

Abstract
Human erythrocyte R-type pyruvate kinase deficiency (PKD) is a disorder caused by mutations in the PKLR gene that produces chronic nonspherocytic hemolytic anemia. Besides periodic blood transfusion and splenectomy, severe cases require bone marrow (BM) transplant, which makes this disease a good candidate for gene therapy. Here, the normal human R-type pyruvate kinase (hRPK) complementary (cDNA) was expressed in hematopoietic stem cells (HSCs) derived from pklr deficient mice, using a retroviral vector system. These mice show a similar red blood cell phenotype to that observed in human PKD. Transduced HSCs were transplanted into myeloablated adult PKD mice or in utero injected into nonconditioned PKD fetuses. In the myeloablated recipients, the hematological manifestations of PKD were completely resolved and normal percentages of late erythroid progenitors, reticulocyte and erythrocyte counts, hemoglobin levels and erythrocyte biochemistry were restored. Corrected cells preserved their rescuing capacity after secondary and tertiary transplant. When corrected cells were in utero transplanted, partial correction of the erythrocyte disease was obtained, although a very low number of corrected cells became engrafted, suggesting a different efficiency of cell therapy applied in utero. Our data suggest that transduction of human RPK cDNA in PKLR mutated HSCs could be an effective strategy in severe cases of PKD.
Introduction
Pyruvate kinase (PK, EC 2.7.1.40) is a metabolic enzyme that catalyses the last step of glycolysis. PK transfers the phosphate group from phosphoenolpyruvate to adenosine diphosphate, generating pyruvate and adenosine triphosphate (ATP). Given the critical nature of this reaction, any loss in PK activity impairs cell metabolism.1 In humans, four PK isoforms are expressed by two structural genes: The PKLR gene (PK liver and red blood cells gene) on Chr1q21, encodes the RPK (R-type pyruvate kinase) and L-type pyruvate kinase tissue specific isoforms expressed in erythroid cells and liver, respectively; and the PKM gene on Chr15q22 codes for isoforms M1-type pyruvate kinase, expressed in brain and skeletal muscle, and M2-type pyruvate kinase, expressed in fetal and most adult tissues except erythroid cells.2 The expression of PKLR- derived isoenzymes is regulated by tissue specific promoters, whereas the two products of the PKM gene are synthesized by alternative mRNA splicing.3
Mutations in the PKLR gene1,3,4 lead to pyruvate kinase deficiency (PKD), an autosomal recessive disorder, which is the most frequently enzymatic defect of the Embden-Meyerhof pathway in erythrocytes. Together with glucose-6-phosphate dehydrogenase deficiency, this is the most common hereditary enzymopathy causing chronic nonspherocytic hemolytic anemia.5 Over 100 different mutations have been identified on the structural PKLR gene.6 In most cases, they are missense mutations, although nonsense mutations, deletions, insertions, and even disruption of the erythroid promoter causing severe deficiency have been reported.6,7,8,9,10 Generally, most patients are compound heterozygous with two different mutant alleles, but homozygous mutations with highly deleterious alleles causing life-threatening anemia have also been described.6,10
Although the global incidence of PKD is unknown, it has been estimated at 51 cases per million in North America.11 Clinical symptoms of PKD vary considerably from mild to severe anemia. Pathological manifestations are usually observed when enzyme activity falls <25% normal PK activity, and severe disease has been associated with a high degree of reticulocytosis.10,12 Currently, there is no definitive treatment for severe PKD (see review in ref. 13) as the PKLR gene is not functionally compensated in the erythrocyte by PKM isoenzymes.10 Although splenectomy can be clinically useful in patients with severe disease, in some cases, allogeneic hematopoietic transplantation is required.14 In these patients, hematopoietic stem cell (HSC) gene therapy might be a good and more effective treatment.
Animal models of PKD have been developed in dogs and mice.15,16,17 In a dog model, bone marrow (BM) transplant without previous recipient conditioning failed to resolve hematological symptoms.15 In mice, attempts have been made to selectively expand normal donor erythrocytes in minimally conditioned recipients.18 Attempts to rescue RPK-deficient mice have been addressed using mouse transgenic cells that expressing the human RPK complementary (cDNA).19 Human L-type pyruvate kinase has been expressed into murine HSC demonstrating long-term expression (3 months) of the human L-type pyruvate kinase protein in peripheral blood.20 Recently we reported that transduction of BM cells using γ-retroviral vectors21 carrying human RPK cDNA22 mediates long-term expression of the human RPK protein in red blood cells obtained from primary and secondary recipients, without any detectable adverse effects.23
Additionally, as PKD is an inherited disease that can be diagnosed before birth,24 the in utero transplant of genetically corrected HSC might be a treatment option, as proposed for other inherited diseases.25 We have recently demonstrated in a mouse model, which genetically modified syngenic hematopoietic cells show efficient engraftment in utero and give rise to mature hematopoietic cells in adulthood,26 thus providing phenotype correction from early development to adulthood. This approach avoids the problems associated to allogeneic transplantation, such us the need of a compatible donor and graft rejection.
In the present work, we show that human RPK expressing vectors are able to fully reverse the hemolytic phenotype in PKD mice. HSCs from these animals can be genetically corrected and transplanted into lethally irradiated adult PKD mice. In addition, our data indicate that the in utero transplant of these genetically corrected cells into nonconditioned PKD fetuses achieves partial correction of the disease.
Results
The anemic phenotype is corrected in adult RPK-deficient mice by the transplantation of genetically corrected HSCs
To assess the efficacy of γ-retroviral vectors carrying human RPK cDNA to treat PKD mice, lineage negative (Lin−) cells from the BM of PKD male mice were transduced with SF11XEG or SF11RPKXEG vectors (see Materials and Methods). At the end of the transduction protocol, transduced cells (2 × 105 cells/mouse) were transplanted into lethally irradiated female PKD mice. Erythroid variables were determined in peripheral blood at 30, 60 and 90 days post-transplant (dpt), and compared to data obtained in wild type and nontransplanted PKD littermates. A marked improvement in red blood cell counts was observed in PKD animals transplanted with genetically corrected cells HSCs as soon as 30 dpt. This recovery was stable for up to 90 dpt and was not observed in the animals transplanted with cells transduced with the control vector (Figure 1a).
Figure 1.

Erythrocyte and reticulocyte levels in animals transplanted with transduced cells. (a) Bars corresponds to peripheral blood erythrocyte levels in each group of pyruvate kinase deficiency (PKD) animals transplanted with enhanced green fluorescent protein or R-type pyruvate kinase at 30 (white bars), 60 (gray bars), and 90 (black bars) days post-transplant. Control groups of wild type and PKD littermates were analyzed in parallel. In brackets appears the number of animals analyzed per group. (b) Representative dot plot of the analysis used to monitor reticulocytes in peripheral blood. Upper row corresponds to a PKD deficient animal transplanted with cells transduced with the control vector. Lower row corresponds to a PKD mouse transplanted with cells transduced with the therapeutic vector. (c) Reticulocyte percentages recorded in the different groups of animals analyzed in a.
