

Abstract
Proximal spinal muscular atrophy (SMA) results from loss of the survival motor neuron 1 (SMN1) gene, with retention of its nearly identical homolog, SMN2. There is a direct correlation between disease severity and SMN2 copy number. Mice do not have a Smn2 gene, and thus cannot naturally replicate the disorder. However, two murine models of SMA have been generated using SMN2-BAC transgenic mice bred onto a mutant Smn background. In these instances mice die shortly after birth, have variable phenotypes within the same litter, or completely correct the SMA phenotype. Both models have been imported to the Jackson Laboratory for distribution to the research community. To ensure that similar results are obtained after importation to The Jackson Laboratory to what was originally reported in the literature, we have begun a molecular and phenotypic evaluation of these mouse models. Here we report our findings for the SMA mouse model that has been deposited by the Li group from Taiwan. These mice, JAX stock number TJL-005058, are homozygous for the SMN2 transgene, Tg(SMN2)2Hung, and a targeted Smn allele that lacks exon 7, Smn1tm1Hung. Our findings are consistent with those reported originally for this line and clarify some of the original data. In addition, we have cloned and mapped the integration site for Tg(SMN2)2Hung to Chromosome 4, and provide a simple genotyping assay that is specific to the junction fragment. Finally, based upon the survival data from our genetic crosses, we suggest that this underused SMA model may be a useful compliment or alternative to the more commonly used “delta7” SMA mouse. We provide breeding schemes in which two genotypes of mice can be generated so that 50% of the litter will be SMA-like pups while 50% will be controls.
Keywords: Spinal muscular Atrophy, SMA, survival motor neuron, SMN, mouse models
Introduction
Proximal spinal muscular atrophy (SMA) is a neuromuscular disease and the leading heritable cause of infant mortality [1]. It is linked at the molecular level to the survival motor neuron (SMN) genes. Due to an inverted duplication at the 5q13 locus, humans contain two nearly identical copies of the SMN gene, SMN1 and SMN2; however, only mutations in SMN1 are responsible for spinal muscular atrophy [2]. The critical difference between SMN1 and SMN2 is a silent, single nucleotide transition within exon 7 that disrupts an exonic splicing enhancer in SMN2 [3]. This results in small amounts of full-length transcripts from SMN2 and high levels of a differentially spliced isoform that lacks exon 7 (Δ7SMN). Consequently, the small amount of functional protein produced from SMN2 is not enough to fully compensate for the loss of SMN1. Family studies have demonstrated that SMN2 modifies disease severity in a dose dependent manner [2, 4]. Thus, SMA is not a true loss of function disease but one of insufficient SMN dosage.
In contrast to humans, mice contain a single copy of the Smn gene that does not alternatively splice Smn exon 7 [5]. Disruption of the locus so that no protein is produced results in embryonic lethality prior to implantation [6]. Mice cannot naturally replicate the disease, so to recapitulate the genetic situation seen in SMA patients and test the hypothesis that human SMN2 acts a disease modifier, two groups generated transgenic mice that contained the entire human SMN2 locus. Both have generously been placed at The Jackson Laboratory (TJL) for distribution to the research community (TJL-005024 and 005058) [7, 8].
The TJL-005024 mice were generated in the Burghes laboratory. The SMN2 transgene, SMN2(89Ahmb), that was used in this model contains only the SMN2 genomic locus and is on a null Smn background (Smn−/−) (TJL-005024) [6]. This model has been used as a base SMA model to breed SMN cDNA transgenes onto to generate additional SMA model mice [9, 10] In contrast, the TJL-005058 mice that the Li laboratory generated used a SMN2 transgene, Tg(SMN2)2Hung, that contained SMN2, the small EDRK-rich factor (SERF1) gene and a portion of the neuronal apoptosis inhibitory protein (NAIP) gene on a Δ7Smn background [7].
