

Abstract
Ras signaling can be modulated by the scaffolding activity of kinase suppressor of Ras-1 (KSR-1) and by the hKSR-2 protein, resulting in diverse phenotypic outcomes. The mitogen-activated protein kinase cascade downstream from Ras and KSRs includes Raf-1 and extracellular signal-regulated kinase 1/2 kinases, known to enhance survival potential of a range of cell types. Because the molecular events that increase survival of HL60 cells induced to differentiate toward monocytic phenotype by 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] are not known, we investigated if KSR proteins provide a survival function in these cells. We found that whereas kinase suppressor of Ras-1 had no detectable effect on cell survival in the system studied here, 1,25-(OH)2D3-induced up-regulation of hKSR-2 enhanced the resistance of HL60 cells to arabinocytosine. Knockdown of hKSR-2 by either small interfering RNA or antisense oligonucleotides increased arabinocytosine-induced apoptosis, which was accompanied by reduced Bcl-2/Bax and Bcl-2/Bad ratios, and increased caspase-3 activating cleavage. In contrast, up-regulation of Mcl-1 was not abrogated by anti-sense (AS) AS-hKSR-2, pointing to a specific role of Bcl-2 in control of 1,25-(OH)2D3-induced increased cell survival. These findings are consistent with the previously shown lack of fully differentiated monocytic cells in HL60 cultures exposed to 1,25-(OH)2D3 in which hKSR-2 was knocked down, suggesting that optimal differentiation of these cells requires enhanced antiapoptotic mechanisms provided, at least in part, by hKSR-2. Collectively, these results suggest that hKSR-2 may offer a new target for novel therapies of acute myelogenous leukemia.
Introduction
Professional phagocytes, such as monocytes and macrophages, require to develop survival mechanisms to counteract the effects of the ingested noxious organisms and particles to withstand the consequences of elevated intracellular levels of the reactive oxygen species (ROS) and digestive enzymes necessary for the disposal of the ingested material (1). Although this is an important physiologic adaptation in normal phagocytes, the increased survival potential may become a therapeutic problem when cells of myeloid lineage become neoplastic, and attempts are being made to eradicate these malignant cells by combination differentiation/chemotherapy. Thus, differentiation therapy could potentially antagonize the action of standard chemotherapeutic agents, such as arabinocytosine (AraC), used for treatment of acute myelogenous leukemia, and this issue presents a barrier to the further development of combined differentiation/cytotoxic antineoplastic treatments for this still difficult-to-treat disease.
Promyeloblastic HL60 cells represent a model of human acute myelogenous leukemia widely used for mechanistic studies of cancer chemotherapy or differentiation therapy (e.g., refs. 2–4). We, and others, have used these cells to investigate the molecular events that underlie monocytic differentiation induced by 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] as potential therapy for the human disease (e.g., refs. 5–7). It was noted that 1,25-(OH)2D3 has an antiapoptotic action associated with the increased expression of the Bcl-2 family prosurvival protein Mcl-1(8). Although this provided a partial basis for the increased survival capacity of differentiated, phagocytic cells, it also suggested the need for further studies to obtain a fuller picture of the underlying phenomena.
Among the recognized extracellularly regulated cellular survival signaling molecules are the kinases Raf-1(e.g., refs. 9, 10) and extracellular signal-regulated kinase 1/2 (e.g., refs. 11, 12), although their precise involvement in maintaining cell survival is not completely understood. Both belong to one of the most studied intracellular signaling cascades, the Ras-Raf-MEK-extracellular signal-regulated kinase pathway, which has been shown to participate in 1,25-(OH)2D3-induced monocytic differentiation (13–15), and in some cell contexts to be regulated by a scaffolding protein, the kinase suppressor of Ras-1 (KSR-1). In earlier work, we have shown that KSR-1 may have an important role in the induction of monocytic differentiation by 1,25-(OH)2D3, as it can amplify the differentiation signal provided by low, near-physiologic, concentrations of 1,25-(OH)2D3 (16). Furthermore, it has been shown that the KSR-1 promoter region harbors a vitamin D response element, which is functional in differentiating HL60 cells, and thereby increases the expression of KSR-1, which then enhances the activity of Raf-1 and its downstream targets (17).
