

Abstract
Clearance of recruited immune cells is necessary to resolve inflammatory reactions. We show here that matrix metalloproteinase 2 (MMP2), as part of an interleukin 13 (IL-13)–dependent regulatory loop, dampens inflammation by promoting the egress of inflammatory cells into the airway lumen. MMP2−/− mice showed a robust asthma phenotype and increased susceptibility to asphyxiation induced by allergens. However, whereas the lack of MMP2 reduced the influx of cells into bronchoalveolar lavage (BAL), numerous inflammatory cells accumulated in the lung parenchyma. BAL of MMP2−/− mice lacked normal chemotactic activity, whereas lung inflammatory cells from the same mice showed appropriate chemotactic responses. Thus, MMP2 establishes the chemotactic gradient required for egression of lung inflammatory cells and prevention of lethal asphyxiation.
The allergic lung is characterized by airway obstruction, including airway hyperresponsiveness (AHR), which is defined as enhanced smooth muscle constrictive responses to provocative challenge. The principal anatomical features of these physiological changes include the accumulation of inflammatory cells, especially eosinophils, within the lung and goblet cell metaplasia of the airway epithelium, which induces a mucus-secreting phenotype1. Although a minor cellular constituent, T helper type 2 (TH2) cells are the immunologically dominant cell type that underlie allergic lung disease2–7.
TH2 cells expand and are recruited to the lung in response to inhaled allergens. All TH2 cytokines contribute to experimental allergic lung disease, however, interleukin 4 (IL-4) is required for TH2 development, immunoglobulin E (IgE) synthesis and atopic reactions based on type 1 hypersensitivity mechanisms8–10. In contrast, IL-13—which is closely related to IL-4, and whose receptor includes the α chain of the IL-4 receptor (IL-4Rα)11–13—induces many of the usual features associated with asthma in mice14. The mechanism(s) by which IL-13 induces the asthma phenotype are unclear, but IL-13 likely represents a bridge that links immune cells with several non-hematopoietic lung tissues15. This suggests that IL-13, and to a lesser extent IL-4, may directly elicit allergic airway disease by stimulating airway epithelial and smooth muscle cells16. Additional communication between immune and parenchymal cells, perhaps via cytokines, blunt harmful immune responses and initiate repair mechanisms. However, the mechanisms that limit allergic inflammatory responses are poorly understood.
Matrix metalloproteinases (MMPs) are up-regulated during allergic inflammation17 and may participate in the pathogenesis of several lung diseases17–21. MMPs also facilitate inflammatory cell recruitment across the endothelial basement membrane22,23. We examined here the immune-mesenchymal cross-talk that occurs during allergic inflammation as well as the anti-inflammatory role of MMP2, which represents an essential link in an IL-13–dependent regulatory loop that dampens allergic inflammation.
Results
MMP2 activity in allergic lung inflammation
We induced stereotypical asthma in BALB/c mice with ovalbumin (OVA)3. BALB/c mice that were immunized and intranasally challenged with OVA to induce the allergic lung phenotype showed exaggerated airway closure, or hyperresponsiveness (AHR), in response to acetylcholine provocation as well as pronounced airway eosinophilia, increased titers of serum antigen-specific IgE and up-regulation of TH2 cytokines in bronchoalveolar lavage (BAL) (Fig. 1). These features are characteristic of human allergic asthma, which is induced by a wide variety of allergens24. Examination of BAL from saline-challenged BALB/c control mice showed that MMP2 was constitutively expressed in the airways of these mice. However, mice with the asthma phenotype showed a fivefold increase in both active and inactive (pro-) MMP2 (Fig. 1b and data not shown). These observations suggested that enhanced expression of MMP2 is a feature of the experimental asthma phenotype.
Figure 1. OVA challenge of BALB/c mice induces a robust asthma phenotype and increased MMP2 activity in BAL.
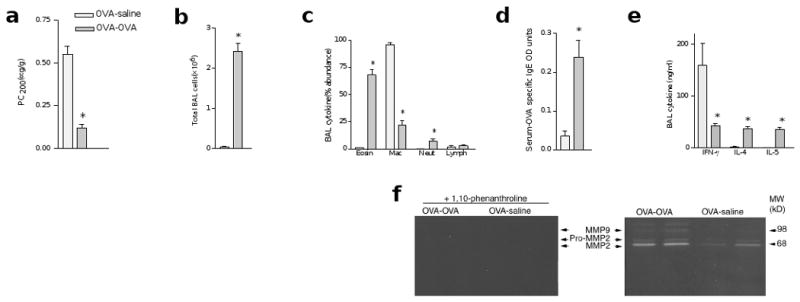
Mice were immunized intraperitoneally with OVA-alum and subsequently challenged intranasally with either saline (OVA-saline) or OVA (OVA-OVA). (a) AHR, assessed as PC200. (b) Total number of cells recovered from BAL. (c) The abundance of eosinophils (Eosin), macrophages (Mac), neutrophils (Neut) and lymphocytes (Lymph) in BAL, as assessed by modified Giemsa staining. (d) Serum OVA-specific IgE, as determined by ELISA. (e) IFN-γ, IL-4 and IL-5 concentrations in BAL fluid, as determined by ELISA. (f) Detection of BAL MMP activity by zymography. (Right) More active MMP2 (68 kD) and MMP9 (98 kD) were expressed in OVA-OVA–treated mice (n = 2 mice for each condition). The higher molecular weight bands indicated the presence of relatively inactive (pro-MMP) zymogens (left). Protease activity was neutralized by the addition of 1,10-phenanthroline, a zinc chelator, which confirmed the identity of the MMPs.