Although there are differences between these transgenic models, the critical observations are the same: 1) SMN2 is able to compliment the embryonic lethality of mutant Smn mice, 2) an increase in SMN2 copy number correlates with a milder disease course, as seen in human family studies [4] and 3) if enough SMN2 copies are introduced into the mouse germline the SMA phenotype can be completely rescued. One major phenotypic difference that exists between these two SMA models is that the Burghes laboratory SMA-like mice present with consistent phenotypes within the same litter [8]. In contrast, the Li group reported severe, intermediate and mild SMA-like mice being born within the same litter [7]. Familial phenotypic heterogeneity occurs infrequently in humans and has not been reported in other SMA mouse models [8-10].
Since the initial reports of these models by both groups, the Li mouse model has been infrequently used. This may be due to confusion of the original molecular and phenotypic characteristics that were reported. Here, we provide a molecular analysis of the SMN2 transgenic line and Smn-targeted allele that the Li group has deposited to the Jackson laboratory for distribution to the research community. We cloned and mapped the integration site of the SMN2 transgene and demonstrate that this is the same SMN2 transgenic mouse that has been distributed to researchers in Europe. Consequently, we believe that all groups that have the Li mouse model have received SMN2 founder line 2 from the original publication [7]. We have performed genetic crosses to re-evaluate the multi-phenotype litters and provide data that clarify the original results. Based on our findings, we suggest that mice generated from our proposed breeding scheme be used to compliment or replace the commonly used “delta 7” SMA mouse model (TJL-005025) in SMA research.
Materials and Methods
Mice
Mice were bred, maintained and used in accordance with approved IACUCs at JAX and CMRC. The generation and initial characterization of the mice used here, Tg(SMN2)2Hung founder line 2, and the Smn targeted allele, Smn1tm1Hung has previously been reported [7]. These mice were imported into the Jackson Laboratory for re-derivation and distribution. After re-derivation the alleles were assembled through intercrosses so that the line maintained and distributed by JAX is homozygous for both Tg(SMN2)2Hung and Smn1tm1Hung (Tg(SMN2)2Hungtg/tg;Smn1tm1Hung/tm1Hung) on a FVB/N background. The JAX strain name is FVB.Cg-Tg(SMN2)2HungSmn1tm1Hung/J and the stock number is 005058. For simplicity we refer to them as TJL-005058.
RT-PCR
RT-PCR was performed on first-strand cDNA to amplify three amplicons: SMN 6 -8, SMNex6For(177):5’-CCCATATGTCCAGATTCTCTTGAT; SMNex8Rev(178):5’-CTACAACACCCTTCTCACAG; Smn 5-8, ex5For (741):5’-TCCCTTCAGGAC CACCAATA; ex8Arev(495):5’-CCACTGATGACGAGGAGACG; Amplicons are 279 bp(+7) and 232 bp (−7). Actin: mActB for(550):5’-GACCCAGATCATGTTTGAGA; mActb#2rev(552):5’-ATGCCACAGGATTCCATACC. The amplicon is 465 bp.
Genotyping
Genotyping the Smn1tm1Hung allele was as described in [7]. Tg(SMN2)2Hung Junction PCR: LiVTSP3C(690):5’-TGTCTTGAGCCAAGTTAGCC, 7LiTg Rev3(698):5’-CCTGCTCCTGCCTATGAAGT and Liunk-Fwd1(737):5’-TTGCTTTATGACTCTTGATACCTG to amplify a wildtype band of 890 bp (737 and 698) and a junction band of 250 bp (690 and 698) using an annealing of 62C and reaction conditions of 5mM MgCl2, 0.25mM dNTP and 50ng of each primer. SMN2 general PCR: F(538):5’-CATATGTCAGAGTGTACAGTGCAG; and R(539);5’-GGTGCTCACATTCCTTAAATTAAG. The amplicon is 380 bp.
Vectorette PCR
Bgl II digested TJL-005058 genomic DNA [2μg] was ligated to Bgl II vectorette linkers and used in sequential rounds of nested PCR. The final 390 bp product was cloned and sequenced and found to be a fusion from BAC7C and Chrm 4. The Chrm 4 sequence is from 2 distinct regions separated by 18.5 kb (NCBI: 6384151-6384307 and 6402953-6402859). PCR amplification is accomplished from the most distal fragment (6402953-6402859) to the Tg(SMN2)2Hung transgene, suggesting a deletion of the 18.5 kb region. Sequence analysis of the transgene specific PCR product confirms this.
