

Abstract
The isolation and structural characterization of metallacyclic allyl (2a) and crotyl (2b) iridium complexes are reported. Complexes 2a and 2b are rare examples of iriduim allyl complexes that undergo nucleophilic attack at terminal position, rather than the central position, of the allyl unit. Structures of 2a and 2b were obtained by X-ray diffraction. Nucleophilic attack was observed at the carbon that is bound to iridium trans to phosphorus through a longer Ir-C bond. However, the effect of the trans phosphine ligand on the Ir-C bond lengths was smaller than the effect of the substituent on the allyl group in 2b. The competence of complexes 2a and 2b to be intermediates in the catalytic asymmetric allylic substitutions was evaluated by studying their reactivity towards stabilized carbon and heteroatom nucleophiles and comparing the rates and selectivities to those of the catalytic reactions. The stereoselectivity and regioselectivity of stoichiometric reactions of 2b were similar to those of reactions catalyzed by the previously reported iridium catalysts, supporting their intermediacy in the catalytic reactions. Based on the structural data, a model is proposed for the origin of stereoselectivity in iridium-catalyzed asymmetric allylic substitution reactions.
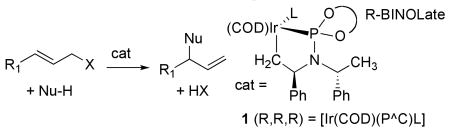
Iridium-catalyzed allylic substitution has become a powerful enantioselective method to form both carbon-heteroatom1-5 and carbon-carbon6-10 bonds (eq 1). When conducted with the proper combination of iridium precursor and phosphoramidite ligands, these reactions form branched substitution products with high regioselectivity and enantioselectivity.
 |
(1) |
These reactions likely occur through an allyliridium intermediate, but the composition and structure of this intermediate has been elusive. Studies from the authors' laboratory led to the isolation of a trapped form of the active catalyst, which possesses a cyclometalated phosphoramidite ligand.11 However, kinetic studies have implied that an allyliridium complex derived from this metalacycle would be a fleeting intermediate in the catalytic system.12 Here, we describe the independent synthesis, characterization, and reactivity of allyliridium complexes that are kinetically and chemically competent to be the elusive allyliridium intermediate. Structural data help reveal the origins of regioselectivity and explain how enantioselectivity is controlled by a stereocenter in the catalyst that is distant from that in the product.
An obvious route to the potential allyliridium intermediates is the reaction of an allylic ester with [Ir(COD)(PˆC)L], a stable adduct of the catalytically active metalacycle. However, treatment of [Ir(COD)(PˆC)L] in which L is a phosphoramidite, PPh3 or ethylene, with allyl methyl carbonate led to no apparent reaction. This observation is consistent with a previous conclusion that the oxidative addition is thermodynamically unfavorable.12
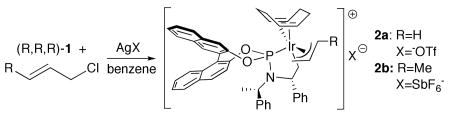
Thus, we tested the reactions of the iridium(I) complex 1 containing an ethylene ligand with more reactive allylic halides (Eq 2). Treatment of complex 1 with allyl chloride in benzene at room temperature led to the rapid formation of a new product corresponding to a singlet resonance at 125 ppm in the 31P NMR spectrum of the crude mixture. Addition of AgOTf in THF to this complex generated in situ formed the stable allyliridium complex 2a. After filtration of the AgCl, this complex deposited as single crystals from the reaction mixture in 55% yield upon standing at room temperature under inert atmosphere for 2 days.
 |
(2) |
An analogous crotyl complex 2b was prepared by similar methods. The reaction of ethylene complex 1 with trans-crotyl chloride in benzene, followed by abstraction of the chloride with AgSbF6, generated samples of crotyl complex 2b. Crystals suitable for X-able for X-ray diffraction formed in 30% yield upon standing at room temperature for 2 days.
The products of these reactions were identified as allyliridium complexes containing an intact metalacycle and a cyclooctadiene ligand by one-dimensional and two-dimensional 1H, 13C and 31P NMR spectroscopy, as well as X-ray diffraction. A set of 1H NMR resonances for the allyl ligand in 2a were observed at 4.63, 4.22, 3.85, 3.14 and 2.71 ppm. 1H resonances for the coordinated olefinic portion of the COD ligand and for the methylene protons of the metalacycle were clearly identified. Similar 1H NMR resonances for 2b were observed, save for a downfield shift of the allylic proton of the substituted terminus at 4.73 ppm. The 31P NMR spectra for 2a and 2b consisted of a singlet near 125 ppm.