More recently, a second KSR family gene was identified in Caenorhabditis elegans and found to be required for germline meiosis in Ras-mediated signaling (18). Its human homologue, hKSR-2, has also been cloned and shown to have 61% amino acid identity with hKSR-1, and although like KSR-1, hKSR-2 lacks the CA1 domain, it does have the CA5 domain, which encompasses 11 conserved kinase subdomains (19). We identified two vitamin D response element–type motifs in the hKSR-2 gene, which bind vitamin D receptor in intact cells in a 1,25-(OH)2D3-dependent manner, that coincides with the recruitment of RNA polymerase II to these motifs, suggesting that hKSR-2 is directly regulated by 1,25-(OH)2D3 (20). We also found that hKSR-2 is required for the expression of the fully mature monocytic phenotype in HL60 cells induced to differentiate by 1,25-(OH)2D3 (20). Because, as indicated above, the mature monocytic phenotype implies phagocytic activity hazardous to cell survival due primarily to generation of ROS (21), we hypothesized that hKSR-2 contributes to the survival functions of cells induced by 1,25-(OH)2D3 to differentiate into monocytes/macrophages. Thus, hKSR-2 knockdown prevents optimal differentiation probably due to death of differentiating cells with low hKSR-2 protein levels, which are producing ROS during phagocytosis. This indeed appears to be the case; we found that depletion of hKSR-2 promotes apoptosis of HL60 cells exposed to AraC, and this is associated with increased Bcl-2 to Bax and to Bad ratios, which function to reduce the activation of caspase-3.
Materials and Methods
Cell Culture
HL60-G cells, derived from a patient with promyeloblastic leukemia (22), were cultured in suspension in RPMI 1640 supplemented with 10% bovine calf serum (Hyclone) in a humidified atmosphere containing 5% CO2 at 37°C. Cells were passaged two to three times a week and were used in the exponential growth phase. Cell numbers and viability were determined by hemocytometer counts and trypan blue (0.4%) exclusion, and routine microbiology testing for Mycoplasma was done by selective culture techniques. For all experiments, the cells were suspended in fresh medium and each experimental condition was repeated at least three times.
The sequence of experimental manipulations was as follows: (a) If indicated, the cells were transfected with small interfering RNA (siRNA) or exposed to antisense oligonucleotides. (b) After 48 h, the cells were exposed to vehicle or the indicated concentration of 1,25-(OH)2D3 for 24 h. (c) The cells were treated with AraC as detailed in individual experiments.
Reagents and Antibodies
1,25-(OH)2D3 was a kind gift from Dr. Milan Uskokovic (Bioxell). AraC and N-acetylcysteine (NAC) were purchased from Sigma. Cell-permeable DEVD-CHO is the “caspase-3 inhibitor I” that was purchased from EMD Chemicals. Complete protease inhibitor cocktail was obtained from Hoffmann-La Roche. For Western blotting studies, anti- bodies Bcl-2, Mcl-1, Bax, Bad, caspase-3, and Crk-L were obtained from Santa Cruz Biotechnology. The hKSR-2 antibody was from Affinity Bioreagents. Anti-rabbit and anti-goat antibodies linked to horseradish peroxidase were purchased from Cell Signaling Technologies. Nitrocellulose membranes were purchased from Amersham Biosciences.
siRNA and Antisense Oligonucleotides
We designed double-stranded 21-mer siRNA targeting hKSR-2 at the sequence 5′-AAUGUCCACAUGGUCAGCACC-3′ (3,068-3,088; Dharmacon). Generic scrambled siRNA was purchased as control (Dharmacon). siRNAs (5 μmol/L) were transfected into HL60 cells using Amaxa nucleofector (solution T and program T-19) according to the manufacturer's protocol. The cells were allowed to recover for 48 h in 6 cm wells in RPMI 1640 supplemented with 10% bovine calf serum and checked for the reduction of mRNA and protein expression. Transfected cells were seeded at a concentration of 2 × 105/mL and exposed to the specified agents for the indicated times. Phosphorothioate antisense oligonucleotides to hKSR-2, 5′-GTCCTCGTTGTCCCTCTCA-3′ (1,724-1,743), and scrambled oligonucleotides were synthesized by Molecular Resource Facility of the New Jersey Medical School. Each oligonucleotide was incubated with the cells at 20 μmol/L (unless indicated otherwise) for 48 h before adding 1,25-(OH)2D3, N-acetylcysteine, or caspase-3 inhibitor I.
Semiquantitative PCR
Semiquantitative measurements of KSR-1, hKSR-2, and β-actin mRNA levels were carried out as described before. Briefly, total RNA was extracted using RNeasy kit, treated with DNase I, reverse transcribed, and amplified by using Eppendorf gradient MasterCycler using 35 cycles of amplification. The primer sequences used were KSR-1 upstream primer (5-AGCAAGTCCCATGAGTCTCA-3) and downstream primer (5-CAACCTGCAATGCTTGCACT-3), hKSR-2 upstream primer (5-CCGACACAGAGGAGGATAAG-3) and downstream primer (5-TTCAAAGGCCCAGCAGAAG-3), and β-actin upstream primer (5-GACGGGGTCACCCACACTGTGCCCAGCTA-3) and downstream primer (5-CTAGAAGCATTTGCCGGTGGACGATGGAGGG-3). The reverse transcription-PCR products were separated in 2% agarose gels containing ethidium bromide (1 μg/mL). The intensities of signals of KSR-1, hKSR-2, and β-actin bands were scanned and quantitated using ImageQuant program (Molecular Dynamics).