Effect of IL-13 on MMP2 and asthma development
We next determined the mechanisms that regulate MMP2 expression during acquisition of the asthma phenotype in mice that were sensitized to OVA or had received intranasal (i.n.) recombinant IL-13 (rIL-13). We found that both OVA-sensitized mice and mice challenged with rIL-13 showed enhanced expression of MMP2 protein in the BAL (Figs. 1b and 2a) and mRNA in lung (Fig. 2b); this was not observed in control tissue. This finding was specific to the lung because northern blot analysis of liver from these mice showed no MMP2 mRNA expression (data not shown). Using in situ hybridization, we found that MMP2 mRNA was primarily expressed in cells of mesenchymal origin (Fig. 2c–f). These observations showed that MMP2 is up-regulated and activated during asthma induction and that IL-13 alone can induce this.
Figure 2. Lung MMP2 is expressed in the presence of allergic inflammation and IL-13.
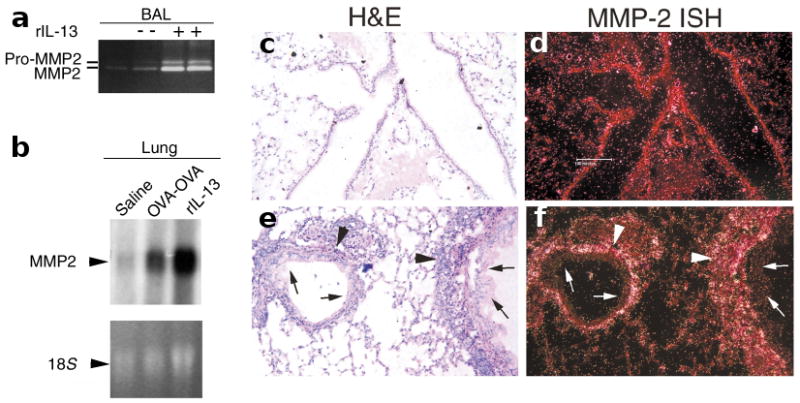
(a) Zymogram showing expression of active MMP2 and pro-MMP2 in BAL in response to i.n. challenge of mice with either saline (−) or rIL-13 (+). (b) Northern blot analysis of MMP2 expression. Total RNA from the lungs of BALB/c mice that were challenged with saline or were immunized and then challenged with either OVA (OVA-OVA) or rIL-13 was probed for MMP2 expression. The MMP2 band (3 kb) and relative loading of total RNA (as indicated by the 18S ribosomal RNA subunit band) are shown. (c–f) Lung MMP2 in situ hybridization (ISH). Airway tissue from (c,d) OVA-saline– or (e,f) OVA-OVA–challenged mice were fixed with formalin and stained with H&E; representative samples (n=3) are shown. MMP2 mRNA was detected in contiguous sections with a radiolabeled antisense MMP2-specific probe. Arrowheads indicate bright ISH signals, which appear as dark grains in H&E-stained sections (c,e) or white grains in the darkfield views (d,f). Significant amounts of MMP2 mRNA were detected only in lung mesenchyme (arrowheads) that showed active allergic inflammation (e,f); it was largely absent from airway epithelium (arrows). Bar, 100 μm.
Lack of MMP exacerbates lung inflammatory cells
To understand the in vivo role MMP2 plays in the asthma model, we first administered a synthetic inhibitor of MMPs, GM6001, to OVA-sensitized BALB/c mice at the time of i.n. OVA challenge. Although GM6001 treatment did not alter the asthma phenotype (Fig. 3a and data not shown), we observed >60% dose-dependent inhibition of inflammatory cell egression with concomitant accumulation of inflammatory cells in the lung parenchyma (Fig. 3b). Because GM6001 inhibits a number of MMPs25 we next examined the lung phenotype of MMP2-deficient mice26. Because MMP2−/− mice were crossed onto the C57BL/6 (B6) background, which is resistant to the OVA-induced asthma phenotype3, we did these experiments using complete aspergillus antigen (CAA)—which is derived from the pathogenic fungus Aspergillus fumigatus—as an allergen. CAA challenge in B6 mice induces all the key features of asthma that are observed in OVA-challenged BALB/c mice5,27: marked increases in AHR; serum total IgE and BAL mucin content (Fig. 4a–e); peribronchovascular inflammation and goblet cell metaplasia (Fig. 4f–i); and MMP2 protein and mRNA expression (data not shown). In addition, compared to saline-challenged controls, CAA-challenged MMP2−/− or GM6001-treated B6 mice showed ∼50% fewer BAL cells, with eosinophils mainly being affected (Fig. 4e). These data suggest that MMP2 accounts for much of the GM6001-induced decrease in BAL inflammatory cells.
Figure 3. Effect of MMP inhibition on AHR and BAL cell egression.

BALB/c mice were immunized with OVA-alum and intranasally challenged with saline or OVA as in Fig. 1. A third immunized group was given the MMP inhibitor GM6001 (150 mg/kg) 1 h before i.n. challenge with OVA (OVA-OVA GM6001). (a) AHR, assessed as PC200. (b) Inhibition of cell egression into BAL relative to OVA-OVA–challenged mice (see Methods). At doses of 250 mg/kg, GM6001 inhibited BAL inflammatory cell recruitment by 65%. *P≤0. 05 relative to (a) saline or (b) 50 mg/kg of GM6001.
Figure 4. Comparison of GM6001 treatment to MMP2 deficiency.