High reticulocyte counts, a pathognomonic sign of PKD in patients, also occur in PKD mice. Through flow cytometry, we observed a steady reduction in reticulocytosis in PKD animals transplanted with genetically corrected cells, but not in PKD animals transplanted with cells transduced with the control vector (Figure 1b). This reduction was observed at 30 dpt and normal reticulocyte counts were recorded in subsequent analyses (Figures 1b,c).
To establish whether gene therapy was capable of restoring other red blood cell variables, hemoglobin, hematocrit, erythrocyte mean corpuscular volume and plasma iron levels were determined at 90 dpt. As shown in Table 1, these variables were restored in PKD mice transplanted with genetically corrected cells. In contrast, in animals transplanted with cells transduced with the control vector the abnormal red cell phenotype was not corrected.
Table 1.
Hematological variables recorded in retroviral transduced RPK-deficient mice

Because of the hemolytic process, PKD patients require constant erythrocyte replenishment and this renders high serum levels of erythropoietin (EPO). We examined this factor in control and deficient animals, as well as in animals transplanted with transduced cells. Plasma EPO concentrations were up to 10 times higher in both deficient animals and deficient animals transplanted with cells transduced with the control vector, compared to normal control littermates (Table 1). However, animals transplanted with RPK-corrected cells, had normal EPO concentration in plasma. Splenomegaly, another characteristic sign of PKD, was also examined in PKD mice at 110 dpt. Animals were killed and their spleens excised for morphometric analysis. As shown in Figure 2, PKD animals transplanted with genetically corrected HSCs showed a marked reduction in spleen weight and size. As expected, anemic mice transplanted with cells transduced with the non-therapeutic vector showed no reduction of splenomegalia.
Figure 2.

Morphometric analysis of spleens and iron deposits in the liver from primary recipients. (a) Pictures of representative spleens from each group of pyruvate kinase deficiency (PKD) animals transplanted with bone marrow cells transduced with enhanced green fluorescent protein (EGFP) or R-type pyruvate kinase (RPK) vectors. One spleen from a wild-type mouse and two spleens from PKD deficient mice (PKD) are also shown. (b) Average spleen weights for each group of PKD transplanted mice. (c) Photomicrographs of Perl's staining to identify iron deposits in liver sections from the same groups of animals shown in a and b. Sections from two different animals are shown for the groups EGFP (EGFP-1, EGFP-2) and RPK (RPK-1, RPK-2).
The phenotytpic changes observed were associated to the increase in activity levels of PK enzyme and ATP in erythroid cells (Table 2). In animals genetically corrected, the erythrocyte PK activity rose ~3.5-fold of the quantified in PKD mice (~75% of wild-type erythrocyte PK activity), and the ATP levels were restored to normal values. Collectively, these data indicate that transgenic expression of the human RPK protein completely resolves the hematological symptoms of this PKD mouse model.
Table 2.
Pyruvate kinase activity and ATP levels in genetically corrected RPK-deficient mice

Additionally, we evaluated iron accumulation in the liver by a Perls' reaction. Due to the hemolytic anemia that PKD animals suffer, there is an iron overload in liver and spleen. We observed that the correction of the disease by gene therapy was able to clear or reduce iron overload in the liver of animals transplanted with cells transduced with the therapeutic vector (Figure 2c). In contrast the animals transplanted with cells transduced with the control vector maintain high amounts of iron deposits in the liver.
RPK expression in late erythroid precursors is needed to correct erythrocyte maturation and generate normal erythroid cells
Next, we examined differentiation patterns of the erythroid cell lineage in control and genetically corrected PKD animals. Expression of TER119 and CD71 antigens in spleen and BM cells were analyzed by flow cytometry. Four erythroid subpopulations: I, early proerythroblasts (Ter119medCD71high); II, basophilic erythroblasts (Ter119highCD71high); III, late basophilic and polychromatophilic erythroblasts (Ter119highCD71med); and IV, orthochromatophilic erythroblasts and mature erythroid cells (Ter119highCD71low) could be identified as previously described23 (Figure 3a). A predominance of immature erythroid precursor cells (proerythroblasts, basophilic erythroblasts, and polychromatophilic erythroblasts) was identified in spleen and BM from PKD mice (Figure 3b). As expected, late erythroid populations were significantly low in PKD mice. The different erythroid subpopulations in BM and spleen were stabilized to normal ranges in animals transplanted with genetically corrected cells but not in those transplanted with cells transduced with the control vector (Figure 3b).
Figure 3.

Flow cytometry analysis of erythroid precursor populations in the bone marrow and spleen of animals transplanted with transduced cells. (a) Representative dot plot and analysis of the different erythroid differentiation steps in bone marrow and spleen. Region I: proerythroblasts; Region II: basophilic erythroblasts; Region III: late basophilic and polychromatic erythroblasts; Region IV: orthochromatic erythroblasts and mature erythroid cells. (b) Percentage of each erythroid precursor population in pyruvate kinase deficiency (PKD) mice transplanted with enhanced green fluorescent protein or R-type pyruvate kinase transduced cells. The bone marrow and spleen of untransduced PKD mice and wild type mice were used as controls. White bars, wild-type mice; gray bars, PKD mice; hatched bars, deficient mice transplanted with cells transduced with the control vector; black bars, deficient mice transplanted with cells transduced with the therapeutic vector. *P < 0.01.
To identify at which erythropoiesis stage of the erythropoiesis anomalies occur in this PKD mouse model and whether altered dynamics of erythroid precursor maturation could be rescued by RPK gene therapy, maturation dynamics of late erythroid precursors were studied in BM and spleen (Figure 4). Maturation dynamics were calculated as the variation in the percentage of precursors corresponding to two consecutive development stages (relative cell variation), as previously reported23,27 and also detailed in Material and Methods. In the BM and spleen of PKD mice, relative increases of 22 and 3%, respectively, were observed in the shift from the basophilic to polycromatophilic erytrhroblasts and a similar relative cell loss between the polycromatophilic and the orthochromatophilic erythroblasts was detected. Overall, in this PKD model, the proliferative increase observed from basophilic (II) to polycromatophilic erytrhroblasts (III), mainly in BM, does not offset the cell loss from polycromatophilic (III) to orthochromatophilic erythroblasts (IV), suggesting that the expression of the erythrocyte PK isoenzyme is essential in the two last differentiation steps of the erythrocyte maturation (Figure 4a).
Figure 4.