Quantitative Real-time PCR
Real-time PCR was carried out by using a LightCycler with FastStart DNA SYBR Green PCR kit (Roche Diagnostics). cDNA was synthesized by using 1 μg DNase I–treated total RNA. Reverse transcription conditions were as follows: 42°C for 15 min, 95°C for 5 min, and 5°C for 5 min (one cycle). Real-time PCR was done following the protocol provided by the manufacturer. Preincubation at 95°C for 7 min was followed by 40 cycles of 95°C for 10 s, 55°C for 10 s, and 72°C for 10 s. Fold changes of mRNA levels in hKSR-2 target gene relative to the RNA polymerase II control were calculated by relative quantification analysis. Primers used for real-time PCR for KSR-1 and hKSR-2 were the same as for semiquantitative PCR primers. Primers for Bcl-2 were upstream 5′-TTCTTTGAGTTCGGTGGGGTC-3′ and downstream 5′-TGCATATTTGTTTGGGGCAGG-3′. Primers for Mcl-1 were upstream 5′-CGGTAATCGGACTCAACCTC-3′ and downstream 5′-CCTCCTTCTCCGTAGCCAA-3′. For RNA polymerase II, the primers were upstream 5-GCACCACGTCCAATGACAT-3 and downstream 5-GTGCGGCTGCTTCCATAA-3. Threshold cycle (Ct) is the cycle number where the fluorescence increases above a background threshold level. Relative expression levels were calculated by equation: ΔÄCt = ΔCtsample - ΔCtcalibrator, ΔCt = CtKSR - CtRNA polymerase II. Ct value for KSR-1 is ∼ 25 cycles and Ct value for hKSR-2 is ∼ 27 cycles, so the real-time PCR was set to 40 cycles. The quality of PCR product was monitored using post-PCR melting curve analysis.
Hematoxylin Staining for Morphologic Observation
HL60 cells were treated with different agents for indicated times, washed briefly with PBS before they were spread onto glass slides, fixed with buffered formalin, and stained with hematoxylin.
Annexin V and Propidium Iodide Staining
Samples were collected and washed once with PBS. The pellet was resuspended in HEPES containing 137 mmol/L NaCl and stained using an Annexin V-FITC kit (Sigma) to detect apoptotic cells generated by treatment with AraC for 24 h. After washing in 1× PBS, 1 × 106 cells were resuspended in binding buffer [10 mmol/L HEPES/NaOH (pH 7.4), 140 mmol/L NaCl, 2.5 mmol/L CaCl2] and Annexin V-FITC was added. Cells were incubated in the dark at room temperature for 10 min, 20 μg/mL propidium iodide (PI) was added for another 20 min, and samples were immediately analyzed by flow cytometry. Annexin V and PI fluorescence of individual cells were measured with an Epics XL-MCL flow cytometer using the FL-1 (Annexin V) and FL-3 (PI) channels. The data for Annexin V–positive/PI-negative cells are presented. Early apoptosis is shown by bottom right-hand quadrants in each panel; late apoptosis is shown by top right-hand quadrants.
Western Blotting
Western blotting was done using whole-cell extracts, except as indicated. The cells were harvested and washed twice with ice-cold 1× PBS. The washed cell pellets were solubilized with a lysis buffer [20 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, and 1 μg/mL aprotinin] followed by centrifugation at 16,000 × g for 15 min. The protein concentrations of the extracts were determined by using Bio-Rad protein assay kit and then incubated in (2:1) 3× SDS sample buffer [150 mmol/L Tris-HCl (pH 6.8), 30% glycerol, 3% SDS, 1.5 mg/mL bromophenol blue dye, and 100 mmol/L DTT]. Proteins (35 μg) in cell extracts were separated on 10% SDS-PAGE gel and then transferred to nitrocellulose membranes (Amersham). The membranes were blocked with 5% milk in TBS/0.1% Tween 20 for 1 h, incubated overnight with primary antibodies, and then blotted with a horseradish peroxidase–linked secondary antibody for 1h. The protein bands were visualized using a chemiluminescence assay system (Pierce Biotechnology), and the absorbance of each band was quantitated using an ImageQuant 5.0 (Molecular Dynamics).
Statistical Methods
Each experiment was done at least three times and the results were expressed as percentages (mean ± SD) of the vehicle controls. Significance of the differences between mean values was assessed by a two-tailed Student's t test and for Fig. 2 curves by the likelihood ratio test (Sigmastat Software). All computations were done with an IBM-compatible personal computer using Microsoft Excel.
Figure 2.
