes is not clear, but it is thought that at some point in evolution, imprinting was lost in a lineage-specific way, at least for Igf2r [40]. Comparative genome analysis has been used to investigate possible explanations for the mouse Impact gene being imprinted and its human orthologue not being imprinted. As expected, the two genes share well-conserved exon–intron organization, but the mouse allele has a CpG island in the first exon that is hypermethylated on the maternal allele and unmethylated on the paternal allele. This island is missing in the human gene [41], and is likely to be involved in the imprinting mechanism of the mouse Impact gene. Whether it was lost in the human gene or arose only in the mouse is not clear.
Summary
The combination of existing genetic tools, new bioinformatics and post-genomic technologies yield tractable systems for the identification of imprinted genes. Making phenotype–genotype correlations by assigning the genes responsible for developmental demise to defined mouse phenotypes ultimately leads to a better understanding of developmental pathways and mechanisms of imprinting. It is noteworthy that imprinted genes might not all contribute to a phenotype. It is thought that some imprinted genes have a specific imprinted expression selected during evolution but for others, imprinting is a consequence of their proximity to imprinted regions [42]. The ‘innocent bystander’ genes can be discriminated by testing the effect of their biallelic expression upon the organism. Those without phenotypic effects will not be informative for studies on imprinting mechanism. Ultimately, knockout and knock-in mice can provide a systematic way of studying the isolated phenotypes of individual imprinted genes. Importantly, human genetic diseases associated with imprinting will be better understood with a greater understanding of imprinted gene control. These include Angelman syndrome (AS), Prader–Willi syndrome (PWS), BWS and Silver–Russell syndrome (SRS), which all show parent-of-origin inheritance effects consistent with imprinting. AS has largely been accounted for by loss of maternal UBE3A brain-specific expression [43] on 15q11–q13. Genetic evidence suggests that PWS arises from functional loss of several paternally expressed genes, including involvement in cis by an imprinting center [44] on 15q11–q13. BWS on 11p15.5 could involve multiple genes, but the underlying mechanism is not completely clarified. SRS characterized by pre- and post-natal growth restriction, has been associated with maternal UpDi for human chromosome 7. Grb10 has been a candidate for SRS but the lack of mutations in patients has prompted continued searches for new imprinted candidates for this disease [45]. The use of mouse models for imprinted gene identification is likely to continue to contribute to the understanding of these human diseases and to help decipher the associated complex detrimental phenotypes.
Acknowledgements
We thank Gavin Kelsey and Rachel Smith for the Me-RDA illustration. We would also like to thank the following individuals for reading parts of this review and for their helpful comments: Jonathan Choi, Michael Malim, Jo Peters, Bruce Cattanach, Lara Underkoffler, Nikos Tripodis, Kathleen Loomes, Patrick Doherty, John Shires, Kenro Kusumi, Peter Botcherby and Chad Swanson.
Glossary
- DNA microarrays
High-density arrays of oligonucleotides or cDNAs that can be used to detect patterns of mRNA expression.
- Expression profile
The differences in expression between two pools of RNA.
- Reciprocal translocation
Exchange of genomic material between non-homologous chromosomes without loss.
- Robertsonian translocation
A chromosome with a single active centromere near the middle of the chromosome resulting from the fusion of two different mouse chromosomes that each had a centromere located near one of the telomeres; that is, having two long arms from nonhomologous chromosomes fused at the centromere.
- Single nucleotide polymorphisms (SNPs)
Single base pair positions in genomic DNA at which different alleles exist in normal individuals in some population(s), wherein the least frequent allele has an abundance of 1% or greater. Single base variants in cDNAs (cSNPs) are also usually classed as SNPs because most of these will reflect underlying genomic DNA variants.
- Temperature-modulated heteroduplex analysis
Also known as denaturing high performance liquid chromatography (DHPLC). A method for detecting polymorphisms between two alleles of a gene, used for SNP analysis.
- Uniparental disomy (UpDi)
Where both copies of a chromosome are inherited from the same parent.
- Uniparental duplication or partial disomy (UpDp)
Where both copies of a specific region of a chromosome inherited from the same parent and the remainder of the genome is inherited from both parents.
Contributor Information
Rebecca J. Oakey, GKT School of Medicine, Division of Medical and Molecular Genetics, 8th Floor Guy’s Tower, London, UK SE1 9RT.