Dynamics of eythrodifferentiation in the bone marrow and spleen of animals transplanted with transduced cells. (a) Schematic diagram of erythroblast maturation indicating in roman numerals the populations studied in b and c. See also Figure 3 for flow cytometry analysis of these populations (b) Relative cell variation between two consecutive erythroid precursor populations is indicated. Steps I–IV are referred to the populations established in Figure 3. Positive and negative values indicate an increase or decrease in cellularity between two consecutive differentiation steps. White bars, wild-type mice; gray bars, PKD mice; hatched bars, deficient mice transplanted with cells transduced with the control vector; black bars, deficient mice transplanted with cells transduced with the therapeutic vector. (c) Proliferative variation observed between two successive erythroid EGFP+ populations in mice transplanted with cells transduced with the control vector (open triangle) or with the therapeutic vector (open diamond). EGFP, enhanced green fluorescent protein.
From the analysis of the enhanced green fluorescent protein (EGFP) expressing population in both groups of animals, we observed that the corrected cells were enriched in the progression of cells from step II to step III (Figure 4b), suggesting a differentiation advantage of corrected cells at this step of the erythropoietic differentiation. These findings support the impact of the therapeutic expression of RPK on the final differentiation and survival of the erythroid precursors.
Overexpression of the RPK protein does not affect metabolic variables in nonerythroid cells
Because of the use of a retroviral promoter, high levels of functional human RPK enzyme were expected not only in erythroid cells but also in other hematopoietic lineages. Analysis of EGFP expression in peripheral leucocytes at 30 dpt showed values around 50% and 40% of white blood cells from animals transplanted with cells transduced with SF11XEG or SF11RPKXEG vectors, respectively, expressing the transgene (data not shown). Increased levels of the PK protein could modify the energy production of blood cells expressing the transgenic human RPK. We thus determined PK activity and ATP levels in both erythrocytes and leukocytes in the different groups of animals (Table 2). As expected, erythrocytes from mice transplanted with genetically corrected cells showed normal PK activity and ATP levels. Interestingly, the different groups of animals showed no significant differences in leukocyte PK activity. Similarly, no differences emerged between leukocyte ATP levels in control mice and mice transplanted with corrected cells, indicating that the transgenic expression of human RPK in these cells did not affect the overall balance of the energy production pathway, probably due to fine regulation of this pathway by other key enzymes, intermediary metabolites and redox coenzymes. Moreover, numbers of peripheral blood leukocytes in genetically corrected mice were not affected, as compared with animals transplanted with the control vector (data not shown).
To check whether the transgenic expression of the human RPK could increase overall plasmatic enzyme activity, we also estimated PK levels in serum samples from the different animal groups. Again, normal levels of serum PK were observed, demonstrating the complete recovery of homeostasis and a lack of side effects in leukocytes produced by the increased PK expression (Table 2).
Long-term correction of the disease is dependent on the level of chimerism
Secondary transplants were performed to explore the stability and efficacy of phenotypic correction. Later 100 days primary transplantation, two-million pooled total BM cells from primary corrected recipients were transplanted into secondary RPK-deficient lethally irradiated mice (n = 7) and erythroid variables monitored in peripheral blood. A mean of 85% of genetically corrected myeloerythroid progenitors, as measure by provirus real-time quantitative PCR (qPCR) detection on methyl cellulose colonies, were transferred to each mouse. In these secondary recipients, erythrocyte and reticulocyte counts were normal up to 120 days postsecondary transplant (Figure 5, group RPK 100%) In addition, hematological variables remained normal, including PK activity in peripheral blood erythrocytes (Supplementary Table S1).
Figure 5.

Analysis of erythrocyte and reticulocyte levels in secondary recipients. Bars correspond to (a) peripheral blood erythrocyte numbers, and (b) percentage reticulocytes for each group of pyruvate kinase deficiency recipients. Three groups with different percentages of primary derived hematopoietic cells (10, 30, and 100%) were evaluated at 30 (white bars), 60 (gray bars), and 90 (black bars) days post-transplant (dpt). A control group of wild type nonmanipulated littermates was also analyzed. In brackets, the number of animals studied per group is given. VCN, number of integrated viral DNA copies per cell analyzed by quantitative PCR. *Statistically significant differences between marked groups, P < 0.05. #Statistically significant referred to 60 and 90 dpt.
To determine the minimum number of corrected cells needed to achieve a therapeutic effect in our model, reduced numbers of corrected cells from primary recipients were transplanted into secondary recipients. Grafts of two-million cells containing 10% (~8.5% of genetically corrected cells as analyzed by qPCR) and 30% (~25% of genetically corrected cells) of cells from primary genetically corrected recipients, groups RPK 10 and 30%, respectively, were transplanted into secondary irradiated recipients (three mice per group). Shortly, after secondary transplant, groups RPK 10 and 30% displayed low numbers of peripheral erythrocytes but reduced numbers of reticulocytes (Figure 5). However, at 90 and 120 dpt this discrete reticulocytosis correction was lost (Figure 5 and Supplementary Table S1). Taking into account that the higher number of corrected cells transplanted without a reverse in the anemic phenotype was 30% (25% corrected cells), we propose that values higher than those would be needed, at least, to rescue RPK-deficient animals from clinical symptoms.
Additionally, when pooled BM from fully corrected secondary animals was again transplanted into tertiary female PKD animals (n = 5), erythroid variables were fully restored up to 2 months after transplantation (data not shown). This data further demonstrates the transduction of very primitive HSCs and the stability of transgene expression in the deficient cells. Moreover, no evidence of leukaemia or myelodisplastic syndrome was observed in primary, secondary, and tertiary transplanted animals followed by flow cytometry and anatomopathologic studies (data not shown).
In utero gene therapy partially rescues anemia in RPK-deficient mice
We next tried to determine whether the treatment of this disease as early as possible might provide additional benefits over the improved clinical manifestations of PKD noted in the treated adult animals. To this end 14.5 day-old fetuses from pregnant PKD female mice were transplanted with 5 × 105 genetically corrected hematopoietic progenitors/fetus. Erythroid variables were followed up to 90 days after birth (Figure 6). At 30 days postbirth, erythrocyte counts were low. However, increased numbers were recorded at 60 days postbirth and these numbers were steady at 90 days postbirth (Figure 6a). Consistently, hematocrit and hemoglobin levels showed similar behavior indicating partial correction of anemia in in utero transplanted animals (Figure 6b). Analysis of late erythroid precursor dynamics showed a partial correction in the BM in the in utero transplanted animals in comparison with deficients animals of the same age (Figure 6c).
Figure 6.

Partial correction of the erythropoietic phenotype in pyruvate kinase deficiency (PKD) mice transplanted in utero with genetically corrected cells. (a) Bars correspond to the number of peripheral blood erythrocytes detected in wild type, PKD and in utero transplanted mice at 30 (white bars), 60 (gray bars) and 90 (black bars) days postbirth (dpb). (b) Erythrocyte, hemoglobin, and hematocrit values recorded in PKD mice transplanted in utero with genetically corrected cells analyzed 90 dpb. WT, wild-type mice; PKD, PKD mice; IUT, mice in utero transplanted with genetically corrected bone marrow (BM) cells. (c) Percentage of cell variations between two consecutive erythroid precursor populations in BM (BM), spleen (SP) and peripheral blood (PB) from the same in utero transplanted animals. Black bars, PKD animals; gray bars, animals in utero transplanted with cells transduced with the therapeutic vector. *Statistically significant differences referred to PKD mice, P < 0.05.