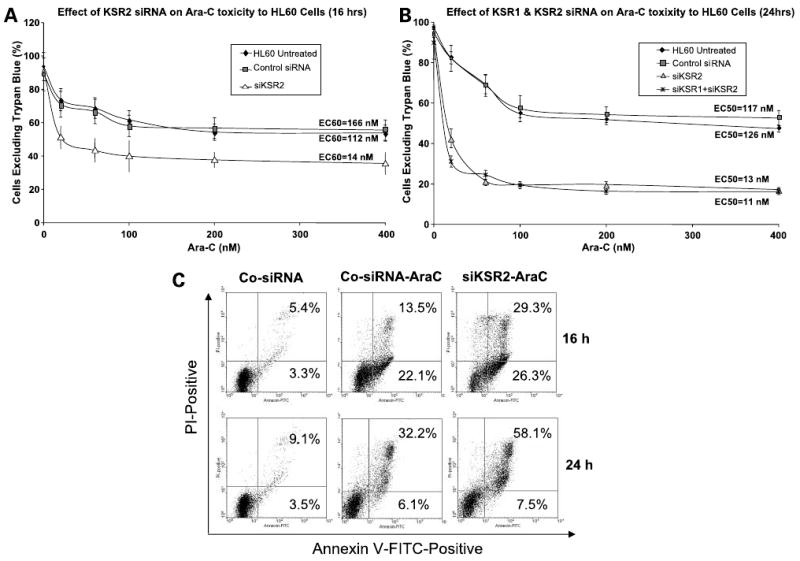
Knockdown of hKSR-2 expression by si-hKSR-2 expression increases the toxicity of AraC to HL60 cells. HL60 cells were transfected with either control scrambled siRNA, siKSR-2, or both siKSR1 and si-hKSR-2, cultured for 48 h, and treated with increasing concentrations of AraC (A and B) for 16 or 24 h. In preliminary experiments, siKSR1 did not affect sensitivity to AraC. Cell viability was determined by the trypan blue exclusion assay. Note that si-hKSR-2 increased the AraC-induced toxicity to HL60 cells, whereas cotransfection of both siKSR1 and siKSR-2 did not significantly increase AraC-induced toxicity when compared with si-hKSR-2 administered alone. EC 50 represents effective concentration for 50% of the cells. C, cell death induced by AraC in this system is principally by apoptosis and is markedly accentuated by a knockdown of hKSR-2.
Results
siRNAto hKSR-2 (siKSR-2) IncreasesToxicityof AraC to HL60 Cells
We reported previously a reduction in the number of differentiated cells in culture in which hKSR-2 levels were knocked down by siKSR-2 (20). To test the hypothesis that this is due to impaired survival of differentiating cells that have increased capacity to produce ROS but have low levels of hKSR-2, which could protect them from cytotoxicity, we treated HL60 cells with siKSR-2 and siKSR-1. Figure 1A shows that this selectively reduced the levels of the cognate mRNAs, showing that there is no overlap in silencing of KSR-1and KSR-2 genes. Figure 1B shows that siKSR-2 reduced the levels of cellular hKSR-2 protein, of principal interest to this study. Additional preliminary experiments indicated that siKSR-1 had no effect on the viability of cells exposed to 100 nmol/L AraC (data not shown). In contrast, exposure to siKSR-2 sensitized the cells to a wide range of concentrations of AraC (Fig. 2A and B). Annexin V staining showed that the AraC-induced cell death was predominantly by apoptosis, as at the earlier time point (16 h) most of the Annexin V–stained cells were PI negative (Fig. 2C). In accord with our hypothesis, a knockdown of hKSR-2 clearly increased the rate of cell death (Fig. 2C). Although, as mentioned above, siKSR-1 had no detectable effect on cell survival, to determine if it can contribute to the effects of hKSR-2 on cell viability, the effect of AraC was tested in the presence of both KSR-1and hKSR-2 mRNA-depleting agents, but no potentiation of the siKSR-2 effect by the addition of siKSR-1was noted (Fig. 2B), confirming that hKSR-2, but not KSR-1, modulates the survival of AraC-treated HL60 cells.
Figure 1.