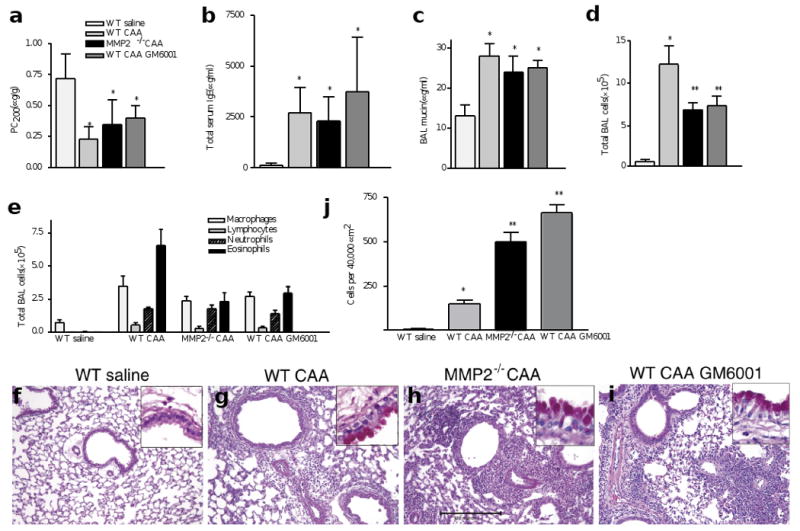
Wild-type and syngeneic MMP2−/− mice were challenged intranasally with saline or CAA and responses were compared to those of allergen-challenged WT mice treated with GM6001. (a) AHR, assessed as PC200. (b) Total serum IgE, measured by ELISA. (c) Total BAL mucin. (d) Total BAL cells. (e) BAL cell differential, which indicates the total numbers of individual inflammatory cells. (f–i) Photomicrographs of representative lung bronchovascular bundles stained with H&E. The insets at the upper right depict higher magnification images of the airway epithelium stained with periodic acid-Schiff. (g–i) Abundant mucus-producing goblet cells (pink cytoplasm) are seen lining the airway epithelium of all mice receiving the CAA allergen. (h,i) Relative to the mice challenged with saline, peri-bronchovascular inflammatory cells were visible in the lungs of allergen-challenged mice but were more prominent in MMP2−/− and GM6001-treated animals. (j) This was confirmed by counting the cells within 40,000 μm2 areas. *P≤0.05 compared to WT saline. **P≤0.05 compared to WT CAA. Bar, 100μm.
MMP2 is required for inflammatory cell egression
There were no marked differences in total blood leukocyte counts during CAA challenge of wild-type and MMP2−/− mice (6.7×103±0.4×103 versus 5.3×103±0.1×103, respectively; n=4). However, MMP2 did play a key role in the regulation of lung inflammatory cell trafficking. If influx of inflammatory cells did not increase, then efflux should decrease. Thus, we counted the total number of inflammatory cells that were present in 40,000 μm2 areas centered on medium-sized bronchovascular bundles (airway diameters ∼100 μm; see Fig. 4h for scale). Compared to wild-type mice, we consistently found a three- to fivefold increase in inflammatory cells present in the parenchyma of CAA-challenged MMP2−/− and GM6001-treated mice (Fig. 4j). Thus, the absence of active MMP2 resulted in excess accumulation of inflammatory cells in the lung.
This increased inflammation in the lung parenchyma was accompanied by excessive amounts of TH2 cytokines. Using RNase protection assay (RPA), we found that the expression of IL-4, IL-5, IL-6, IL-9 and IL-13 mRNA was markedly up-regulated in wild-type mice that were challenged with antigen. In the absence of MMP2, expression of IL-4 and IL-13 mRNA was 2.7- and 4.2-fold higher, respectively, than in MMP2-sufficient mice (Fig. 5a and data not shown). Similarly, using quantitative real-time polymerase chain reaction (PCR), we found that expression of IL-4 and IL-13 mRNA was highly up-regulated in the lungs of MMP2−/− mice that had been challenged with antigen (Fig. 5b). Together, these data indicate that a principal function of MMP2 during allergic lung inflammation is to facilitate the egression of allergic inflammatory cells into the airway lumen and, in its absence, these cells accumulate massively in the lung parenchyma.
Figure 5. MMP2−/− mice aberrantly accumulate TH2 cytokine mRNA in the lung and show increased susceptibility to lethal asphyxiation.
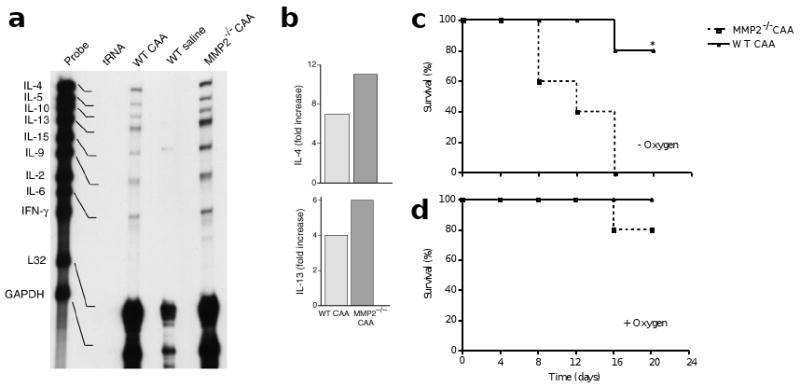
(a) Total RNA was obtained from lungs of mice treated as in Fig. 4. Equal quantities of RNA (10 ng) were then analyzed for the indicated TH2 cytokines and housekeeping genes (L32 and GAPDH) by RPA. (b) Specific mRNA species are indicated by dark bands that are shifted slightly downward in the gels relative to the gene probes. Relative to wild-type, all mRNA species were over-represented in CAA-challenged MMP2−/− mice, but IL-4 and IL-13 were disproportionately affected. This was confirmed by quantitative real-time PCR which indicates an increase in expression of IL-4 and IL-13 in the absence of MMP2. Mice were given i.n. CAA every 4 days in the (c) absence (n=10) or (d) presence (n=20) of 100% supplemental oxygen (given for 10 min before and after CAA challenge). Survival was assessed within 30 min of each antigen challenge. tRNA, transfer RNA (used as negative control). *P≤0.05 compared to MMP2−/−.