Colin V. Beechey, Mammalian Genetics Unit, Harwell, Didcot, Oxon, UK OX11 0RD
References
- 1.Silver L. Mouse Genetics. Oxford University Press; 1995. [Google Scholar]
- 2.Mills AA, Bradley A. From mouse to man: generating megabase chromosome rearrangements. Trends Genet. 2001;17:331–339. doi: 10.1016/s0168-9525(01)02321-6. [DOI] [PubMed] [Google Scholar]
- 3.Cattanach BM, Beechey CV. Genomic imprinting in the mouse: possible final analysis. In: Reik W, Surani A, editors. Genomic Imprinting: Frontiers in Molecular Biology. Vol. 18. IRL Press; 1997. pp. 118–145. [Google Scholar]
- 4.Cleary MA, et al. Disruption of an imprinted gene cluster by a targeted chromosomal translocation in mice. Nat. Genet. 2001;29:78–82. doi: 10.1038/ng715. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt JV, et al. The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 2000;14:1997–2002. [PMC free article] [PubMed] [Google Scholar]
- 6.Barlow DP. Gametic imprinting in mammals. Science. 1995;270:1610–1613. doi: 10.1126/science.270.5242.1610. [DOI] [PubMed] [Google Scholar]
- 7.Bartolomei MS. The search for imprinted genes. Nat. Genet. 1994;6:220–221. doi: 10.1038/ng0394-220. [DOI] [PubMed] [Google Scholar]
- 8.Tilghman SM. The sins of the fathers and mothers: genomic imprinting in mammalian development. Cell. 1999;96:185–193. doi: 10.1016/s0092-8674(00)80559-0. [DOI] [PubMed] [Google Scholar]
- 9.Mann JR, et al. Androgenetic mouse embryonic stem cells are pluripotent and cause skeletal defects in chimeras: Implications for genetic imprinting. Cell. 1990;62:251–260. doi: 10.1016/0092-8674(90)90363-j. [DOI] [PubMed] [Google Scholar]
- 10.Fundele RH, et al. Temporal and spatial selection against parthenogenetic cells during development of fetal chimeras. Development. 1990;108:203–211. doi: 10.1242/dev.108.1.203. [DOI] [PubMed] [Google Scholar]
- 11.Barton SC, et al. Influence of paternally imprinted genes on development. Development. 1991;113:679–688. doi: 10.1242/dev.113.2.679. [DOI] [PubMed] [Google Scholar]
- 12.Guillemot F, et al. Genomic imprinting of Mash2, a mouse gene required for trophoblast development. Nat. Genet. 1995;9:235–241. doi: 10.1038/ng0395-235. [DOI] [PubMed] [Google Scholar]
- 13.Kelsey G, Reik W. Analysis and identification of imprinted genes. Methods. 1998;14:211–234. doi: 10.1006/meth.1997.0579. [DOI] [PubMed] [Google Scholar]
- 14.Kuroiwa Y, et al. Peg3 imprinted gene on proximal chromosome 7 encodes for a zinc finger protein. Nat. Genet. 1996;12:186–190. doi: 10.1038/ng0296-186. [DOI] [PubMed] [Google Scholar]
- 15.Kaneko-Ishino T, et al. Peg1/Mest imprinted gene on chromosome 6 identified by cDNA subtraction hybridization. Nat. Genet. 1995;11:52–59. doi: 10.1038/ng0995-52. [DOI] [PubMed] [Google Scholar]
- 16.Kagitani F, et al. Peg5/Neuronatin is an imprinted gene located on sub-distal chromosome 2 in the mouse. Nucleic Acids Res. 1997;25:3428–3432. doi: 10.1093/nar/25.17.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piras G, et al. Zac1 (Lot1), a potential tumor supressor gene, and the gene for ε-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol. Cell. Biol. 2000;20:3308–3315. doi: 10.1128/mcb.20.9.3308-3315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagiwara Y, et al. Screening for imprinted genes by allelic message display: Identification of a paternally expressed gene Impact on mouse chromosome 18. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9249–9254. doi: 10.1073/pnas.94.17.9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgiades P, et al. Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development. 2000;127:4719–4728. doi: 10.1242/dev.127.21.4719. [DOI] [PubMed] [Google Scholar]
- 20.Takada S, et al. Delta-like and Gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr. Biol. 2000;10:1135–1138. doi: 10.1016/s0960-9822(00)00704-1. [DOI] [PubMed] [Google Scholar]
- 21.Velculescu VE, et al. Serial analysis of gene expression. Science. 1995;270:368–369. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 22.Velculescu VE, et al. Analysing uncharted transcriptomes with SAGE. Trends Genet. 2000;16:423–425. doi: 10.1016/s0168-9525(00)02114-4. [DOI] [PubMed] [Google Scholar]