Results and Discussion
Molecular characterization of TJL-005058 mice
The generation and initial characterization of the mice used here, TJL-005058, (Tg(SMN2)2Hungtg/tg; Smn1tm1Hung/tm1Hung) has previously been reported [7] and their importation into the Jackson laboratory is detailed in the Material and Methods section. As a first step to molecularly characterize this model, we assessed the expression pattern of the SMN2 transgenic allele, Tg(SMN2)2Hung, as well as the endogenous Δ7Smn allele, Smn1tm1Hung. For this, tissues were harvested from one month old mice that were homozygous for both the transgene and the targeted Smn allele. SMN2 expression was analyzed by RT-PCR using human specific primers in SMN exons 6 and 8. Consistent with previous results, we found SMN2 to be expressed in all tissues and to produce a greater amount of transcripts that lack SMN2 exon 7 than those that include exon 7 (Figure 1A and data not shown). We also confirmed using murine specific Smn primers in exons 5 and 8 that only transcripts that lack Smn exon 7 are produced from the Smn1tm1Hung allele (Figure 1B). These results confirm the RNA expression data originally reported by Hsieh-Li et al. (2000) [7] and dispel any suggestion that a small amount of full-length transcripts are produced from the murine Smn1tm1Hung allele.
Figure 1.

Analysis of human and murine SMN transcripts. A) RT-PCR analysis of SMN exons 6-8 in wildtype and TJL-005058 mice. Total reference human cDNA from Stratagene was used as a positive control. Only TJL-005058 mice express human SMN and demonstrate the specificity of the primer set for human SMN sequence. Transcripts lacking exon 7 are more abundant than those that include exon 7 in TJL-005058 mice as would be expected from SMN2 expression. B) RT-PCR analysis of wild type and TJL-005058 mice using murine specific primers in Smn exons 5-8. Wild type mice do not alternatively splice Smn exon 7 and all transcripts contain exon 7, whereas TJL-005058 mice, which are homozygous for the Smn1tm1Hung allele, only express transcripts that lack exon 7. Actin was used as a positive control for RT-PCR reactions. Abbreviations: FL denotes transcripts containing exon 7, Δ7 denotes transcripts lacking exon 7.
Since the original report described five different founder SMN2 lines, we focused our attention on characterizing the SMN2 transgenic allele, Tg(SMN2)2Hung. The five founder SMN2 lines were generated by microinjection of a 115 kb DNA fragment from BAC clone, 7C. This genomic fragment contains the entire SMN2 locus as well as upstream and downstream sequences that encompass an intact centromeric SERF1 gene and a portion of the NAIP gene [7]. The SMN2 founder line that was donated to the Jackson laboratory was founder line 2, which has two copies of SMN2. We performed sequence tagged site (STS) content mapping using human specific primer pairs to determine that Tg(SMN2)2Hung is at least 112 kb in size and has a maximal 5’ and 3’ deletion of >1 kb and 3 kb, respectively (data not shown). Since two tandem copies of Tg(SMN2)2Hung have integrated into the mouse genome and STS content mapping only scores the presence or absences of sequences, it may very well be that each copy is not 112kb in size and one fragment may be larger than the other.
To determine the integration site of Tg(SMN2)2Hung, we used the STS content information as a starting point to design a series of primers for use in vectorette PCR. We were able to clone one side of the integration site using this method. A BLAST search of the non-redundant database using the unique sequence that we obtained indicated that Tg(SMN2)2Hung integrated into an intergenic region of Chrm4 between the Toll-like receptor (Tlr4) gene and an EST sequence located ~165 kb and 47 kb proximal and distal to the transgene, respectively (Figure 2A and B). The integration site was confirmed by PCR analysis after designing primers, one of which matched BAC 7C genomic sequence, and two within the genomic region where the transgene integrated (Figure 2C). A wild type allele amplifies an 890 bp fragment while a transgenic hemizygous allele amplifies two products, a 250 bp junction fragment as well as an 890 bp wild type fragment. This assay is unable to differentiate whether Tg(SMN2)2Hung is present in a hemizygous or homozygous state. We believe this is due to a genomic duplication has also occurred at the integration site since we consistently amplify a wild type fragment. In addition, it is clear that an 18.5 kb deletion has occurred 154 bp downstream of the junction site (exact nucleotides are provided in the material and methods).