Lethal susceptibility to allergic lung inflammation
We next examined the physiological relevance of our findings by monitoring the survival of MMP2−/− and wild-type mice after i.n. CAA challenge. We observed a sharp rise in the mortality of MMP2−/− mice after the second challenge (Fig. 5c). Mortality rates continued to increase in MMP2−/− mice after the third i.n. administration of CAA and resulted in 100% mortality by the fourth challenge (Fig. 5c). The susceptibility of MMP2−/− mice to death after acute allergen challenge was markedly different from that of wild-type mice (P<0.008). Asphyxiation was the cause of death, as treatment with oxygen for 10 min before and after challenge reduced mortality to 20% and 0% in MMP2−/− and wild-type mice, respectively (Fig. 5d).
Chemotactic activity in CAA-challenged MMP2−/− mice
We reasoned that reduced inflammatory cell egression in MMP2−/− mice was caused either by defective chemotaxis of the recruited lung inflammatory cells or a lack of chemotactic activity in the MMP2−/− BAL. We found the latter to be the case because inflammatory cells extracted from whole-lung homogenates from MMP2−/− or wild-type mice showed similar chemotaxis in response to wild-type BAL (Fig 6a). However, a marked reduction in chemotaxis was observed when the same cell populations were tested with BAL from MMP2−/− mice, which indicated that chemotactic activity was deficient in MMP2−/− BAL (Fig. 6a). In addition, the recombinant mouse chemokines CCL7 (also known as MCP-3, 100 ng/ml) and CCL11 (also known as eotaxin, 10–100 ng/ml) induced similar chemotactic responses from MMP2−/− or wild-type lung inflammatory cells (Fig. 6a and data not shown). Finally, we determined the concentration in BAL of three chemokines—CCL7, CCL11 and CCL17 (also known as TARC)—relevant to allergic lung disease28,29. We found comparable concentrations of CCL7 and CCL17 in the BAL of MMP2−/− mice and wild-type controls. However, the amount of CCL11 detected in the BAL of MMP2−/− was one-third that of wild-type mice (Fig. 6b).
Figure 6. Aberrant chemotaxis of lung inflammatory cells in response to MMP2−/− BAL.

(a) The migratory responses of lung inflammatory cells from CAA-challenged wild-type and MMP2−/− mice in response to RPMI (media), BAL from CAA-challenged MMP2−/− (BAL MMP2−/−) or wild-type (BAL WT) mice and recombinant MCP-3 were determined by transfilter assays. The total number of cells that migrated into the chamber filters are shown. Note the normal chemotactic response of MMP2−/− cells in response to wild-type BAL. *P≤0.05 compared to the media control. **P≤0.05 compared to MMP2−/− BAL. (b) Concentrations of CCL11, CCL7 and CCL17 in BAL from wild-type and MMP2−/− mice challenged with CAA, as measured by ELISA. *P≤0.05 compared to wild-type mice.
Discussion
Our data highlight the importance of luminal clearance of inflammatory cells in preventing lethal asphyxiation and the role that MMP2 plays in this process. Both fibroblasts and smooth muscle cells—which are lung cells of mesenchymal origin—participate in the asthmatic phenotype, which is regulated by the signaling cascade initiated by TH2 cytokines, in particular IL-13. Lack of MMP2 during allergen challenge resulted in increased expression of several TH2 cytokines, exacerbated inflammatory cell infiltration of the lung and a decrease in the chemoattractant activity of these inflammatory cells in the BAL. Paradoxically, these findings were accompanied by decreased recruitment of the same inflammatory cells into the BAL, with eosinophils representing the inflammatory cell that was most affected by this aberrant cellular trafficking pattern. However, consistent with these findings, we discovered a selectively decreased concentration of CCL11—a potent chemoattractant for eosinophils30,31—in the BAL of MMP2−/− mice. Thus, IL-13 initiates secondary gene-activation programs in the mesenchyme, which are essential for the clearance of eosinophils, by providing the necessary chemotactic gradient required to prevent the harmful accumulation of these cells.
The term airway remodeling is often applied to describe the range of anatomical changes observed in the lungs during acute and chronic allergic inflammation1,32. Although variously defined, airway remodeling includes changes such as goblet cell metaplasia and mucus gland hyperplasia, subepithelial collagen deposition, smooth muscle hyperplasia and/or hypertrophy, airway mucus plugging, peribronchovascular accumulation of inflammatory cells and other changes that are believed to contribute to the airway obstruction of asthma. Our results establish a key contribution made by the lung parenchyma to the lung-remodeling paradigm. We found that the clearance of allergic inflammatory cells from the lung parenchyma was critically influenced by parenchymally derived MMP2. Our findings were confirmed in two different mouse strains, using different allergens and methods for inactivating MMP2. Our data define a unique mechanism by which parenchymal inflammatory cells egress into the airway lumen that requires generation of an MMP2-dependent trans-epithelial chemotactic gradient. Because the phenotype of MMP2−/− mice largely reproduced that of GM6001-treated mice, MMP2 is likely the most important MMP regulating parenchymal inflammatory cell flux via this mechanism. However we cannot exclude contributions made by MMP9 (also known as gelatinase B) and MMP14 (also known as MT1-MMP), which are also expressed in the lung. Lung neutrophil emigration and egression induced by lipopolysaccharide remain intact in the absence of MMP933. However this finding does not exclude a potential role played by MMP9 in the allergic setting. Indeed, whereas CCL11 is particularly affected by a lack of MMP2, deficiency in other MMPs (for example MMP9) reduces formation of the airway trans-epithelial gradient of distinct chemokines (F. K and K. R., unpublished data). This work provides the impetus to evaluate the role of other MMPs in allergic and other lung inflammatory settings.
