

The hydroamination of olefins1 is one of the simplest and most atom-economical methods to prepare alkylamines, and much effort has now been spent to develop catalysts for this process. Some of most reactive catalysts contain lanthanides2 and group IV transition metals.3 However, the high sensitivity of these catalysts toward air and moisture and their low tolerance of polar functional groups have limited their use and motivated efforts to develop late-metal hydroamination catalysts. Although several catalysts based on late metals have been published for additions of amines to vinylarenes4–6 and for the addition of amides and sulfonamides to alkenes, allenes, and dienes,7 few catalysts for additions of amines to alkenes, arguably the most important donors and acceptors, have been reported.
The existing late-metal catalyst for the intramolecular additions of amines to olefins8 is generated from [PtCl2(H2CdCH2)]2 and PPh3. This system operates at 120 °C, catalyzes only reactions of secondary amines, not primary amines, and catalyzes additions to only terminal olefins, not internal olefins.9 Moreover, this catalyst requires substituents that bias the substrate toward cyclization. Alterative palladium catalysts for additions of amides and carbamates at lower temperature have been reported,10 but these N−H bond donors require installation and removal of the activating group in most synthetic sequences and do not provide products that would result from addition of a secondary amine. Thus, a catalyst that tolerates polar functional groups and that induces additions of both primary and secondary amines without activating groups to both terminal and internal alkenes is needed.
We report the identification of such a system. We report a rhodium catalyst for the hydroamination of terminal and internal alkenes with primary and secondary amines, with or without substituents that favor cyclization to form five- or six-membered ring products. In addition to spanning a broader scope of amine and alkene than other late metal catalysts, this system operates in the presence of functional groups, including esters and free hydroxyl groups, which do not tolerate early metal and lanthanide catalysts. These results demonstrate that complexes of group 9 metals, which are classic catalysts for alkene hydrogenation and hydrosilylation, can also be made useful for alkene hydroamination.
 |
(1) |
Stimulated by our previous studies on rhodium-catalyzed additions of amines to vinylarenes,5,6 we investigated combinations of rhodium precursors and dative ligands as catalysts for cyclization of the N-methyl aminopentene in eq 1. Experiments to develop conditions for the cyclization of aminoalkene 1 are summarized in Table 1. These reactions were conducted at 70 °C over 7 h, unless otherwise stated.
Table 1.
Effect of Ligand on the Selectivity for Rhodium-Catalyzed Intramolecular Hydroamination of the Aminoalkene in Equation 1a
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | ligand b | %Ac | %Bc | %Cc | %Dc |
| 1 | [Rh(COD)2]BF4 | DPEphos | 0 | 0 | 93 | 0 |
| 2 | [Rh(COD)2]BF4 | DPPB | 10 | 40 | 10 | 30 |
| 3 | [Rh(COD)2]BF4 | PPh3 | 0 | 0 | 92 | 0 |
| 4 | [Rh(COD)2]BF4 | PCy3 | 0 | 30 | 30 | 30 |
| 5 | [Rh(COD)2]BF4 | DPPF | 20 | 30 | 10 | 30 |
| 6 | [Rh(COD)2]BF4 | t-BuXantphos | 60 | 10 | 10 | 10 |
| 7 | [Rh(COD)2]BF4 | L1 | 86 | 0 | 0 | 0 |
| 8 | [Rh(COD)2]BF4 | L2 | 93 | 0 | 0 | 0 |
| 9 | [Rh(MeCN)2(COD)]BF4 | L2 | 92 | 0 | 0 | 0 |
| 10d | [Rh(COE)2Cl]2 | L2 | 28 | 0 | 0 | 0 |
| 11d | [Rh(COD)Cl]2 | L2 | 9 | 0 | 0 | 0 |
| 12 | 5% HBF4·OEt2 | L2 | 0 | 0 | 0 | 0 |
Reaction conditions: 0.5 mmol of amine 1, 2.5 mol % of Rh, and 2.5 mol % of biphosphine or 5 mol % monophosphine in 0.5 mL of dioxane at 70 °C for 7 h unless otherwise specified.