To estimate the engraftment efficiency of corrected cells, we determined the number of proviral insertions in BM by quantitative real-time PCR. A mean of 0.8% of transduced EGFP+ cells was detected in the in utero transplanted animals 3 months after birth. It is worthy to point out that similar partial corrections were observed in the in utero transplanted animals than in the secondary recipients transplanted with tenfold more corrected primary cells (group RPK 10%).
Discussion
The treatment of genetic erythroid deficiencies, such us hemoglobinopathies, or metabolic diseases like glucose-6-phosphate dehydrogenase deficiency by gene therapy has yielded promising results in animals models.28 Here, we demonstrate that our previously developed retroviral vectors23 are capable of fully resolving the pathological symptoms of PKD in a mouse model, including anemia, reticulocytosis, and splenomegalia, and of normalizing EPO, PK, and ATP levels, without modifying the metabolic energy balance in white blood cells. Moreover, we show that values above 25% of genetically corrected cells are needed to fully rescue the deficiency. The lack of sufficient therapeutic effects observed in adult chimerism and in in utero assays discarded a possible proliferative advantage from corrected HSC population. Also, we suggest that the in utero transplant of RPK-corrected cells could be a therapeutic strategy when fetal anemia due to PKD is early diagnosed to prevent hydrops fetalis due to this disorder.29
Transplantation of RPK-corrected cells resolved erythroid pathological symptoms as early as 30 dpt, indicating that the normal function of human RPK in corrected erythrocytes is rapidly restored. Splenomegalia and iron deposits in the liver, common signs of hemolytic anemia were also reversed in response to the gene therapy protocol, further demonstrating that the damage on these organs is also reversed after anemia rescue. As expected, other relevant erythroid variables, including hemoglobin levels, hematocrit, mean corpuscular volume and iron plasma levels, were fully restored in PKD animals transplanted with the therapeutic vector, demonstrating that the expression of human RPK in the mouse erythrocytes completely rescues this metabolic pathway in the erythroid lineage and allows the generation of normal erythrocytes in peripheral blood. In addition, EPO levels dramatically drop indicating that a normal steady state of hematopoiesis is attained after gene therapy.
Although the use of retroviral vectors to drive the ubiquitous expression of the hRPK gene could disturb the physiological generation of ATP in white blood cells, ATP levels measured in peripheral blood leukocytes from genetically corrected PKD mice were not increased as a result of the ectopic expression of the human RPK in these cells, indicating a fine regulation of this pathway either by direct ATP inhibition30 or by metabolic and transcriptional control.31
In humans, pathological signs of PKD appear when enzyme activity drops below 25% normal levels.32 A mouse model of PKD has revealed that 10% normal BM renders red blood cells expressing nearly normal RPK protein levels.18 As a result of the transduction protocol used here, the viral dosage was high, up to 11 viral DNA copies per cell in the case of the group RPK 100%. However, due to the variability of the transplanted animals some mice with phenotypic correction receive no more than 2.5 viral copies per cell (data not shown). To estimate the minimal amount of genetically corrected cells needed for rescue therapy, we transplanted 30 and 10% of BM cells harvested from primary recipient animals, with 25 and 8.5% of corrected cells, respectively. Although in the short-term reticulocytosis was significantly corrected, these reduced proportions of transduced HSCs were unable to solve the hematological defects of PKD mice in the long term. These data indicate that, in our PKD mice model the relevant parameter to obtain a phenotypic correction is the amount of corrected cells transplanted.
Interestingly, a low correction of the phenotype was observed in PKD mice injected in utero with a 0.8% of engraftment of transduced cells. These data suggest that the administration of corrected cells at very early development stages could be therapeutically a more efficient strategy to address the correction of this disease. This strategy should be very efficient for PKD cases in which fetal symptoms are detected. In these cases fetal liver biopsys for the isolation of hematopoietic progenitors33,34 that could be genetically corrected and reinfused into the fetus could be applied.
In gene therapy for metabolic diseases, the expression of the normal version of the dysfunctional protein could confer the corrected cells an advantage over noncorrected cells. In our system, we observed an increased capacity of basophilic erythroblasts to differentiate into polychromatic erythroblasts, suggesting the dependence of this step on correct RPK expression. Thus, the expression of the human RPK enzyme in deficient erythroid precursors confers a significant differentiation advantage over noncorrected deficient erythroid precursors. However, when reduced numbers of corrected HSC are transplanted as occurred in secondary transplants of the group RPK 10%, or when corrected cells were transplanted in utero, the therapeutic effect is lost. These evidences would indicate that the differentiation advantage observed in the genetically corrected erythroid precursors might be insufficient to completely rescue the deficiency in the presence of low numbers of corrected cells. Alternatively, the transfer of reduced numbers of cells could result in the absence of genetically corrected hematopoietic stem cells in the graft that will end in the loss of anemia recovery in the long term.
Attempts to selectively expand normal donor erythrocytes have included the use of chemical inducers of dimerization in minimally conditioned, BM transplanted pyruvate kinase-deficient mice, but the proliferative advantage observed in normal erythroid progenitors was found to be dependent on regular chemical inducers of dimerization administration.18 Thus, RPK transfer protocols will always require a significant extent of donor chimeric hematopoiesis. In our system, high and stable expression levels of the RPK gene after myeloablative conditioning in vivo was achieved, with no deleterious effects observed even after tertiary transplant. The use of autologous genetically corrected cells along with a submyeloablative conditioning protocol to allow significant engraftment of genetically corrected HSCs, as it is undertaken in adenosine deaminase deficient patients,35 would probably be sufficient for successful RPK gene therapy in humans.
Insertional mutagenesis leading to leukaemia has been associated with the transactivation of oncogenes due to insertion of retroviral vectors in the vicinity of the affected genes.36,37 Although we have not observed adverse effects associated to the gene therapy protocol up to tertiary transplanted animals, improved vectors such as self-inactivated retroviral vectors, either γ- or lenti-vectors, which have shown lower risks of leukaemogenesis,38,39 should be used. In addition, the obvious benefits of lineage-specific transgene expression determine that the ideal vector for RPK gene therapy would be a self-inactivating lentiviral vector carrying an erythroid-specific promoter40 or, at least, a weak promoter driving the expression of the hRPK gene. We have recently evaluated the potential use of weak promoters such as the vav proto-oncogene, which shows weak and stable protein expression.41 New lentiviral vectors using this promoter for the treatment of PKD are presently being developed in our laboratory.