Figure 2.

Tg(SMN2)2Hung transgene integration structure. A) Schematic of the transgene integration on murine Chromosome 4. The integration site is 165kb downstream of Toll Like Receptor 4 (Tlr4) gene and 47kb upstream of novel bladder cancer associated gene ENSMUSG00000064154. Dots surround the second copy of SMN2 as we do not know how much of this trangene is present outside of the SMN2 region. B) Sequence of vectorette PCR clone 2B4 used to identify the genomic integration site. Bold letters are sequence derived from BAC7C, and regular letters are those found on Chromosome 4. The grey italicized letters are the 154bp found in Chromosome 4 proximal to the 18.5kb deletion and those not shaded are distal. C) Genotyping assay specific for the Tg(SMN2)2Hung-Chr 4 junction. Note that the WT band persists in the homozygous transgenic mice. This is most likely due to a duplication of sequence that occurred during transgene integration.
Several publications that utilized the Li mouse model for their studies obtained the line directly from the Li Laboratory [11, 12]. To determine whether those studies used the same SMN2 transgenic line that has been imported to the Jackson laboratory, we performed our integration specific assay with DNA obtained from those mice. Based on our results, we concluded that the same SMN2 transgene, Tg(SMN2)2Hung founder line 2, was used (Supplementary Figure 1).
Phenotypic characterization of Tg(SMN2)2Hung founder line 2 bred onto a Δ7 Smn background
It was originally described by Hsieh-Li [7] that SMA-like mice representing severe, intermediate and mild forms of the disease could be found within the same litter when Tg(SMN2)2Hung founder line 2 was bred onto the Δ7Smn mutant background (Smn1tm1Hung/tm1Hung) [7]. Familial heterogeneity of SMA occurs infrequently in humans and large phenotypic differences have not been reported in other SMA model mice [8-10]. Hence to phenotypically characterize the mice that were imported to The Jackson Laboratory and further investigate the large phenotypic heterogeneity from the original report, we performed a series of genetic crosses and monitored the resulting progeny for survival, phenotype and general appearance.
First, we evaluated TJL-005058 mice as they are homozygous at both loci (Tg(SMN2)2Hungtg/tg;Smn1tm1Hung/tm1Hung). They were able to breed normally and 100% of the offspring were normal except for a short thick tail that was noticeable at about two weeks of age. As with other long-lived SMA model mice or those with induced survival from therapeutic treatment, the tail became necrotic from the tip around or just after weaning and usually fell off by one month of age [8, 10, 13]. The tail necrosis was not immediately progressive but extended to the pinnae of the ears and feet later in life. The results were consistent and there was no variability in phenotype. It is these mice, which have a total of 4 copies of SMN2 (2 from each Tg(SMN2)2Hung allele) being expressed on a mutant Δ7Smn background, that Hsieh-Li et al. (2000) [7] reported as having mild SMA. His group has used these mice in studies that involve very late onset SMA symptoms after more than 9 months of age [14-16].