Ligand structures are shown above the table.
GC yields.
Reaction run using 1.25 mol % catalyst.
Reaction of aminoalkene 1 in the presence of 2.5 mol % [Rh-(COD)2]BF4 and 2.5 mol % DPEphos, which catalyzed the anti-Markovnikov hydroamination of vinylarenes,6 formed none of the cyclized pyrrolidine product. The major product of this reaction resulted from isomerization of the olefin from the terminal to the internal position.11 Reactions catalyzed by 2.5% [Rh(COD)2]BF4 and 2.5% DPPB (1,4-bis(diphenylphosphino)butane), which catalyzed the intramolecular hydroamination of vinylarenes,5 led to a mixture of cyclic amine, isomerized olefin, cyclic enamine, and alkylamine, with the cyclic amine comprising only 10% of the material. Reactions catalyzed by complexes of other ligands (entries 3–6) also formed mixtures, but two systems formed exclusively cyclic amine. The catalyst generated from [Rh(COD)2]BF4 and an amino analogue of Xantphos L1 or the biarylphosphine ligand L2 developed in the Buchwald laboratory for cross-coupling12 formed the cyclic amine as the sole detectable product in 86 and 93% yield by GC (entries 7 and 8). Studies with various rhodium precursors (entries 8–11) showed that [Rh(COD)2]BF4 formed the most active catalyst. Reactions with protic acid as catalyst, which could be generated from the rhodium precursor,13a formed no product (entry 12).13b
The scope of the hydroamination of 5- and 6-N-alkyl aminoalkenes to form pyrrolidines and piperidines under these reaction conditions are summarized in Table 2. Cyclizations to form five-membered rings with aminoalkenes containing not only N-methyl (Table 1), but more bulky N-cyclohexylmethyl and N-benzyl amines (Table 2, entries 1–2) generated high yields of cyclized products. These cyclizations occurred for substrates with (entries 1–4) or without (entries 5–7) gem-disubstitution on the linker chain. Likewise, cyclizations to form six-membered rings occurred with substrates possessing (entry 8) or fully lacking substituents in the linking chain (entry 9).
Table 2.
Rhodium Catalyzed Intramolecular Hydroamination of Secondary Aminoalkene a
| entry | alkenyl amine | product | yield %b |
|---|---|---|---|
| 1 | 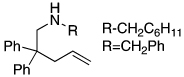 |
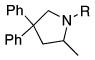 |
91 |
| 2 | 91 | ||
| 3 | 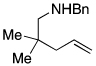 |
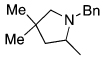 |
83 |
| 4 | 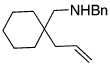 |
 |
92 |
| 5 |  |
 |
69 |
| 6 | 62 | ||
| 7 | 72 | ||
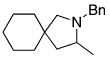
| 8 |  |
 |
86 |
| 9 |  |
 |
74 |
| 10 |  |
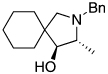 |
65 (11:1) |
| 11 |  |
 |
96 |
| 12 |  |
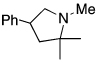 |
80c |
| 13 | 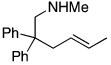 |
 |
76d |
Reaction conditions: 0.5 mmol aminoalkene, 2.5 mol % of [Rh(COD)2]-BF4, and 3 mol % of L2 in 0.5 mL of dioxane at 70 °C for 7 h unless otherwise specified.
Isolated yield (average of two runs).
Reaction run using 7.5 mol % of catalyst and 9 mol % of ligand at 120 °C.
Reaction run using 5 mol % of catalyst and 6 mol % of ligand at 100 °C.
In addition, these reactions occurred in the presence of a variety of functional groups, such as halides, nitriles, and esters (entries 5–7). Perhaps most remarkable, reaction of the substrate in entry 10 containing an allylic alcohol function occurred in good yield with high diastereoselectivity without significant decomposition of the alcohol or deactivation of the catalyst. The reaction of N-benzyl-2,2-diphenylpent-4-en-1-amine even occurred in an 82% yield in the presence of 5.0 equiv of added water.