In summary, we show here that murine erythroid PKD can be treated by gene therapy with HSCs. Due to its long lasting nature and the fact that it solves all hematological symptoms, this strategy may be an option for the management of severe cases of PKD patients (RPK activity below 20% of normal levels) in which splenectomy and periodical red cell infusions fail to control the anemia and the only therapeutic alternative should be allogeneic BM transplant. Patients with these characteristics lacking a human leukocyte antigen matched donor should be the ideal target. Also, in utero transplantation of corrected cells could be a helpful approach in order to diminish the severe fetal and childhood clinical manifestation, such as hydrops fetalis, stillbirth and early neonatal death once a clinically relevant in utero engraftment could be obtained.
Materials and Methods
Animals. AcB55 (C57Bl/6 × A/J) mice (also recorded as PKD mice) carrying a point mutation at nucleotide 269 (T→A) of the pklr gene16 were obtained from Emerillon Therapeutics (Montreal, Quebec, Canada) and bred at the animal house of the CIEMAT (registration number 28079-21A). Animals were allowed food and water ad libitum, and were routinely screened for pathogens in accordance with Federation of European Laboratory Animal Science Associations procedures. All experimental procedures were conducted according to Spanish and European legislation (Spanish RD 223/88 and OM 13-10-89 of the Ministry of Agriculture, Food and Fisheries, for the protection and use of animals in scientific research; and European convention ETS-123, for the use and protection of vertebrate mammals used for experiments or other scientific purposes). Eight-to-ten week-old PKD mice were used as BM cell donors and recipients.
Vectors and cell lines. The SF11XEG vector, expressing the EGFP, and SF11RPKXEG vector, expressing the human RPK and the EGFP proteins in a single mRNA transcript containing an internal ribosome entry site sequence between both cDNAs, vectors used here are modifications of pSF11γ (GenBank accession no. AJ132035) as described previously.23 These vectors contain the 3′ LTR of spleen focus-forming virus and the leader sequence of murine embryonic stem cell virus.42 Phoenix 293T cell-based ecotropic packaging cells (Phoenix-eco; kindly provided by Dr Nolan, Stanford University, Stanford, CA) were maintained in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Intergen, Purchase, NY).
Viral supernatant production and cell line transduction. To establish ecotropic packaging cells for the SF11XEG and SF11RPKXEG vectors, 20 µg of the corresponding retroviral plasmid DNA were transfected using FuGENE reagent (Roche Diagnosis, Indianapolis, IN) on Phoenix-eco cells following the manufacturer's recommendations. EGFP+ cells were sorted in an EPICS Elite ESP flow cytometer (Coulter Electronics, Hialeah, FL) and kept as producer pools.
Purification, transduction, and transplant of murine HSCs. BM was harvested from 8 to 10 weeks old PKD male mice and subjected to red blood cell lysis. Lin− progenitors were sorted using the Lin− magnetic sort kit (Miltenyi Biotec, Gladbach, Germany) according to the manufacturer's instructions. On average, 80–90% pure populations of Lin− were obtained. Transduction of fresh Lin− cells were performed as previously described.23 Briefly, Lin− cells were cultured in vitro for 48 hours in Iscove's Modified Dulbecco's Medium (BioWhitacker, Walkersville, MD) plus 20% fetal bovine serum, 100 ng/ml murine stem cell factor and 100 ng/ml human interleukin-11. Cells were then harvested and resuspended in fresh supernatant from retroviral producer cells supplemented with the same growth factors and plated in retronectin (Takara, Shiga, Japan) coated dishes previously loaded with the corresponding retroviral vectors (3 incubations for 30 minutes of freshly supernatants). Hematopoietic cells were cultured for another 48 hours with 12 hour changes of viral supernatant containing factors. Cells harvested 4 hours after the last infection cycle were washed twice in phosphate-buffered saline (1× + 0.1% bovine serum albumin + 0.02% NaN3), and 2 × 105 hematopoietic cells/mouse were injected into the tail vein of 8 to 12 week-old female PKD mice, previously irradiated with 9.5 Gy in two split doses of 4.75 Gy, 24 hour apart, using a Philips MG324 X-ray instrument (Philips, Hamburg, Germany) set at 300 kV, 10 mA, and a delivery dose rate of 1.03 Gy/minute.
Hematological variables. Anticoagulated blood samples were collected for haematology analysis and to determine PK activity, ATP, and EPO. Complete blood counts, hematocrits, hemoglobin contents, and red blood cells indices were obtained using an automated blood cell analyzer (Abacus Junior vet; CMV Analitica, Spain). PK activity was determined by the lactate dehydrogenase-coupled spectrophotometric assay43 using the PKD Kit (Greiner diagnostic, Bahlingen, Germany). PK activity was expressed as microkatals per microgram of total protein (µKat/µg protein). Basal rates of activity were measured at all times.
ATP was quantified using a spectrophotometric ATP detection assay system (ATP Hexokinase FS; DiaSys, Holzhelm, Germany). This assay is based on the reaction of glucose with ATP catalyzed by hexokinase and Mg2+ ions. ATP concentration was expressed as micromoles per microgram of total protein (µmol/µg protein). Plasma EPO concentrations were determined through an enzyme-linked immunosorbent assay to detect mouse EPO using an anti-mouse EPO antibody (Quantikine Mouse Epo Immunoassay; R&D Systems, Minneapolis, MN).
Colony-forming unit cell assays. BM-derived cells (3 × 104) were seeded on 35-mm plastic tissue culture dishes (Nunc, Roskilde, Denmark) in triplicate in methyl cellulose (MethoCult GF M3534 culture medium, StemCell Technologies, Vancouver, British Columbia, Canada) containing recombinant murine stem cell factor (50 ng/ml), recombinant murine interleukin-3 (10 ng/ml), recombinant murine interleukin-6 (20 mg/ml), recombinant murine granulocyte-macrophage colony-stimulating factor (granulocyte macrophage–colony stimulating factor; 0.01 ng/ml), and EPO (recombinant mouse EPO; 3 U/ml). Cultures were incubated at 37 °C in a 95% humidified atmosphere with 5% CO2 in air. Later 7 days plating, overall colony numbers and EGFP+ colonies were scored under a light and epifluorescence microscope, respectively.
Flow cytometry and quantification of cell variation at specific erythroid subpopulation. The transgene expression and variation in the percentage of erythroid precursors in each erythroid development stages was measured as described elsewhere.23,27 Briefly, to analyze the most related erythroid precursors obtained from BM and spleen, 1 × 106 cells were stained with biotinylated anti-CD71 murine (Pharmingen, Palo Alto, CA) and TER-119-phycoerythrin (PE) (Pharmingen) antibodies, washed and incubated with streptavidin-tricolor (Caltag, Burlingame, CA). Cells were resuspended in phosphate-buffered saline with 2 µg/ml propidium iodide, and analyzed. A minimum number of 3 × 105 viable cells were acquired using a EPICS XL flow cytometer (Coulter Electronics, Hialeah, FL).