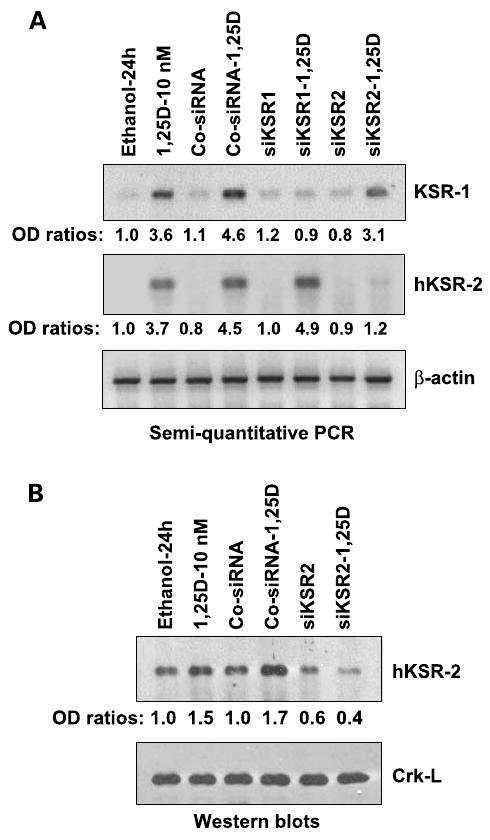
siRNA to hKSR-2 selectively reduces the levels of the cognate mRNA and protein. A, HL60-G cells were transfected with either control siRNA (Co-siRNA), siKSR1, or si-hKSR-2 (all 5 μmol/L), incubated for 48 h, and treated with 10 nmol/L 1,25-(OH)2D3 for another 24 h. mRNA levels of KSR1 and hKSR-2 were determined by semiquantitative reverse transcription-PCR. Note that both KSR1 and si-hKSR-2 mRNA levels were induced by 1,25-(OH)2D3 treatment, and siKSR-1 or si-hKSR-2 selectively reduced the cognate mRNA expression. β-Actin RNA was used as a housekeeping gene control. The ratios of absorbance of each gene to β-actin are shown below each lane. B, protein levels of hKSR-2 in cells transfected with si-hKSR-2. Crk-L was used as a loading control. The experiments were repeated three times with similar results.
Morphologic Evidence That hKSR-2 Modulates Apoptosis in AraC-Treated Cells
One of the clearest means to determine if cell death occurred by apoptosis or necrosis is by light microscopic examination of cell morphology (23, 24). As shown in Fig. 3, AraC at the concentration used in these experiments produced morphologic changes such as small cells with condensed nuclei (examples indicated by narrow black arrows) indicative of apoptosis and smaller numbers of disintegrating rather large cells with indistinctive borders and fading nuclei, indicating necrosis (wide arrows). In AraC-treated cultures, the cells undergoing apoptosis were clearly increased in number by pretreatment with siKSR-2. Thus, these experiments further show that low levels of hKSR-2 can sensitize cells to induction of cell death and thus that hKSR-2 serves to promote the survival of human leukemia HL60 cells.
Figure 3.
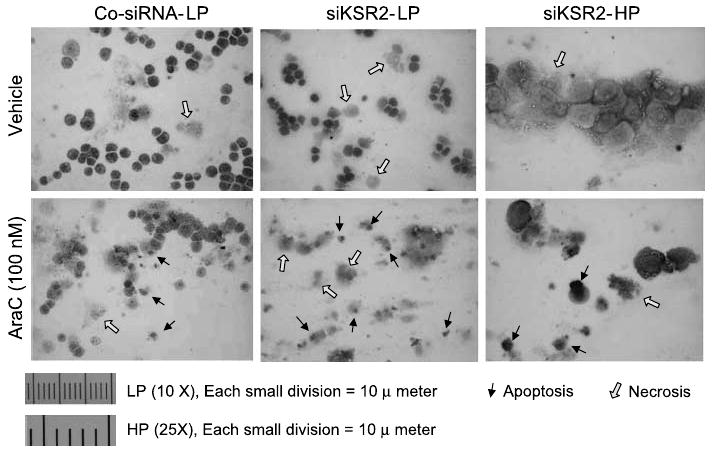
Knockdown of hKSR-2 increases both apoptosis and necrosis in AraC-treated HL60 cells. HL60 cells were transfected with either control siRNA or si-hKSR-2, cultured for 48 h, and treated with AraC (100 nmol/L) for 24 h. The cells were spread on slides and stained with hematoxylin. Apoptosis or necrosis was determined by cell morphology. Black arrows, cells undergoing apoptosis; open arrows, necrotic cells, which were principally induced by the transfection procedure because they were also seen in the absence of AraC. Left, cells under low power (×10 objective); bottom, cells under high power (×25 objective).
Depletion of hKSR-2 by Antisense Oligonucleotides Abrogates the Protection by 1,25-(OH)2D3 or by the Antioxidant N-Acetylcysteine of the Apoptotic Effects of AraC
Because transfection of siKSR-2 is rather traumatic to hematopoietic cells, thus complicating the interpretation of the experimental outcomes, we used antisense strategy for hKSR-2 depletion for further studies of the role of hKSR-2 in AraC-induced apoptosis. Antisense oligonucleotides do not require transfection as they are taken up by the cells from the medium spontaneously and reduce the hKSR-2 mRNA (Fig. 4A) and protein (shown previously in ref. 20) levels to a considerable extent. We used this approach to determine if hKSR-2 is required for the previously known protection from AraC-induced apoptosis by the exposure of the cells to 1,25-(OH)2D3 (8). This protection and its abrogation by transfection with AS-hKSR-2 are shown in Fig. 4B, which summarizes three experiments, one of which is also illustrated in Fig. 4C, the primary data from Annexin V/PI assay. In this figure, the right-side quadrants in each panel show early (bottom quadrants) and late (top quadrants) apoptotic cells. It is apparent that AraC induces apoptosis in this experimental system, which is reduced by preexposure of the cells to 1,25-(OH)2D3, known to increase the expression of hKSR-2, and that the protection from apoptosis provided by 1,25-(OH)2D3 is negated by hKSR-2 knockdown. Interestingly, a similar protection from apoptosis by the antioxidant N-acetylcysteine was also abrogated by hKSR-2 knockdown (Fig. 4B). Given that ROS are produced in AraC-treated leukemia cells and contribute to AraC toxicity (25), this suggests that hKSR-2 may play an important role in protecting differentiating HL60 cells from ROS-induced toxicity.