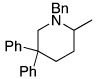
Most additions of amines to alkenes have been conducted with terminal, monosubstituted olefins. However, reactions with disubstituted olefins catalyzed by this rhodium system also formed cyclized products. Additions of amines occurred across geminally substituted olefins (entries 11 and 12), including those in relatively unbiased substrates, and across an unstrained internal olefin (entry 13).14
The rhodium catalyst also increased the scope of amines that would add to alkenes. For example, the species generated from 5% Rh(COD)2BF4 and 6% of L2 catalyzed the cyclization of aminoalkenes containing primary amine units (Table 3). Reactions of primary aminoalkenes to form five- and six-membered rings occurred in good yields.
Table 3.
Rhodium Catalyzed Intramolecular Hydroamination of Primary Aminoalkene a

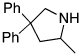

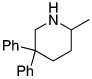
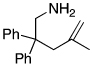
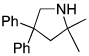

Reaction conditions: 0.5 mmol aminoalkene, 5 mol % of rhodium, and 6 mol % of L2 in 0.5 mL of dioxane at 100 °C for 10 h unless otherwise specified.
Isolated yield.
Reaction was run for 1 d.
NMR yield.
In summary, we report a rhodium complex that catalyzes cyclizations of aminoalkenes under mild conditions with substrates containing primary or secondary amines, terminal or internal alkenes, and linkers that possess or lack substituents that bias the substrate toward cyclization. This reaction tolerates a variety of common functional groups. Studies on the mechanism of this process and development of enantioselective versions of this process are ongoing.
Supplementary Material
Acknowledgment
We thank the NIH (NIGMS GM-55382) for support of this work and Johnson-Matthey for rhodium.
Footnotes
Supporting Information Available: All experimental procedures and characterization data of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Beller M, Seayad J, Tillack A, Jiao H. Angew. Chem., Int. Ed. 2004;43:3368. doi: 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]; (b) Hultzsch KC. Org. Biomol. Chem. 2005;3:1819. doi: 10.1039/b418521h. [DOI] [PubMed] [Google Scholar]; (c) Muller TE, Beller M. Chem. ReV. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]; (d) Brunet JJ, Neibecker D. In: Catalytic Heterofunctionalizaiton from Hydroamination to Hydrozirconation. Togni A, Gruetzmacher H, editors. New York: Wiley-VCH; 2001. p. 91. [Google Scholar]
- 2.(a) Hong S, Marks TJ. Acc. Chem. Res. 2004;37:673. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]; (b) Stubbert BD, Marks TJ. J. Am. Chem. Soc. 2007;129:6149. doi: 10.1021/ja0675898. [DOI] [PubMed] [Google Scholar]; (c) Stubbert BD, Marks TJ. J. Am. Chem. Soc. 2007;129:4253. doi: 10.1021/ja0675898. [DOI] [PubMed] [Google Scholar]; (d) Yu X, Marks TJ. Organometallics. 2007;26:365. [Google Scholar]; (e) Ryu JS, Marks TJ, McDonald FE. J. Org. Chem. 2004;69:1038. doi: 10.1021/jo035417c. [DOI] [PubMed] [Google Scholar]; (f) Gribkov DV, Hultzsch KC, Hampel F. J. Am. Chem. Soc. 2006;128:3748. doi: 10.1021/ja058287t. [DOI] [PubMed] [Google Scholar]; (g) Kim JY, Livinghouse T. Org. Lett. 2005;7:1737. doi: 10.1021/ol050294z. [DOI] [PubMed] [Google Scholar]; (h) Kim YK, Livinghouse T. Angew. Chem., Int. Ed. 2002;41:3645. doi: 10.1002/1521-3773(20021004)41:19<3645::AID-ANIE3645>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]; (i) Molander GA, Pack SK. J. Org. Chem. 2003;68:9214. doi: 10.1021/jo035205f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bytschkov I, Doye S. Eur. J. Org. Chem. 2003;2003:935. [Google Scholar]; (b) Gribkov DV, Hultzsch KC. Angew. Chem., Int. Ed. 2004;43:5542. doi: 10.1002/anie.200460880. [DOI] [PubMed] [Google Scholar]; (c) Thomson RK, Bexrud JA, Schafer LL. Organometallics. 