Quantification of cell variation at specific subpopulation stages was calculated following the mathematical development described elsewhere.27 Percentage of cell gain or loss (CL) in a subpopulation was calculated using the equation:
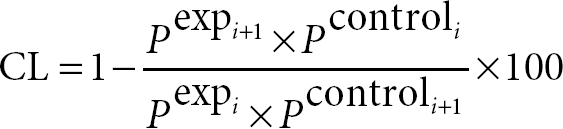 |
Where Pcontroli is the percentage of cells in the initial population in control mice, Pexpi the percentage of cells in the initial population in RPK-deficient mice and Pcontroli+1 the percentage of cells in the subsequent population in controls. To calculate the relative cell variation between two stages the following formula was applied: relative cell variation = −(CL × Pexpi/100), where a positive value corresponded to relative cell gain and a negative value corresponded to a relative cell loss. For reticulocyte analysis, 5 µl total blood was added to 2 ml phosphate-buffered saline containing 0.1 µg/ml of Acridine Orange (Invitrogen, Carlsbad, CA), incubated for 30 minutes at room temperature and directly analyzed by flow cytometry. Reticulocytes were identified as RNA positive cells within the erythrocyte population. A total of 106 erythrocytes were analyzed.
Chimerism. Chimerism was quantified as described44 through a real-time qPCR approach using Rotor Gene RG-3000 (Corbett Research Products, Foxboro, MA). Genomic DNA was extracted using the DNAeasy Tissue Kit (Qiagen, Duesseldorf, Germany) according to the manufacturer's instructions. Primers for the male-specific sequence were as follows: SRY-F: 5′-TGTTCAGCCCTACAGCCACA-3′ and SRY-R: 5′-CCTCTCACCACGGGACCAC and detected with the TaqMan probe SRY-T: 5′-FAM-ACAATTGTCTAGAGAGCATGGAGGGCCA-BHQ1. The murine genomic β-actin sequence was amplified using the primers: β -actin-MF: 5′-ACGGCCAGGTCATCACTATTG-3′ and β -actin-MR: 5′-ACTATGGCCTCAAGGAGTTTTGTCA- 3′; and detected with the TaqMan probe β-actin-T: 5′-TR-AACGAGCGGTTCCGATGCCCT-BHQ2-3′. Real-time qPCR was carried out in a multiplex reaction. For Amplification, 5 µl of genomic DNA (to 5 ng/µl) were mixed with 20 µl of a PCR master mix consisting of 1 × TaqMan universal master mix NoAmpErase (UNG Applied Biosystems, Roche, Nutley, NJ), 200 nmol/l of each primer and 200 nmol/l of each probe. The thermal profile was one hold of 10 minute at 95 °C, and 55 cycles of 20 seconds at 95 °C, and 30 seconds at 58 °C.
Proviral DNA analysis. To monitor provirus copy numbers, the EGFP sequence from the retroviral engineered vectors was assayed in transduced cells by real-time PCR. Primers and the TaqMan MGB probe were designed with the aid of the Primers Express software program (Applied Biosystems, Carlsbad, CA). The specific EGFP detector was composed of the forward primer: F1EGFP 5′GTAAACGGCCACAAGTTCAGC; reverse primer R1EGFP 5′TGGTGCAGATGAACTTCAGGG and the TaqMan MGB probe PEGFP 5′6-FAM-CTTGCCGTAGGTGGC-MGB. For cell lines, DNA was isolated from 1 × 106 cultured cells or from 5 × 105 BM cells using the Puregene kit (Promega, Madison, WI). Genomic DNA was resuspended in 200 µl of TE Buffer (10 mmol/l Tris with 0.1 mmol/l EDTA), incubated at 65 °C for 30 minutes, vortexed, and stored at 4 °C. For real-time PCR analysis, 5 µl of genomic DNA (to 5 ng/µl) were mixed with 20 µl of a PCR master mix consisting of 1 × TaqMan universal master mix, 200 nmol/l of each primer (F1EGFP/R1EGFP) and 200 nmol/l of the MGB probe (PEGFP). For negative controls, we used 5 µl of H2O and 5 µl of genomic DNA from untransduced cells. All reactions were conducted in triplicate and amplifications were performed as one cycle of 95 °C for 10 minutes, and 40 cycles of 95 °C for 30 seconds and 58 °C for 30 seconds. The average number of provirus copies per cell was quantified using a standard curve for the retroviral plasmid as described previously.45
To measure the transduction efficiency in hematopoietic progenitors we followed the protocol for Villella et al.46 with several modifications. Single colonies growing in methylcellulose were harvested and transferred directly into the PCR tubes, containing 50 µl crude cell lysing buffer (1× PCR buffer, 0.5% NP-40; 0.5% Tween 20; and 0.91 mg/ml proteinase K). Approximately 80 colonies from each sample were collected and labeled. Nontranduced BM and methylcellulose samples obtained from noncolony areas of the plates and samples containing cell lysis buffer alone were analyzed as negative controls. After all colonies were transferred, the tubes were placed in a thermal for 1 hour at 60 °C followed by 15 minute at 95 °C. Then, to detect EGFP sequences from the retroviral engineered vectors, 10 microliters of cell lysate from each sample was then transferred to each corresponding tube previously loaded with 40 µl per well PCR master mix, consisting of 1 × TaqMan universal master mix, 200 nmol/l of each primer (F1EGFP/R1EGFP) and 200 nmol/l of the MGB probe (PEGFP). All reactions were conducted in duplicate and amplifications were performed as one cycle of 95 °C for 10 minutes, and 55 cycles of 95 °C for 30 seconds and 58 °C for 30 seconds. Real-time qPCR assay on the murine genomic β-actin sequence using similar thermal profile and primers/probe concentration was done as an internal control for DNA content. Samples that did not contain the β-actin sequence were removed from the analysis.
In utero transplantation. Retrovirally transduced Lin− progenitors from PKD mice BM were injected in PKD fetuses on gestation day 14.5. Briefly, under isoflurane anesthesia, the uterine horns were exposed through a midline laparotomy. Under a dissecting microscope, the fetal liver was identified, and each foetus was injected with 5 × 105 Lin− cells transduced with the human RPK oncoretroviral therapeutic vector in a total volume of 5 µl using a 100 µm bevelled-glass micropipette. After this, the uterine horns were returned to the maternal peritoneal cavity, the abdomen closed by two running reabsorbable 5-0 vicryl sutures and each mother injected subcutaneously with 0.15 mg/weight mg of buprenorphine (Buprex; Reckitt Benckiser Healthcare, Hull, UK) as analgesic. After birth, hematological variables were monitored for up to 3 months in the surviving offspring.
Statistical analysis. Data are represented as the mean ± standard deviation of the mean. The significance of differences between groups was determined by using the nonparametric Wilcoxon Mann–Whitney W test. The processing and statistical analysis of the data was performed by using the Statgraphics Plus 5.0 software package (Manugistics, Rockville, MD).