Like most other features of experimental asthma, MMP2 expression is controlled through IL-13. Lung fibroblasts, smooth muscle and other parenchymal cells contribute significantly to the total production of lung MMP2. Importantly, rIL-13 directly stimulated MMP2 mRNA and protein, which provides an essential link between the pulmonary mesenchyme and cytokine-producing TH2 cells that underlie allergic lung disease. Such a connection further establishes IL-13 as a general mediator of TH2 -dependent physiological effects14,15,18.
Our findings suggest that both eosinophils and cells that produce TH2 cytokine accumulate in the lungs in the absence of MMP2. Although excess eosinophils could account for the aberrant cytokine expression patterns observed, lymphocytes are the major producers of TH2 cytokine mRNA in murine lungs after antigen challenge (D. C., unpublished data) and are, therefore, a more likely source. TH2 cells and eosinophils are strongly implicated in the pathogenesis of allergic diseases such as asthma, and their accumulation—together with their excess cytokine products—would be expected to correlate with more severe disease.
The mechanisms that regulate clearance of recruited leukocytes from the lung are not fully understood, but may include several routes, including apoptosis and phagocytosis, lymphatic recirculation and cell egression into the airway lumen34,35. In vitro, clearance of apoptotic neutrophils is mediated through phagocytosis by macrophages36. Both lung and gut are frequently involved with allergic inflammation and, not surprisingly, their lumens are sites of inflammatory cell clearance37,38. In acute allergic airway inflammation, eosinophils and macrophages constitute >80% of total recovered leukocytes3,5,14. Although allergic inflammation may be harmful to any organ, the host is particularly susceptible to lung involvement due to the potentially lethal effects of the inflammatory cells on gas exchange39. We showed that 100% mortality occurred in MMP2−/− mice compared to 20% in wild-type mice (P<0.008). The vastly improved survival chances that administration of oxygen conferred showed that death resulted from asphyxiation, which was consistent with the aberrant accumulation of lung parenchymal inflammatory cells observed in MMP2−/− mice. Asphyxiation is the major cause of mortality in human asthma40,41, which highlights the relevance of this asthma model and the critical role played by MMPs in clearing inflammatory cells and preventing death. Because we measured similar AHR in allergen-challenged MMP2−/− and wild-type mice, differences in smooth muscle or other contractile cell function cannot explain the increased mortality observed.
Inflammatory cell concentration in BAL diminished in MMP2−/− mice despite peribronchovascular cell accumulation, thus we conclude that MMP2 is essential for the egression of recruited inflammatory cells into the airway lumen. This impairment was due to the failure to establish an appropriate chemokine gradient that extended into the BAL of MMP2−/− mice. The MMP2−/− inflammatory cells responded normally to the BAL from wild-type mice, which showed that the appropriate chemokines were lacking. MMP2 may also indirectly contribute to altered cell recruitment. Several chemokines, including CCL7, are physiological substrates for MMP242 and the cleavage products of CCL7 behave as receptor antagonists for the chemokine receptors CCR1, CCR2 and CCR342. Although we did not find marked differences in CCL7 concentrations between the BAL of wild-type and MMP2−/− mice, this suggests that other MMPs may contribute to cleavage of CCL7.
Our data highlight the importance of luminal clearance of inflammatory cells in preventing lethal asphyxiation and the role that MMP2 plays in this process. In the absence of MMP2, the rapid accumulation of cells in the lung parenchyma—as early as 5 days after i.n. antigen challenge—showed that the luminal route may be the most important route for clearing lung allergic inflammatory cells. MMP inhibitors block transmigration of IL-5–activated eosinophils through an artificial basement membrane43. Our data show that extravasation of inflammatory cells across the endothelial basement membrane occurs independently of the MMPs required for luminal clearance. Thus, the processes of extravasation and luminal clearance, although obviously related, rely on distinct mechanisms, with MMP2 playing a key role only in luminal clearance.
Our results are in agreement with a published study of the role played by MMPs in limiting BAL cell recruitment19; however, we have extended this observation to the entire lung and demonstrate the accumulation of cells in the parenchyma. Our data show that the principal effect of MMP inhibition is reduced trafficking of parenchymal inflammatory cells into the airway lumen, with little or no effect on inflammatory cell recruitment or production. Thus, inhibition of MMPs does not preclude lung inflammatory cell recruitment, only cell egress. Neither the presence nor absence of MMPs markedly hinders the development of the pathognomonic features of asthma. Because parenchymal inflammatory cells continue to contribute to tissue damage, allergic features may actually worsen in the absence of MMPs, obviating MMP inhibition as a useful therapy for asthma and other allergic diseases19. Indeed, after only 3 weeks of allergic lung inflammation, MMP2−/− mice showed greatly increased susceptibility to the asphyxiating potential of acute allergen challenge. Clarification of the role of other MMPs will further elucidate the pathogenesis of allergic inflammation and may provide additional therapeutic insight into diseases such as asthma.
Methods
Mice
MMP2−/−26 (on a B6 background), BALB/c and B6 mice were bred in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited transgenic animal facility at Baylor College of Medicine.
Allergens and other reagents
Chicken egg OVA (Sigma, St. Louis, MO) was precipitated in alum (OVA-alum) as described3. CAA was prepared from a clinical isolate of Aspergillus fumigatus as described5. Mice were given 50, 150 or 250 mg/kg of GM6001 (AMS Scientific, Concord, CA)25 by i.p injection 1 h before i.n. challenge with OVA or CAA. Murine rIL-13 was from Peprotech (Rocky Hill, NJ).