- 23.Choi JD, et al. Microarray expression profiling of tissues from mice with uniparental duplications of chromosomes 7 and 11 to identify imprinted genes. Mamm. Genome. 2001;12:758–764. doi: 10.1007/s00335-001-3027-5. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi S, et al. Mouse Peg9/Dlk1 and human PEG9/DLK1 are paternally expressed imprinted genes closely located to the maternally expressed imprinted genes: mouse Meg3/Gtl2 and human MEG3. Genes Cells. 2000;5:1029–1037. doi: 10.1046/j.1365-2443.2000.00390.x. [DOI] [PubMed] [Google Scholar]
- 25.Mizuno Y, et al. Asb4, Ata3 and Dcn are novel imprinted genes identified by high-throughput screening using RIKEN cDNA microarray. Biochem. Biophys. Res. Commun. 2002;290:1499–1505. doi: 10.1006/bbrc.2002.6370. [DOI] [PubMed] [Google Scholar]
- 26.Lehner B, et al. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- 27.Coghill EL, et al. A gene-driven approach to the identification of ENU mutants in the mouse. Nat. Genet. 2002;30:255–256. doi: 10.1038/ng847. [DOI] [PubMed] [Google Scholar]
- 28.Li E, et al. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 29.Peters J, et al. A cluster of oppositely imprinted transcripts at the Gnas locus in the distal imprinting region of mouse chromosome 2. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3830–3835. doi: 10.1073/pnas.96.7.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelsey G, et al. Identification of imprinted loci by methylation-sensitive representational difference analysis: application to mouse distal chromosome 2. Genomics. 1999;62:129–138. doi: 10.1006/geno.1999.6022. [DOI] [PubMed] [Google Scholar]
- 31.Wroe SF, et al. An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse Gnas locus. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3342–3346. doi: 10.1073/pnas.050015397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cattanach BM, et al. Two imprinted genes: three phenotypes. Hum. Mol. Genet. 2000;9:2263–2273. doi: 10.1093/oxfordjournals.hmg.a018917. [DOI] [PubMed] [Google Scholar]
- 33.Bartolomei MS, et al. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 34.Prawitt D, et al. Identification and characterization of MTR1, a novel gene with homology to melastatin (MLSN1) and the trp gene family located in the BWS-WT2 critical region on chromosome 11p15.5 and showing allele-specific expression. Hum. Mol. Genet. 2000;9:203–216. doi: 10.1093/hmg/9.2.203. [DOI] [PubMed] [Google Scholar]
- 35.Paulsen M, et al. Sequence conservation and variability of imprinting in Beckwith–Wiedemann syndrome gene cluster in human and mouse. Hum. Mol. Genet. 2000;9:1829–1841. doi: 10.1093/hmg/9.12.1829. [DOI] [PubMed] [Google Scholar]
- 36.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 37.Onyango P, et al. Sequence and comparative analysis of the mouse 1-Megabase region orthologous to the human 11p15 imprinted domain. Genome Res. 2000;10:1697–1710. doi: 10.1101/gr.161800. [DOI] [PubMed] [Google Scholar]
- 38.Paulsen M, et al. Comparative sequence analysis of the imprinted Dlk1-Gtl2 locus in three mammalian species reveals highly conserved genomic elements and refines comparison with the Igf2-H19 region. Genome Res. 2001;11:2085–2094. doi: 10.1101/gr.206901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smilinich NJ, et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith–Wiedemann syndrome. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8064–8069. doi: 10.1073/pnas.96.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Killian JK, et al. Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the Quaternary. Hum. Mol. Genet. 2001;10:1721–1728. doi: 10.1093/hmg/10.17.1721. [DOI] [PubMed] [Google Scholar]
- 41.Okamura K, et al. Comparative genome analysis of the mouse imprinted gene Impact and its nonimprinted human homolog IMPACT: toward the structural basis for species-specific imprinting. Genome Res. 2000;10:1878–1889. doi: 10.1101/gr.139200. [DOI] [PubMed] [Google Scholar]
- 42.Barlow DP. The origins of genomic imprinting in mammals. In: Hall JC, et al., editors. Advances in Genetics. Vol. 46. Academic Press; 2002. pp. 119–163. [DOI] [PubMed] [Google Scholar]
- 43.Rougelle C, et al. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 44.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader–Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 45.Hitchins MP, et al. Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver-Russell syndrome. Eur. J. Hum. Genet. 2001;9:82–90. doi: 10.1038/sj.ejhg.5200583. [DOI] [PubMed] [Google Scholar]