SUPPLEMENTARY MATERIALTable S1. Relevant hematological variables recorded in secondary PKD recipients of genetically corrected BM cells obtained from transduced RPK-deficient mice.
Supplementary Material
Relevant hematological variables recorded in secondary PKD recipients of genetically corrected BM cells obtained from transduced RPK-deficient mice.
Acknowledgments
We thank E. López, A. de la Cal and M.D. López for their technical assistance, S. García for irradiating the animals and I. Orman for his expert help with the flow cytometry procedures at the CIEMAT/CIBER-ER. This work was funded by grants from the Ministerio de Educación y Ciencia (SAF2008-1883; SAF2005-02381; SAF2005-00058), Fondo de Investigaciones Sanitarias (RD06/0010/0015), the CONSERT European project, private support by the Botin Foundation given to J.C.S. and J.A.B., a grant from the Comunidad de Madrid (No. 08.2/0039/2001) awarded to J.M.B. and a grant by the Committee for Scientific, Humanistic, and Technological Development of the University of Los Andes (CDCHT: M-941-08-07-A) given to N.W.M. We also thank the Fundación Marcelino Botín for promoting translational research at the Hematopoiesis and Gene Therapy Division-CIEMAT/CIBERER.
REFERENCES
- Fothergill-Gilmore LA., and , Michels PA. Evolution of glycolysis. Prog Biophys Mol Biol. 1993;59:105–235. doi: 10.1016/0079-6107(93)90001-z. [DOI] [PubMed] [Google Scholar]
- Takenaka M, Noguchi T, Sadahiro S, Hirai H, Yamada K, Matsuda T, et al. Isolation and characterization of the human pyruvate kinase M gene. Eur J Biochem. 1991;198:101–106. doi: 10.1111/j.1432-1033.1991.tb15991.x. [DOI] [PubMed] [Google Scholar]
- Miwa S, Kanno H., and , Fujii H. Concise review: pyruvate kinase deficiency: historical perspective and recent progress of molecular genetics. Am J Hematol. 1993;42:31–35. doi: 10.1002/ajh.2830420108. [DOI] [PubMed] [Google Scholar]
- Kanno H, Fujii H, Tsujino G., and , Miwa S. Molecular basis of impaired pyruvate kinase isozyme conversion in erythroid cells: a single amino acid substitution near the active site and decreased mRNA content of the R-type PK. Biochem Biophys Res Commun. 1993;192:46–52. doi: 10.1006/bbrc.1993.1379. [DOI] [PubMed] [Google Scholar]
- Tanaka KR., and , Zerez CR. Red cell enzymopathies of the glycolytic pathway. Semin Hematol. 1990;27:165–185. [PubMed] [Google Scholar]
- Bianchi P., and , Zanella A. Hematologically important mutations: red cell pyruvate kinase (Third update) Blood Cells Mol Dis. 2000;26:47–53. doi: 10.1006/bcmd.2000.0276. [DOI] [PubMed] [Google Scholar]
- Lenzner C, Nürnberg P, Jacobasch G, Gerth C., and , Thiele BJ. Molecular analysis of 29 pyruvate kinase-deficient patients from central Europe with hereditary hemolytic anemia. Blood. 1997;89:1793–1799. [PubMed] [Google Scholar]
- Kanno H, Fujii H, Wei DC, Chan LC, Hirono A, Tsukimoto I, et al. Frame shift mutation, exon skipping, and a two-codon deletion caused by splice site mutations account for pyruvate kinase deficiency. Blood. 1997;89:4213–4218. [PubMed] [Google Scholar]
- van Wijk R, van Solinge WW, Nerlov C, Beutler E, Gelbart T, Rijksen G, et al. Disruption of a novel regulatory element in the erythroid-specific promoter of the human PKLR gene causes severe pyruvate kinase deficiency. Blood. 2003;101:1596–1602. doi: 10.1182/blood-2002-07-2321. [DOI] [PubMed] [Google Scholar]
- Diez A, Gilsanz F, Martinez J, Pérez-Benavente S, Meza NW., and , Bautista JM. Life-threatening nonspherocytic hemolytic anemia in a patient with a null mutation in the PKLR gene and no compensatory PKM gene expression. Blood. 2005;106:1851–1856. doi: 10.1182/blood-2005-02-0555. [DOI] [PubMed] [Google Scholar]
- Beutler E., and , Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood. 2000;95:3585–3588. [PubMed] [Google Scholar]
- Miwa S., and , Fujii H. Molecular basis of erythroenzymopathies associated with hereditary hemolytic anemia: tabulation of mutant enzymes. Am J Hematol. 1996;51:122–132. doi: 10.1002/(SICI)1096-8652(199602)51:2<122::AID-AJH5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Cazzola M. Pyruvate kinase deficiency. Haematologica. 2005;90:1–2. [PubMed] [Google Scholar]
- Tanphaichitr VS, Suvatte V, Issaragrisil S, Mahasandana C, Veerakul G, Chongkolwatana V, et al. Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant. 2000;26:689–690. doi: 10.1038/sj.bmt.1702576. [DOI] [PubMed] [Google Scholar]
- Zaucha JA, Yu C, Lothrop CD, Nash RA, Sale G, Georges G, et al. Severe canine hereditary hemolytic anemia treated by nonmyeloablative marrow transplantation. Biol Blood Marrow Transplant. 2001;7:14–24. doi: 10.1053/bbmt.2001.v7.pm11215693. [DOI] [PubMed] [Google Scholar]
- Min-Oo G, Fortin A, Tam MF, Nantel A, Stevenson MM., and , Gros P. Pyruvate kinase deficiency in mice protects against malaria. Nat Genet. 2003;35:357–362. doi: 10.1038/ng1260. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Kanno H, Asai H, Tsujimura T, Fujii H, Moriyama Y, et al. Pyruvate kinase deficiency of mice associated with nonspherocytic hemolytic anemia and cure of the anemia by marrow transplantation without host irradiation. Blood. 1995;86:4323–4330. [PubMed] [Google Scholar]
- Richard RE, Weinreich M, Chang KH, Ieremia J, Stevenson MM., and , Blau CA. Modulating erythrocyte chimerism in a mouse model of pyruvate kinase deficiency. Blood. 2004;103:4432–4439. doi: 10.1182/blood-2003-10-3705. [DOI] [PubMed] [Google Scholar]
- Kanno H, Utsugisawa T, Aizawa S, Koizumi T, Aisaki K, Hamada T, et al. Transgenic rescue of hemolytic anemia due to red blood cell pyruvate kinase deficiency. Haematologica. 2007;92:731–737. doi: 10.3324/haematol.10945. [DOI] [PubMed] [Google Scholar]
- Tani K, Yoshikubo T, Ikebuchi K, Takahashi K, Tsuchiya T, Takahashi S, et al. Retrovirus-mediated gene transfer of human pyruvate kinase (PK) cDNA into murine hematopoietic cells: implications for gene therapy of human PK deficiency. Blood. 1994;83:2305–2310. [PubMed] [Google Scholar]