Allergen and IL-13 challenge
All mice on the BALB/c background were sensitized with 25 μg of OVA-alum, which was administered intradermally three times at 7-day intervals. Starting on day 21, sensitized mice received 25 μg of i.n. OVA daily for 5 days, as described14. Because mice on the B6 background are insensitive to OVA3, we used CAA instead of OVA to immunize MMP2−/−, MMP2−/+ or wild-type B6 mice every 4 days, as described5. Unless otherwise specified, mice were treated in an inhalation chamber with 100% oxygen for 10 min before and after i.n. CAA challenges. Because MMP2−/+ and wild-type B6 mice showed identical responses (data not shown), data from wild-type mice was compared to that from MMP2−/− mice. Additional groups of mice were given 3.5 μg of i.n. rIL-13 twice daily for 3 days14.
Analysis of the asthma phenotype
All data were collected 24 h after the final allergen challenge. AHR was measured based on the provocative concentration of acetylcholine that caused a 200% increase in lung resistance (PC200)3. Bronchoalveolar lavage cytology, OVA-specific and total IgE, lung histopathology and lung cytokine profiles obtained by ELISA were determined as described3. Mucin was quantified using the mucin-binding lectin Jacalin (Calbiochem, La Jolla, CA). Aliquots (40 μl) of BAL diluted 1:1000 and 1:10,000 were added to the individual wells of microtiter ELISA plates and incubated for 2 h at 37 °C. Plates were washed, blocked with 5% bovine serum albumin (BSA) and 0.002% biotinylated Jacalin added. After incubation for 1 h at 37 °C, plates were washed, then developed with alkaline phosphatase–conjugated streptavidin (Jackson Immunoresearch, Westgrove, PA) and nitrophenylphosphate (Sigma) and quantified by comparison with a mucin (Sigma) standard curve.
Gelatin gel zymography
Zymography was used to detect the presence of MMP2 in BAL with standard protocols33. Briefly, 5 μl samples of BAL were added to nondenaturing loading buffer and separated in 10% SDS–polyacrylamide gels that contained 0.02% gelatin. SDS was removed by three 20-min washes with 2.5% Triton X-100 before incubation for 24 h at 37 °C in developing buffer (50 mM TrisCl at pH 8, 5 mM CaCl2 and 0.02% NaN3) with or without 10 mM of 1′10-phenanthroline (Sigma). Gelatin gels were then fixed and stained with 50% methanol and 10% acetic acid that contained 0.3% w/v Coomassie Blue. The optical density of clear bands was determined by densitometry with Quantity 1 software (Bio-Rad, Richamond, CA).
RPA and quantitative PCR
Cytokine mRNA was measured using total RNA (10 μg) by RNAse protection with a commercial kit (Pharmingen, San Diego, CA). Briefly, mouse cytokine plasmid-1 (mCK-1, Pharmingen) was used as a template to synthesize the antisense RNA probe and was labeled with [α-32P]-UTP. A total of 1×106 cpm of radiolabeled probe was used to hybridize to 15 μg of total RNA extracted from lungs. A two-step reverse-transcription PCR was used to determine the relative expression of mRNA with the Perkin Elmer Prism 5700 Sequence Detection System (Applied Biosystems, Norwalk, CT). Ten nanograms of cDNA per sample were generated from 1 μg of RNA; this was analyzed for the expression of IL-4, IL-13 and 18S rRNA with Taqman predeveloped assay reagents (Applied Biosystems). Relative quantities of 18S rRNA and IL-4 and IL-13 mRNA were calculated using the comparative threshold cycle number for each sample fitted to a five-point standard curve. The expression of mRNA for each sample was normalized to 18S rRNA. The samples were analyzed three times for the target genes, and the average of the readings was used to calculate the relative abundance of IL-4 and IL-13 mRNA in the tissue.
Northern blot analysis and in situ hybridization
Northern blot analysis of total RNA isolated from lungs of mice after i.n. immunization with allergens, rIL-13 (Peprotech) or saline was done as described44. We used RNA probes prepared from the plasmid vector pSP6545 for MMP2. The labeling of the probes and in situ hybridization on prepared paraffin sections of lung tissue were done as described46.
Inflammatory cell egression
BAL samples were collected and cell counts done as described above. Immunized BALB/c mice (n=5) received either PBS (vehicle) or 50, 150 or 250 mg/kg of the MMP inhibitor GM6001 intraperitoneally 1 h before i.n. OVA challenge. For each dose of GM6001, the BAL cell count was expressed relative to that of the vehicle-treated group according to the following formula: (total number of BAL inflammatory cells from GM6001- and OVA-challenged mice/total number of BAL inflammatory cells from PBS- and OVA-challenged mice)×100.
Peribronchovascular cell infiltration assay
Total inflammatory cells within a 200×200 μm2 area of bronchovascular bundles (a standard anatomical landmark of the lung) were counted by light microscopic analysis of hematoxylin and eosin (H&E)-stained sections. To ensure comparable analyses between different groups, we analyzed randomly selected medium-sized airways (∼100 μm airway diameter). A total of at least ten different lung fields from three mice in each experimental group were visualized under high power.
Survival experiment
CAA was given to MMP2−/− and wild-type mice every 4 days as described5 without (n=10) or with (n=20) 100% oxygen, which was administered in an inhalation chamber for 10 min both before and after treatment. Mortality within 30 min of antigen challenge was recorded for each group and the percentage survival recorded with Kaplan-Meier coordinates. No additional mortality beyond the 30-min period was observed.