Figure 4.
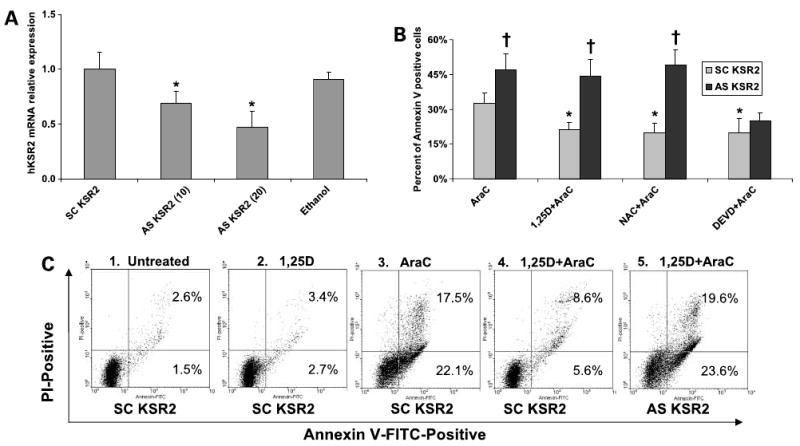
Protection from the proapoptotic effects of AraC by 1,25-(OH)2D3 or by N-acetylcysteine (NAC), but not by DEVD, is abrogated by a down-regulation of hKSR-2 expression using antisense oligonucleotides. HL60 cells were incubated for 48 h with either scrambled (SC) or antisense (AS) oligonucleotides to hKSR-2 at 20 μmol/L, except as indicated in A, and then treated with the indicated agents. A, partial depletion of hKSR-2 mRNA by antisense oligonucleotides at 10 or 20 μmol/L concentrations was confirmed by quantitative real-time PCR to hKSR-2. *, P < 0.05, significant reduction from scrambled KSR-2 control (n = 3). Error bars represent SD of the mean value. The “Ethanol” group represents vehicle-treated but untransfected cells. B, summary of several types of experiments showing abrogation by antisense oligonucleotide hKSR-2 knockdown of the protection from apoptosis by 1,25-(OH)2D3 or N-acetylcysteine, but not by the caspase-3 inhibitor 1 (DEVD), as determined by the Annexin V assay. *, P < 0.05, significant reduction from the AraC-only group; †, P < 0.05, significant increase in sensitivity to AraC in antisense KSR-2-treated groups compared with scrambled oligonucleotide-treated groups (n = 3). C, illustrations of the primary data used in part to calculate the first data cluster in B. For instance, panel 4 shows reduced apoptosis in cells pretreated for 24 h with 10 nmol/L 1,25-(OH)2D3 before the addition of 100 nmol/L AraC for 24 h, whereas panel 5 shows that 1,25-(OH)2D3-induced protection is abrogated by AS-KSR-2. Note that unlike transfection of siRNA (Fig. 2C), the exposure to antisense oligonucleotide in this experiment did not contribute to cell death, so AraC-induced apoptosis values are lower than the values shown in Fig. 2C.
Further, DEVD-CHO, a caspase-3 inhibitor, also blocked AraC-induced apoptosis (Fig. 4B), and caspase-3 cleavage, indicative of its activation, induced by AraC as shown in Fig. 5A (lane 3) was markedly reduced when the cells were pretreated with 1,25-(OH)2D3 (lane 4). However, in cells in which hKSR-2 was depleted by AS-KSR-2, pretreatment with 1,25-(OH)2D3, which in the absence of hKSR-2 knockdown increases hKSR-2 protein level (Fig. 1B), did not reduce AraC-induced caspase-3 cleavage (Fig. 5, lane 5). These results were paralleled by the results of flow cytometric determination of fragmented DNA measured as the sub-G1-G0 fraction of PI-stained cells. As shown in Fig. 5B, a large quantity of such fragments were found in cells treated with AraC (group 3), but this was reduced by pretreatment of the cells with 1,25-(OH)2D3 (group 4). However, the knockdown of hKSR-2 by antisense oligonucleotides abrogated the protective effect of 1,25-(OH)2D3 (group 5). Thus, by several complementary approaches, we showed that hKSR-2 plays a role in survival of HL60 cells.