2006;25:4069. [Google Scholar]; (d) Wood MC, Leitch DC, Yeung CS, Kozak JA, Schafer LL. Angew. Chem., Int. Ed. 2007;46:354. doi: 10.1002/anie.200603017. [DOI] [PubMed] [Google Scholar]
- 4.(a) Beller M, Trauthwein H, Eichberger M, Breindl C, Herwig J, Muller TE, Thiel OR. Chem. Eur. J. 1999;5:1306. [Google Scholar]; (b) Utsunomiya M, Hartwig JF. J. Am. Chem. Soc. 2003;125:14286. doi: 10.1021/ja0375535. [DOI] [PubMed] [Google Scholar]
- 5.Takemiya A, Hartwig JF. J. Am. Chem. Soc. 2006;128:6042. doi: 10.1021/ja058299e. [DOI] [PubMed] [Google Scholar]
- 6.Utsunomiya M, Kuwano R, Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 2003;125:5608. doi: 10.1021/ja0293608. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bender CF, Widenhoefer RA. Chem. Commun. 2006:4143. doi: 10.1039/b608638a. [DOI] [PubMed] [Google Scholar]; (b) Brouwer C, He C. Angew. Chem., Int. Ed. 2006;45:1744. doi: 10.1002/anie.200504495. [DOI] [PubMed] [Google Scholar]; (c) Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]; (d) Komeyama K, Morimoto T, Takaki K. Angew. Chem., Int. Ed. 2006;45:2938. doi: 10.1002/anie.200503789. [DOI] [PubMed] [Google Scholar]; (e) Liu XY, Li CH, Che CM. Org. Lett. 2006;8:2707. doi: 10.1021/ol060719x. [DOI] [PubMed] [Google Scholar]; (f) Taylor JG, Whittall N, Hii KK. Org. Lett. 2006;8:3561. doi: 10.1021/ol061355b. [DOI] [PubMed] [Google Scholar]; (g) Zhang J, Yang CG, He C. J. Am. Chem. Soc. 2006;128:1798. doi: 10.1021/ja053864z. [DOI] [PubMed] [Google Scholar]; (h) Zhang Z, Bender CF, Widenhoefer RA. Org. Lett. 2007;9:2887. doi: 10.1021/ol071108n. [DOI] [PubMed] [Google Scholar]; (i) Zhang Z, Liu C, Kinder RE, Han X, Qian H, Widenhoefer RA. J. Am. Chem. Soc. 2006;128:9066. doi: 10.1021/ja062045r. [DOI] [PubMed] [Google Scholar]
- 8.For intermolecular addition to hexene at high temperature in modest yields, see: Brunet JJ, Chu NC, Diallo O. Organometallics. 2005;24:3104.; For intermolecular addition to ethylene, see: Brunet JJ, Cadena M, Chu NC, Diallo O, Jacob K, Mothes E. Organometallics. 2004;23:1264. Coulson DR. Tetrahedron Lett. 1971;12:429.
- 9.Bender CF, Widenhoefer RA. J. Am. Chem. Soc. 2005;127:1070. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]
- 10.Michael FE, Cochran BM. J. Am. Chem. Soc. 2006;128:4246. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]
- 11.The side products were identified by signals in the crude 1H NMR spectrum and by GC–MS.
- 12.Gaertzen O, Buchwald SL. J. Org. Chem. 2002;67:465. doi: 10.1021/jo0107756. [DOI] [PubMed] [Google Scholar]
- 13.(a) Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org. Lett. 2006;8:4179. doi: 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]; (b) Ackermann L, Kaspar LT, Althammer A. Org. Biomol. Chem. 2007;5:1975. doi: 10.1039/b706301f. [DOI] [PubMed] [Google Scholar]
- 14.(a) Casalnuovo AL, Calabrese JC, Milstein D. J. Am. Chem. Soc. 1988;110:6738. [Google Scholar]; (b) Dorta R, Egli P, Zurcher F, Togni A. J. Am. Chem. Soc. 1997;119:10857. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.