Lung inflammatory cell extraction and chemotaxis assay
Inflammatory cells were extracted from lungs of sensitized wild-type or MMP2-deficient mice and used in chemotaxis assays. Briefly, BAL inflammatory cells were discarded by injecting into the lung and withdrawing, via the tracheal cannula, 1 cc aliquots of PBS for a total of 3 cc of BAL. The lung was then minced and pressed through a fine nylon mesh. The crude cell suspension was purified by lysing the red blood cells, which was followed by the selection of live inflammatory cells by differential centrifugation through 1-Step lymph-prep media (Accurate Chemical & Scientific Corporation, Westbury, NY). Viable inflammatory cells (macrophages, eosinophils, lymphocytes and neutrophils) were resuspended to a final working concentration of 1×106 cell/ml in RPMI and were used in the chemotaxis assays. Inflammatory cell chemotaxis was measured by transfilter assays in 48-well chemotaxis chambers (Neuro Probe, Gaithersburg, MD) as described47,48. The chemotactic activity of the BAL fluid from wild-type or MMP2−/−mice (n=5) was assessed against each of the lung inflammatory cell populations in duplicate. Serum-free media, BAL from wild-type and MMP2−/− mice, CCL7 (100 ng/μl) and CCL11 (100 ng/μl, both from R&D Systems, Minneapolis, MN) were placed in the lower wells of the chemotaxis chambers and separated from cell suspension in the upper wells. The total number of cells that migrated into the chamber filters was counted by microscopy and data were expressed as the total cells/filter.
Chemokine concentration assay
Standard antibody-based ELISAs were used to measure concentrations of CCL7 (polyclonal mouse anti-CCL7), CCL11 (polyclonal mouse anti-CCL11) and CCL17 (monoclonal mouse anti-CCL17) in the BAL fluid of wild-type or MMP2−/− (n=5) mice. All capture antibodies, their corresponding biotin-conjugated detection antibodies and recombinant proteins were from R&D Systems.
Statistics
Data are mean±s.e.m and are representative of at least three in vivo independent experiments that used four or five mice in each. Significant differences are expressed relative to saline-challenged control (*P≤0.05) or CAA-challenged wild-type (**P≤0.05) mice using Student's t-tests for the PC200 values or Mann-Whitney U-tests for all other data. Cell migration was expressed as the mean±s.e.m. of the numbers cell observed per ten high-power fields (×400). Results were assessed for statistical significance with ANOVA and the Bonferroni multiple comparison tests.
Acknowledgments
We thank A. C. White and B. Dickey for helpful comments and T. Itoh and S. Itohara for providing the MMP2−/− mice. Supported by the Caroline Weiss Law Fund for Molecular Medicine; National Institutes of Health grants K08 HL03344 and R01 HL69585 (to D. C.), K08 HL03732 and R01 HL64061 (to F. K.); and the Sandler Family Asthma Fund (to Z. W.).
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Barnes PJ. Pathophysiology of asthma. Br J Clin Pharmacol. 1996;42:3–10. doi: 10.1046/j.1365-2125.1996.03721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coyle AJ, et al. Interleukin-4 is required for the induction of lung Th2 mucosal immunity. Am J Respir Cell Mol Biol. 1995;13:54–59. doi: 10.1165/ajrcmb.13.1.7598937. [DOI] [PubMed] [Google Scholar]
- 3.Corry DB, et al. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corry DB, et al. Requirements for allergen- induced airway hyperreactivity in T and B cell– deficient mice. Mol Med. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson DS, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ, Page C. Mediators of asthma: a new series. Pulm Pharmacol Ther. 2001;14:1–2. doi: 10.1006/pupt.2000.0263. [DOI] [PubMed] [Google Scholar]
- 8.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 9.Kopf M, et al. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 10.Finkelman FD, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol. 1988;141:2335–2341. [PubMed] [Google Scholar]
- 11.Murata T, Obiri NI, Debinski W, Puri RK. Structure of IL-13 receptor: analysis of subunit composition in cancer and immune cells. Biochem Biophys Res Commun. 1997;238:90–94. doi: 10.1006/bbrc.1997.7248. [DOI] [PubMed] [Google Scholar]
- 12.Miloux B, et al. Cloning of the human IL-13Rα chain and reconstitution with the IL4Rα of a functional IL-4/IL-13 receptor complex. FEBS Lett. 1997;401:163–166. doi: 10.1016/s0014-5793(96)01462-7. [DOI] [PubMed] [Google Scholar]
- 13.Hilton DJ, et al. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci USA. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunig G, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corry DB. IL-13 in allergy: home at last. Curr Opin Immunol. 1999;11:610–614. doi: 10.1016/s0952-7915(99)00025-4. [DOI] [PubMed] [Google Scholar]
- 16.Doucet C, et al. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest. 1998;101:2129–2139. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becky Kelly EA, Busse WW, Jarjour NN. Increased matrix metalloproteinase-9 in the airway after allergen challenge. Am J Resp Crit Care Med. 2000;162:1157–1161. doi: 10.1164/ajrccm.162.3.9908016. [DOI] [PubMed] [Google Scholar]
- 18.Zheng T, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumagai K, et al. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J Immunol. 1999;162:4212–4219. [PubMed] [Google Scholar]
- 20.Holla LI, Vasku A, Stejskalova A, Znojil V. Functional polymorphism in the gelatinase B gene and asthma. Allergy. 2000;55:900–901. doi: 10.1034/j.1398-9995.2000.00736.x. [DOI] [PubMed] [Google Scholar]
- 21.Cataldo D, et al. MMP2- and MMP-9-linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 2000;123:259–267. doi: 10.1159/000024452. [DOI] [PubMed] [Google Scholar]