Figure 5.
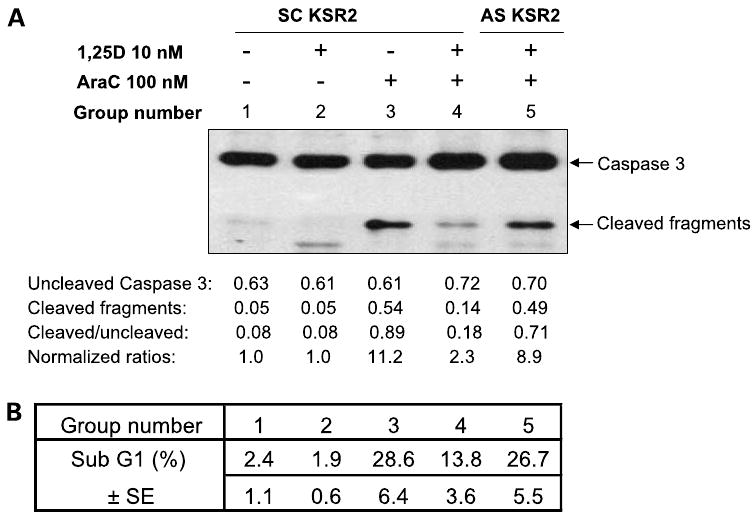
Abrogation of 1,25-(OH)2D3-induced protection from AraC toxicity by knockdown of hKSR-2. A, Western blot showing that 1,25-(OH)2D3 reduces AraC-induced caspase-3 activating cleavage (lane 4) and its abrogation by hKSR-2 knockdown (lane 5). B, changes in caspase-3 activation are paralleled by DNA degradation as shown by the sub-G1-G0 compartment of the cell cycle distribution measured by flow cytometry of PI-stained cells. The group numbers correspond to the lanes in A.
Bcl-2, but Not Mcl-1, Is Regulated by Both 1,25-(OH)2D3 and hKSR-2
Mcl-1, a member of the Bcl-2 family, has an important role in the survival of hematopoietic stem cells (26) and has been reported to be induced in ML-1 and HL60 human myeloblastic leukemia cells in response to signals for monocytic differentiation (8, 27). This was apparent when 1,25-(OH)2D3 was used to induce differentiation of HL60 cells (Fig. 6A, lanes 3 and 4), but depletion of hKSR-2 by AS-KSR-2 had no significant effect (P > 0.05) on Mcl-1 protein levels (Fig. 6A, lanes 7 and 8). In contrast, pretreatment with AS-hKSR-2 markedly reduced the Bcl-2 protein levels, which were increased by 1,25-(OH)2D3 (Fig. 6A, compare lanes 3 and 4 with lanes 7 and 8). Determination of the protein levels of proapoptotic Bax and Bad did not show significant changes in these experiments (Fig. 6A). Accordingly, there was a markedly reduced Bcl-2/Bax ratio, as shown in a quantitative summary of this and additional experiments presented in Fig. 6B, which correlated with the activating cleavage of caspase-3 (Fig. 5A), as well as with the increased DNA fragmentation (Fig. 5B), an increase in AraC-induced Annexin V positivity (Fig. 4B), and the reduced overall survival of these cells (Figs. 2 and 3). Similar results were observed when Bcl-2/Bad ratio was calculated (data not shown). Thus, it appears that 1,25-(OH)2D3 increases cell survival of these leukemia cells, at least in part, through the increased hKSR-2 expression (8), which regulates downstream death effectors that include the Bcl-2/Bax and Bcl-2/Bad apoptosis rheostats and the activity of caspase-3.
Figure 6.