We next intercrossed mice that were hemizygous for Tg(SMN2)2Hung and heterozygous for the Smn1tmHung allele to determine if severe, intermediate and mild SMA mice could be generated in the same litter using only this SMN2 transgene. The resulting pups could potentially have either 0, 2 or 4 copies of SMN2 depending on whether the mouse was wild type, hemizygous or homozygous for Tg(SMN2)2Hung, respectively. For this intercross we used F1 progeny from a 10th mating of a 3rd generation TJL-005058 mouse (F10N3) that had been crossed to a FVB/N mouse. In total, 20 matings were set that represented an intercross between Tg(SMN2)2Hungtg/0;Smn1tm1Hung/WT mice. A total of 245 live born pups from 29 litters were monitored for survival from birth to adulthood and this represented an average litter size of just over 8 pups (8.4 exactly). Table 1 provides the expected vs. actual number of pups obtained for each of the nine genotypes that could be generated. We obtained no pups that were solely homozygous for the targeted Smn allele (Smn1tm1Hung/tm1Hung), this was consistent with previous results of mice that express Δ7Smn [7, 17]. In addition, the actual number of pups that were homozygous for both the SMN2 transgene and the targeted Smn allele (N=17; Tg(SMN2)2Hungtg/tg;Smn1tm1Hung/tm1Hung) were approximately equal to that expected for this genotype (N=16). This survival result was consistent with our breeding stock of TJL-005058 mice (Tg(SMN2)2Hungtg/tg;Smn1tm1Hung/tm1Hung) that have 4 copies of SMN2 and we verified that all 17 mice had tail necrosis. Interestingly, based upon mendelian ratios we expected ~33 mice to be born that were hemizygous for the SMN2 transgene and homozygous for the targeted allele (Tg(SMN2)2Hungtg/0;Smn1tm1Hung/tm1Hung), but we obtained only 12 (Table 1). The deviation from the expected ratio could be the result of in utero death. It could also be that pups were born but died and were cannibalized prior to morning observation on P0. The maximal survival of the pups that were born was 17 days, with an average and median survival of 12 and 13 days, respectively. A Kaplan-Meir survival graph of mice hemizygous for Tg(SMN2)2Hung and homozygous for Smn1tm1Hung alleles is shown in Figure 4. All of these mice have a total of 2 copies of SMN2 (from a single SMN2 transgene allele) expressed on a Δ7Smn background.
Table 1.
Expected vs actual outcome from Tg(SMN2)2Hungtg/0;Smn1tm1Hung/WT intercross.
| Tg(SMN2)2Hung | Smn1tm1Hung/J | Expected | Actual |
|---|---|---|---|
| wildtype | homozygous | 16.25 | 0 |
| hemizygous | homozygous | 32.5 | 12a |
| homozygous | homozygous | 16.25 | 17 |
| wild type | heterozygous | 32.5 | 36 |
| hemizygous | heterozygous | 65 | 73 |
| homozygous | heterozygous | 32.5 | 36 |
| wild type | wild type | 16.25 | 11 |
| hemizygous | wild type | 32.5 | 30 |
| homozygous | wild type | 16.25 | 19 |
| TOTAL | 260 | 235 |
denotes deviation from expected number and signifies loss either in utero or cannibalization prior to first morning observation on the day of birth.
Figure 4.

Breeding scheme to generate SMA litters with a lifespan similar to “delta 7” SMA mice. A) Breeding scheme for researchers that have access to TJL-005058 mice from The Jackson Laboratory. Genotypes of mice that are boxed represent progenitor lines or breeding stocks that are used for the final cross of obtaining SMA-like mice. B) Breeding scheme for those that do not have access to TJL-005058 and maintain their Li mouse model as a SMN2 hemizygote and Smn heterozygote. Note in both schemes the expected value of 50% control and 50% SMA-like pups are shown, but the actual value may differ based upon data presented in Table 1.
Overall, the survival results that we obtained from our F1 intercross were consistent with the original report by Hsieh-Li et al. (2000) [7]. Severe SMA pups die within 1-7 days of birth, intermediate pups die between 12-17 days of age, and finally mild SMA mice have a necrotic tail and late in life present with muscle weakness. We believe a lack of clarity of how the genetic crosses were performed and the reporting of the subsequent results generated confusion in the original and subsequent publications by others.
It remains unclear why mice of the identical genotype (Tg(SMN2)2Hungtg/0;Smn1tm1Hung/tm1Hung) fall into disease categories of either severe (death 1-7 days) or intermediate (death 12-17 days). The simplest explanation would be that the cross originally reported by Hsieh-Li [7], as well as the one we performed was not on a fully congenic FVB/N background. This is similar to our experience using a hybrid FVB/N background with the most commonly used SMA mouse model, the “delta7” SMA mouse (TJL-005025; SMN2Ahmb89tg/tg;SMNΔ7tg/tg:Smn1−/−)[9]. We can find SMA pups that are significantly weaker than other mutants within the same litter. They fail to gain weight and die by P5-P7 while their mutant siblings survive to P13 (unpublished data and [18]). Hence we feel that Tg(SMN2)2Hungtg/0;Smn1tm1Hung/tm1Hung mice are equivalent to “delta 7” SMA mice and should be used to compliment or replace them for pre-clinical and pathological studies. It should be noted that the extensive necrosis that occurs in “delta7” SMA mice that have extended survival through some therapeutic means, will most likely occur with these mice as well.