- 22.Haas TL, Madri JA. Extracellular matrix-driven matrix metalloproteinase production in endothelial cells: implications for angiogenesis. Trends Cardiovasc Med. 1999;9:70–77. doi: 10.1016/s1050-1738(99)00014-6. [DOI] [PubMed] [Google Scholar]
- 23.Madri JA, Graesser D, Haas T. The roles of adhesion molecules and proteinases in lymphocyte transendothelial migration. Biochem Cell Biol. 1996;74:749–757. doi: 10.1139/o96-082. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein RA, Paul WE, Metcalfe DD, Busse WW, Reece ER. NIH conference. Asthma. Ann Intern Med. 1994;121:698–708. doi: 10.7326/0003-4819-121-9-199411010-00011. [DOI] [PubMed] [Google Scholar]
- 25.Galardy RE, Grobelny D, Foellmer HG, Fernandez LA. Inhibition of angiogenesis by the matrix metalloprotease inhibitor N-[2R-2-(hydroxamidocarbonymethyl)-4-methylpentanoyl)]-l-tryptophan methylamide. Cancer Res. 1994;54:4715–4718. [PubMed] [Google Scholar]
- 26.Itoh T, et al. Unaltered secretion of β-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- 27.Grunig G, et al. Interleukin-10 is a natural suppressor of cytokine production and inflammation in a murine model of allergic bronchopulmonary aspergillosis. J Exp Med. 1997;185:1089–1099. doi: 10.1084/jem.185.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baggiolini M. Reflections on chemokines. Immunol Rev. 2000;177:5–7. doi: 10.1034/j.1600-065x.2000.17722.x. [DOI] [PubMed] [Google Scholar]
- 29.Mathew A, et al. Signal transducer and activator of transcription 6 controls chemokine production and T helper cell type 2 cell trafficking in allergic pulmonary inflammation. J Exp Med. 2001;193:1087–1096. doi: 10.1084/jem.193.9.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lilly CM, et al. Eotaxin expression after segmental allergen challenge in subjects with atopic asthma. Am J Respir Crit Care Med. 2001;163:1669–1675. doi: 10.1164/ajrccm.163.7.9812044. [DOI] [PubMed] [Google Scholar]
- 31.Lamkhioued B, et al. Increased expression of eotaxin in bronchoalveolar lavage and airways of asthmatics contributes to the chemotaxis of eosinophils to the site of inflammation. J Immunol. 1997;159:4593–4601. [PubMed] [Google Scholar]
- 32.Sears MR. Consequences of long-term inflammation. The natural history of asthma. Clin Chest Med. 2000;21:315–329. doi: 10.1016/s0272-5231(05)70269-0. [DOI] [PubMed] [Google Scholar]
- 33.Betsuyaku T, Shipley JM, Liu Z, Senior RM. Neutrophil emigration in the lungs, peritoneum, and skin does not require gelatinase B. Am J Respir Cell Mol Biol. 1999;20:1303–1309. doi: 10.1165/ajrcmb.20.6.3558. [DOI] [PubMed] [Google Scholar]
- 34.Milik AM, et al. Lung lymphocyte elimination by apoptosis in the murine response to intratracheal particulate antigen. J Clin Invest. 1997;99:1082–1091. doi: 10.1172/JCI119236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savill J. Apoptosis in resolution of inflammation. Kidney Blood Press Res. 2000;23:173–174. [PubMed] [Google Scholar]
- 36.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 37.Bancroft AJ, Artis D, Donaldson DD, Sypek JP, Grencis RK. Gastrointestinal nematode expulsion in IL-4 knockout mice is IL-13 dependent. Eur J Immunol. 2000;30:2083–2091. doi: 10.1002/1521-4141(200007)30:7<2083::AID-IMMU2083>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 38.Dubois GR, Bruijnzeel PL. IL-4-induced migration of eosinophils in allergic inflammation. Ann NY Acad Sci. 1994;725:268–273. doi: 10.1111/j.1749-6632.1994.tb39809.x. [DOI] [PubMed] [Google Scholar]
- 39.Azzawi M, Johnston PW, Majumdar S, Kay AB, Jeffery PK. T lymphocytes and activated eosinophils in airway mucosa in fatal asthma and cystic fibrosis. Am Rev Respir Dis. 1992;145:1477–1482. doi: 10.1164/ajrccm/145.6.1477. [DOI] [PubMed] [Google Scholar]
- 40.Kikuchi Y, et al. Chemosensitivity and perception of dyspnea in patients with a history of near-fatal asthma. N Engl J Med. 1994;330:1329–1334. doi: 10.1056/NEJM199405123301901. [DOI] [PubMed] [Google Scholar]
- 41.Molfino NA, Nannini LJ, Martelli AN, Slutsky AS. Respiratory arrest in near-fatal asthma. N Engl J Med. 1991;324:285–288. doi: 10.1056/NEJM199101313240502. [DOI] [PubMed] [Google Scholar]
- 42.McQuibban GA, et al. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 43.Okada S, Kita H, George TJ, Gleich GJ, Leiferman KM. Migration of eosinophils through basement membrane components in vitro: role of matrix metalloproteinase-9. Am J Respir Cell Mol Biol. 1997;17:519–528. doi: 10.1165/ajrcmb.17.4.2877. [DOI] [PubMed] [Google Scholar]
- 44.Reponen P, et al. Molecular cloning of murine 72-kDa type IV collagenase and its expression during mouse development. J Biol Chem. 1992;267:7856–7862. [PubMed] [Google Scholar]
- 45.Alexander CM, et al. Expression and function of matrix metalloproteinases and their inhibitors at the maternal-embryonic boundary during mouse embryo implantation. Development. 1996;122:1723–1736. doi: 10.1242/dev.122.6.1723. [DOI] [PubMed] [Google Scholar]
- 46.Chin JR, Werb Z. Matrix metalloproteinases regulate morphogenesis, migration and remodeling of epithelium, tongue skeletal muscle and cartilage in the mandibular arch. Development. 1997;124:1519–1530. doi: 10.1242/dev.124.8.1519. [DOI] [PubMed] [Google Scholar]
- 47.Chen S, et al. In vivo inhibition of CC and CX3C chemokine-induced leukocyte infiltration and attenuation of glomerulonephritis in Wistar-Kyoto (WKY) rats by vMIP-II. J Exp Med. 1998;188:193–198. doi: 10.1084/jem.188.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu JY, et al. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001;410:948–952. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]