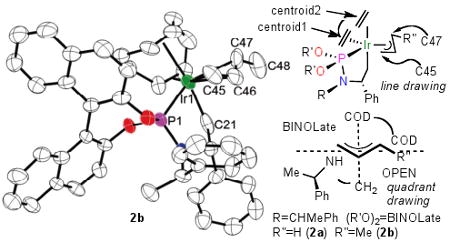
Table 1 contains an ORTEP diagram, line drawing, and quadrant diagram of the crotyl complex 2b and metric parameters for the core of both 2a and 2b. An ORTEP diagram for the nearly identical structure of allyl complex 2a is included in supporting information. These complexes can be considered to be severely distorted octahedra with the allyl ligand, an olefinic portion of the COD ligand, and the phosphorus of the metalacycle occupying one plane, and the other olefinic unit of the COD and a methylene of the metalacycle lying apical to this plane. The four angles involving the olefin centroid, two allyl termini and phosphorus atom of parent complex 2a total 361.9°, indicating the relative planarity of these groups with the metal. The angles between the termini of the allyl group and the metalacycle methylene carbon are 90.9° (C21-Ir-C47) and 102.9° (C21-Ir-C45), indicating the apical positioning of this methylene group, and the angles between one of the olefin centroids and the terminal carbons of the allyl unit are 90.0° (cent2-Ir-C45) and 95.7° (cent2-Ir-C47), indicating the apical positioning of this olefin unit in the opposite direction. The metal-carbon distances to the two allyl termini are 2.204(3)Å and 2.274(3)Å. This 0.07Å difference in distance reflects the trans influence of the phosphoramidite and olefin ligands on the metal-carbon bond lengths. The effect of a trans ligand on bond distances was stated to be small in the classic review on trans influence,13 and our data are consistent with this assertion.
Table 1.
ORTEP diagram of 2b and bond distances and angles around Iridium for complexes 2a and 2b.
 | |||||
|---|---|---|---|---|---|
| Distances (Å) | 2a | 2b | Angles (°) | 2a | 2b |
| Ir-P | 2.2685(6) | 2.280(3) | cent1-Ir-C47 | 102.2 | 103 |
| Ir-C45 | 2.204(3) | 2.240(10) | C47-Ir-C45 | 66.4(1) | 66.0(5) |
| Ir-C47 | 2.274(3) | 2.377(11) | cent2-Ir-P | 103.8 | 104.6 |
| Ir-C21 | 2.125(3) | 2.114(15) | cent2-Ir-C45 | 90.0 | 90.9 |
| Ir-cent1 | 2.099 | 2.097 | cent2-Ir-cent1 | 82.3 | 81.4 |
| Ir-cent2 | 2.244 | 2.215 | cent2-Ir-C47 | 95.8 | 97.3 |
| C21-Ir-P | 75.73(8) | 75.0(4) | |||
| Angles (°) | 2a | 2b | C21-Ir-C45 | 102.9(1) | 102.5(5) |
| P-Ir-C45 | 86.96(8) | 85.9(3) | C21-Ir-C47 | 90.9(1) | 89.9(5) |
| P-Ir-cent1 | 106.6 | 107.6 | C21-Ir-cent1 | 85.6 | 86.4 |
The structure of crotyl complex 2b is similar to that of 2a. The major differences between the two structures involve the substituted terminal carbon (C47) of the allyl unit. The iridium-C47 bond length in 2b is 0.1 Å longer than the corresponding bond in 2a. The two angles involving the allyl termini, the iridium and the two apical ligands indicate that the allyl group in 2b is rotated by 1.5-2°, relative to the allyl group in 2a. These structural differences presumably result from greater steric interactions of the COD ligand with C47 in methylallyl 2b than with C47 of allyl complex 2a, greater electron donation by the methyl substituent, or both. Other bond lengths and angles in 2a and 2b agree within 0.04 Å and 1° respectively.
The competence of allyl complexes 2a and 2b in catalytic asymmetric allylic substitutions was assessed by studying their reactions with carbon and heteroatom nucleophiles. After reaction of 2a and 2b with the nucleophiles, PPh3 was added to displace the 1-alkene product and form the known [Ir(COD)(PˆC)-(PPh3)].11 The yields, rates, and selectivities of these stoichiometric reactions were compared to those of the reactions of methallyl methyl carbonate catalyzed by [Ir(COD)(PˆC)(C2H4)]. The reactions of 2a and 2b with eight different nucleophiles are shown in Scheme 1. The reactions occurred rapidly at room temperature. The regioselectivities, as well as the enantioselectivities, of the stoichiometric reactions of 2b were within 5% and 10%, respectively, of the values for the analogous catalytic reactions (see supporting information). Thus, 2a and 2b are chemically and kinetically competent to be intermediates in the catalytic processes.
Scheme 1. Reactions of Allyl Complexes 2a,b with nucleophilesa,b.

a Yields and selectivities determined for reactions at room temperature for 0.5 h in THF-d8; b Reactions of amines were conducted with triethylamine or Bu4N[OC(O)CH3] as base.
The reaction of parent allyl complex 2a with a soluble carboxylate (Scheme 1) was also studied to test the reversibility of the oxidative addition of allylic esters to 1. Consistent with the proposed endothermicity of the oxidative addition, sodium-2-methylvaleriate reacted with 2a to form Ir(I) and the allylic ester.