- Klein C., and , Baum C. Gene therapy for inherited disorders of haematopoietic cells. Hematol J. 2004;5:103–111. doi: 10.1038/sj.thj.6200366. [DOI] [PubMed] [Google Scholar]
- Wang C, Chiarelli LR, Bianchi P, Abraham DJ, Galizzi A, Mattevi A, et al. Human erythrocyte pyruvate kinase: characterization of the recombinant enzyme and a mutant form (R510Q) causing nonspherocytic hemolytic anemia. Blood. 2001;98:3113–3120. doi: 10.1182/blood.v98.10.3113. [DOI] [PubMed] [Google Scholar]
- Meza NW, Quintana-Bustamante O, Puyet A, Rio P, Navarro S, Diez A, et al. In vitro and in vivo expression of human erythrocyte pyruvate kinase in erythroid cells: a gene therapy approach. Hum Gene Ther. 2007;18:502–514. doi: 10.1089/hum.2006.052. [DOI] [PubMed] [Google Scholar]
- Steiner LA., and , Gallagher PG. Erythrocyte disorders in the perinatal period. Semin Perinatol. 2007;31:254–261. doi: 10.1053/j.semperi.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surbek D, Schoeberlein A., and , Wagner A. Perinatal stem-cell and gene therapy for hemoglobinopathies. Semin Fetal Neonatal Med. 2008;13:282–290. doi: 10.1016/j.siny.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Rio P, Martinez-Palacio J, Ramirez A, Bueren JA., and , Segovia JC. Efficient engraftment of in utero transplanted mice with retrovirally transduced hematopoietic stem cells. Gene Ther. 2005;12:358–363. doi: 10.1038/sj.gt.3302419. [DOI] [PubMed] [Google Scholar]
- Beauchemin H, Blouin MJ., and , Trudel M. Differential regulatory and compensatory responses in hematopoiesis/erythropoiesis in alpha- and beta-globin hemizygous mice. J Biol Chem. 2004;279:19471–19480. doi: 10.1074/jbc.M309989200. [DOI] [PubMed] [Google Scholar]
- Persons DA., and , Tisdale JF. Gene therapy for the hemoglobin disorders. Semin Hematol. 2004;41:279–286. doi: 10.1053/j.seminhematol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Gilsanz F, Vega MA, Gómez-Castillo E, Ruiz-Balda JA., and , Omeñaca F.1993Fetal anaemia due to pyruvate kinase deficiency Arch Dis Child 695 Spec No): 523–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood T. The inhibition of pyruvate kinase by ATP. Biochem Biophys Res Commun. 1968;31:779–785. doi: 10.1016/0006-291x(68)90630-x. [DOI] [PubMed] [Google Scholar]
- Xu J, Christian B., and , Jump DB. Regulation of rat hepatic L-pyruvate kinase promoter composition and activity by glucose, n-3 polyunsaturated fatty acids, and peroxisome proliferator-activated receptor-alpha agonist. J Biol Chem. 2006;281:18351–18362. doi: 10.1074/jbc.M601277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Kanno H, Hirono A., and , Fujii H. Red cell enzymopathies as a model of inborn errors of metabolism. Southeast Asian J Trop Med Public Health. 1995;26 Suppl 1:112–119. [PubMed] [Google Scholar]
- Murotsuki J, Uehara S, Okamura K, Yajima A, Oura T., and , Miyabayashi S. Fetal liver biopsy for prenatal diagnosis of carbamoyl phosphate synthetase deficiency. Am J Perinatol. 1994;11:160–162. doi: 10.1055/s-2007-994579. [DOI] [PubMed] [Google Scholar]
- Golbus MS, Simpson TJ, Koresawa M, Appelman Z., and , Alpers CE. The prenatal determination of glucose-6-phosphatase activity by fetal liver biopsy. Prenat Diagn. 1988;8:401–404. doi: 10.1002/pd.1970080603. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Kustikova O, Fehse B, Modlich U, Yang M, Düllmann J, Kamino K, et al. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science. 2005;308:1171–1174. doi: 10.1126/science.1105063. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Laufs S, Guenechea G, Gonzalez-Murillo A, Zsuzsanna Nagy K, Luz Lozano M, del Val C, et al. Lentiviral vector integration sites in human NOD/SCID repopulating cells. J Gene Med. 2006;8:1197–1207. doi: 10.1002/jgm.958. [DOI] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knoss S, Schambach A, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity 10.1182/blood-2005-08-024976. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard E, Mendez M, Mazurier F, Morel C, Costet P, Xia P, et al. Gene therapy of a mouse model of protoporphyria with a self-inactivating erythroid-specific lentiviral vector without preselection. Mol Ther. 2001;4:331–338. doi: 10.1006/mthe.2001.0467. [DOI] [PubMed] [Google Scholar]
- Almarza E, Río P, Meza NW, Aldea M, Agirre X, Guenechea G, et al. Characteristics of lentiviral vectors harboring the proximal promoter of the vav proto-oncogene: a weak and efficient promoter for gene therapy. Mol Ther. 2007;15:1487–1494. doi: 10.1038/sj.mt.6300213. [DOI] [PubMed] [Google Scholar]
- Hildinger M, Abel KL, Ostertag W., and , Baum C. Design of 5' untranslated sequences in retroviral vectors developed for medical use. J Virol. 1999;73:4083–4089. doi: 10.1128/jvi.73.5.4083-4089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Fujii H, Takegawa S, Nakatsuji T, Yamato K, Ishida Y, et al. Seven pyruvate kinase variants characterized by the ICSH recommended methods. Br J Haematol. 1980;45:575–583. doi: 10.1111/j.1365-2141.1980.tb07181.x. [DOI] [PubMed] [Google Scholar]
- Navarro S, Meza NW, Quintana-Bustamante O, Casado JA, Jacome A, McAllister K, et al. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol Ther. 2006;14:525–535. doi: 10.1016/j.ymthe.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Meza NW, Puyet A, Pérez-Benavente S, Quintana-Bustamante O, Diez A, Bueren JA, et al. Functional analysis of gammaretroviral vector transduction by quantitative PCR. J Gene Med. 2006;8:1097–1104. doi: 10.1002/jgm.951. [DOI] [PubMed] [Google Scholar]
- Villella AD, Yao J, Getty RR, Juliar BE, Constantin Y, Hartwell JR, Cai S, Sadat MA, Cornetta K, Williams DA., and , Pollok KE. Real-time PCR: an effective tool for measuring transduction efficiency in human hematopoietic progenitor cells. Mol Ther. 2005;11:483–491. doi: 10.1016/j.ymthe.2004.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relevant hematological variables recorded in secondary PKD recipients of genetically corrected BM cells obtained from transduced RPK-deficient mice.