There are two advantages for using TJL-005058 mice. The first is that Tg(SMN2)2Hungtg/0;Smn1tm1Hung/tm1Hung offspring can survive to 12-17 days without over-expressing high levels of Δ7SMN protein as does the “delta7” SMA mouse model. The second is the potential to generate mice in which only two genotypes, mutants or controls, will be born at a 1:1 ratio. We provide two different breeding schemes by which this can be achieved (Figure 4A and 4B).
The first breeding scheme is for those who have access to TJL-005058 mice (Figure 4A). In this strategy, TJL-005058 mice can be outcrossed to FVB/N mice to generate F1 progeny that are all hemizygous for SMN2 and heterozygous for the Smn1tm1Hung allele. Subsequently, these F1s can be backcrossed to FVB/N mice to generate F2 progeny. These F2s should be genotyped, and only those mice that are positive for the Smn1tm1Hung allele and negative for the SMN2 transgene should be maintained and developed into a stock line. A cross between TJL-005058 mice that are homozygous for both Tg(SMN2)2Hung and Smn1tm1Hung to mice that are heterozygous for the Smn1tm1Hung allele should yield litters in which 50% of the pups are either controls or SMA mutants. The actual percentage may deviate from this based upon our intercross data (Table 1).
The second strategy (Figure 4B) is for those investigators that either cross Smn1tm1Hung heterozygotes to Tg(SMN2)2Hung tg/0;Smn1tm1Hung/wt mice or intercross Tg(SMN2)2Hungtg/0;Smn1tm1Hung/wt mice and use qPCR to determine SMN2 copy number. In this scenario an intercross between mice that are hemizygous for SMN2 and heterozygous for Smn1tm1Hung results in F1 progeny that are scored phenotypically. Only those mice that survive and lose their tail should be kept. These mice are homozygous for both Tg(SMN2)2Hung and Smn1tm1Hung. In contrast, mice that are either heterozygous for the Smn1tm1Hung allele or wildtype at the endogenous Smn locus will have a tail and should be sacrificed. Finally, SMA mutant mice that are hemizygous for SMN2 and homozygous for Smn1tm1Hung alleles will die. The mice that survive and lose their tail are equivalent to the TJL-005058 line and can be crossed to heterozygous Smn1tm1Hung mice to generate litters in which half of the pups that are born alive should be mutants.
In summary we have evaluated molecularly the SMN2 transgene, (SMN2)2Hung, and the targeted Smn allele, Smn1tm1Hung. We find that these lines, which have been deposited at the Jackson Laboratory, are consistent with the original report by Hsieh-Li [7] for the SMN2 founder line 2 and the targeted Smn mutation. Our results also demonstrate that this is the same SMA model reported by the Charbonnier laboratory in France [11, 12]. Finally our phenotypic results confirm and clarify the original results reported by Hsieh-Li [7]. We suggest that this underused SMA model serve as a compliment or possible alternative to the “delta 7” SMA model and provide two different breeding schemes that can be used to generate litters in which 50% of the pups that are born alive will be mutants.
Supplementary Material
Supplementary Figure 1. Tg(SMN2)2Hung junction specific PCR on Li-SMA model mice from France. Tail biopsies from Li-SMA model mice that are used in France were analyzed with junction specific primers and found to be positive (noted as France 5058). TJL-005058 mice were used as a positive control and TJL-005024, which contain a different SMN2 BAC transgene, (SMN2)Ahmb89, was used as a negative control along with wild type FVB/N DNA. A primer set that amplifies all SMN2 BAC transgenes was used as a control to demonstrate the presence of SMN2 in all DNA samples except wild type FVB/N mice.
Figure 3.