The reactivity of 2a and 2b contrasts with that of previous allyliridium complexes. In contrast to 2a and 2b, previous allyliridium complexes have undergone reactions with nucleophiles at the central carbon of the allyl unit.14,15 The structural data provide insights into the regioselectivity of attack on the allyl intermediates. Consistent with data on allylpalladium complexes,16-18 attack on allyliridium 2b occurs at the carbon atom containing the longer M-C bond. This carbon is located trans to the ligand with the stronger trans influence.13 However, a comparison of the structures of 2a and 2b imply that the effect of the trans ligand on the bond distances is smaller than the effect of the methyl substituent.
Finally, the structure of 2b helps explain the origin of enantioselectivity. The stereochemistry of the allyl ligand and products corresponding to aniline and sodium dimethylmalonate nucleophiles is consistent with attack of the nucleophile anti to the metal center. The enantioselectivity by iridium catalyst 1 has been shown to originate from the stereochemistry of the β-carbon in the metalacycle,19 but this stereocenter is located far from the expected trajectory of the nucleophile. The quadrant diagram in Table 1 implies that the relay of stereochemistry from this site to the metal center to the allyl ligand occurs by orientation of the aryl group of the phenethyl substituent by the aryl group on the β-carbon. The phenethyl aryl group then blocks, in combination with the binolate group, adoption of the stereoisomer in which the methyl group of the allyl ligand would be oriented in either of the two western quadrants. The difference in steric properties of the COD and metalacycle methine hydrogen causes the allyl methyl group to occupy the southeastern quadrant that contains the less sterically demanding methine hydrogen. The allyl methyl substituent extends into solution, and this structural feature allows the reaction to occur in a similar fashion with many allylic electrophiles. Studies on the relationship between the dynamics of these allyl complexes and enantioselectivity, and the preparation of further derivatives of 2a and 2b, are ongoing.
Supplementary Material
Acknowledgments
We thank the NIH (NIH-GM55382 and 58108) for support and Johnson-Matthey for a gift of [Ir(cod)Cl]2.
Footnotes
Supporting Information Available: Experimental procedures for the synthesis and reactions of 2a and 2b and for the catalytic reactions of trans-methyl crotyl carbonate. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ohmura T, Hartwig JF. J Am Chem Soc. 2002;124:15164. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]
- 2.Shu CT, Hartwig JF. Angew Chem Int Ed. 2004;43:4794. doi: 10.1002/anie.200460214. [DOI] [PubMed] [Google Scholar]
- 3.Lyothier I, Defieber C, Carreira EM. Angew Chem, Int Ed. 2006;45:6204. doi: 10.1002/anie.200602408. [DOI] [PubMed] [Google Scholar]
- 4.Pouy MJ, Leitner A, Weix DJ, Ueno S, Hartwig JF. Org Lett. 2007;9:3949. doi: 10.1021/ol701562p. [DOI] [PubMed] [Google Scholar]
- 5.Satoshi Ueno JFH. Angew Chem Int Ed. 2008;47:1928–1931. doi: 10.1002/anie.200705267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tissot-Croset K, Polet D, Alexakis A. Angew Chem, Int Ed. 2004;43:2426. doi: 10.1002/anie.200353744. [DOI] [PubMed] [Google Scholar]
- 7.Alexakis A, Polet D. Org Lett. 2004;6:3529. doi: 10.1021/ol048607y. [DOI] [PubMed] [Google Scholar]
- 8.Graening T, Hartwig JF. J Am Chem Soc. 2005;127:17192. doi: 10.1021/ja0566275. [DOI] [PubMed] [Google Scholar]
- 9.Weix DJ, Hartwig JF. J Am Chem Soc. 2007;129:7720. doi: 10.1021/ja071455s. [DOI] [PubMed] [Google Scholar]
- 10.Helmchen G, Dahnz A, Duebon P, Schelwies M, Weihofen R. Chem Commun. 2007:675. doi: 10.1039/b614169b. [DOI] [PubMed] [Google Scholar]
- 11.Kiener CA, Shu C, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]
- 12.Markovic D, Hartwig JF. J Am Chem Soc. 2007;129:11680. doi: 10.1021/ja074584h. [DOI] [PubMed] [Google Scholar]
- 13.Appleton TG, Clark HC, Manzer LE. Coord Chem Rev. 1973;10:335. [Google Scholar]
- 14.Wakefield JB, Stryker JM. J Am Chem Soc. 1991;113:7057. [Google Scholar]
- 15.Garcia-Yebra C, Janssen JP, Rominger F, Helmchen G. Organometallics. 2004;23:5459. [Google Scholar]
- 16.Hayashi T, Kawatsura M, Uozumi Y. Chem Commun. 1997:561. [Google Scholar]
- 17.Helmchen G, Pfaltz A. Acc Chem Res. 2000;33:336. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 18.Pretot R, Pfaltz A. Angew Chem, Int Ed. 1998;37:323. doi: 10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J Am Chem Soc. 2005;127:15506. doi: 10.1021/ja054331t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.