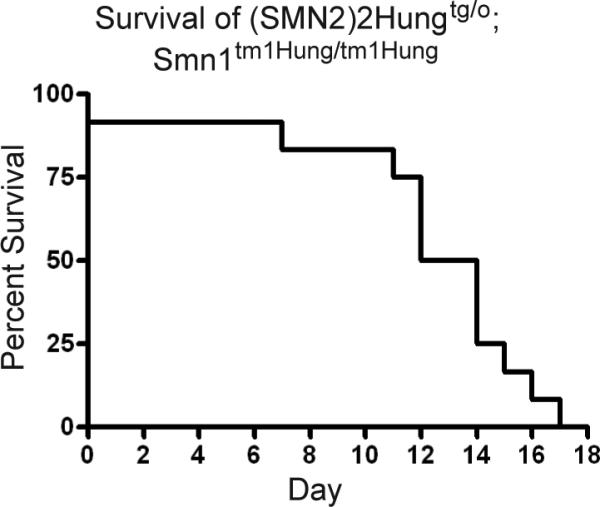
Kaplan-Meier survival analysis of SMA-like mice that are hemizygous for Tg(SMN2)2Hung and homozygous for the Smn1tm1Hung allele.
Acknowledgements
We would like to acknowledge the scientific contributions that Dr. Hung Li and his laboratory have contributed to the SMA research field over the past 10 years. It is our hope that this manuscript supports his legacy and provides benefit to the scientific community to more fully utilize the resource that he and Academia Sinica have provided. We also thank Dr. Frédéric Charbonnier for generously providing tail biopsies to assess the SMN2 transgene in his Li-SMA model mice.
Funding
R.G. is supported by a NIH training grant [T32 AG000260] “Drug Discovery Training in Age-related Disorders”. Previous and current research support for these studies was provided by The Muscular Dystrophy Association #3572, Families of Spinal Muscular Atrophy DiD0809, the NIH grants #1ROIN5060926 (NINDS), and 1R21HD058311-01A1 (NICHD) to CJD and the SMA Foundation to C.L. During a portion of this work C.J.D. was an American Academy of Neurology/SMA Foundation Young Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiology of disease. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy- determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 3.Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Human molecular genetics. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 5.DiDonato CJ, Chen XN, Noya D, et al. Cloning, characterization, and copy number of the murine survival motor neuron gene: homolog of the spinal muscular atrophy-determining gene. Genome Res. 1997;7:339–352. doi: 10.1101/gr.7.4.339. [DOI] [PubMed] [Google Scholar]
- 6.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 8.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Human molecular genetics. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 9.Le TT, Pham LT, Butchbach ME, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Human molecular genetics. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 10.Monani UR, Pastore MT, Gavrilina TO, et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biondi O, Grondard C, Lecolle S, et al. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2008;28:953–962. doi: 10.1523/JNEUROSCI.3237-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grondard C, Biondi O, Armand AS, et al. Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci. 2005;25:7615–7622. doi: 10.1523/JNEUROSCI.1245-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. The Journal of clinical investigation. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai LK, Tsai MS, Ting CH, et al. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. Journal of molecular medicine (Berlin, Germany) 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 15.Tsai LK, Tsai MS, Ting CH, et al. Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiology of disease. 2008;31:361–367. doi: 10.1016/j.nbd.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Tsai LK, Tsai MS, Lin TB, et al. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiology of disease. 2006;24:286–295. doi: 10.1016/j.nbd.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Frugier T, Tiziano FD, Cifuentes-Diaz C, et al. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Human molecular genetics. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 18.Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside genetecin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Human molecular genetics. 2009 doi: 10.1093/hmg/ddp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Tg(SMN2)2Hung junction specific PCR on Li-SMA model mice from France. Tail biopsies from Li-SMA model mice that are used in France were analyzed with junction specific primers and found to be positive (noted as France 5058). TJL-005058 mice were used as a positive control and TJL-005024, which contain a different SMN2 BAC transgene, (SMN2)Ahmb89, was used as a negative control along with wild type FVB/N DNA. A primer set that amplifies all SMN2 BAC transgenes was used as a control to demonstrate the presence of SMN2 in all DNA samples except wild type FVB/N mice.