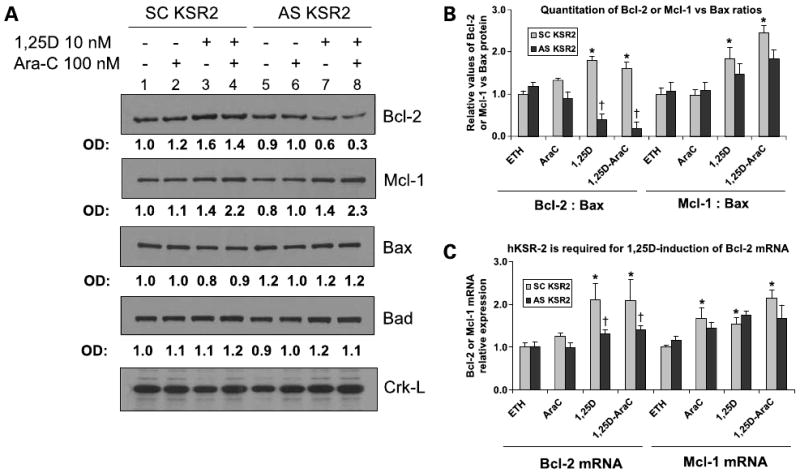
Bcl-2 expression is enhanced by 1,25-(OH)2D3 in a hKSR-2-dependent manner. A, illustration of the reduced expression of Bcl-2 protein, but not Mcl-1 protein, in cells which hKSR-2 expression was inhibited by antisense oligonucleotides. HL60 cells were incubated for 48 h with either scrambled or antisense oligonucleotides to hKSR-2 for 48 h and treated with the indicated agents. Protein levels of Bcl-2, Mcl-1, Bax, and Bad were determined by Western blot analysis. Absorbance of each protein band is shown below each blot. Crk-L served as a loading control. B, reduction of Bcl-2 protein by AS-hKSR-2 results in reduced Bcl-2 to Bax ratios. Quantitation of three experiments, one of which is illustrated in A, showing a significant (*, P < 0.05) effect of AS-hKSR-2 (versus scrambled KSR-2) protein signals for Bcl-2 or Mcl-1 versus Bax. †, P < 0.05, significant of differences from vehicle control (ETH). C, quantitative reverse transcription-PCR determination of Bcl-2 and Mcl-1 levels showing that Bcl-2 or Mcl-1 mRNA basal levels are not altered by an exposure to AS-KSR-2, whereas the 1,25-(OH)2D3-induced increase in Bcl-2, but not Mcl-1, mRNA is abrogated by AS-KSR-2. Note the reduction in the antiapoptotic Bcl-2 rheostat from basal as well as 1,25-(OH)2D3-enhanced protein levels (n = 3). Error bars in panels B and C represent SD of the mean.
Because the data presented here identify the upstream event controlling caspase activation as the reduction in the level of Bcl-2 protein (Fig. 6A), already known to be able to reduce ROS cytotoxicity (25), we investigated at which level of gene expression hKSR-2 regulates Bcl-2. It appears that this regulation is complex, in that hKSR-2 is required for the 1,25-(OH)2D3-induced up-regulation of Bcl-2 mRNA levels but not for the basal expression of Bcl-2 (Fig. 6C). In addition, there appears to be post-transcriptional regulation of Bcl-2 expression; as in hKSR-2 knockdown, the basal levels of Bcl-2 protein were also reduced (Fig. 6A, top). This suggests that hKSR-2 regulates Bcl-2 expression by two pathways; one controls 1,25-(OH)2D3-induced Bcl-2 transcription, whereas another controls translation or stability of the Bcl-2 protein. The nature of these pathways is currently being investigated.
Discussion
hKSR-2 was first identified in C. elegans as a positive regulator of Ras-dependent extracellular signal-regulated kinase phosphorylation and was found to function both nonredundantly, in germ-line meiosis, and redundantly with KSR-1 in the development of the genital and excretory systems of the worm (18). Human KSR-2 has been reported previously to negatively regulate Tpl2/Cot-mediated mitogen-activated protein kinase and nuclear factor-κB pathway signaling (19, 28), and we show here that it can inhibit apoptosis induced by AraC-generated ROS and perhaps also by DNA damage, although the DNA damage component remains to be clearly shown. We also show that an up-regulation of Bcl-2 and inhibition of caspase-3 are a part of this process. This function of hKSR-2 is nonredundant with the function of KSR-1 in 1,25-(OH)2D3-treated HL60 cells, although KSR-1 has been reported to enhance cell survival in intestinal cells of mice (29) or in neuronal cells (30) but to increase cell sensitivity to cisplatin-induced apoptosis (31). Also, the notion that hKSR-2 inhibits Tpl2/Cot is not likely to be applicable to this system, as the mitogen-activated protein kinase pathways, considered to be downstream from Tpl2/Cot1(19), are up-regulated by 1,25-(OH)2D3 in parallel with hKSR-2 in 1,25-(OH)2D3-treated HL60 cells (20). It seems, therefore, that functions of KSRs are highly cell type and cell context dependent, as would be expected of scaffolding proteins (32), although a scaffolding role has only been documented for KSR-1 (33, 34).
The initial mode of action of hKSR-2 in the system studied here remains to be established, but a scenario can be envisaged in which 1,25-(OH)2D3-enhanced hKSR-2 expression increases Bcl-2 expression. The functional domains of hKSR-2 correspond to the domains of KSR-1, with the exception of the NH2-terminal CA-1 domain, which is unique to KSR-1 (35, 36). Thus, hKSR-2 has four functional domains: CA-2 with Src homology, the cysteine-rich CA-3, and serine/threonine-rich region resembling the CR-2 domain of c-Raf-1, and the COOH-terminal CA-5 domain, which contains 11 conserved kinase subdomains that resemble the subdomains found in other kinases (19, 28). It is likely that the CA-3 domain regulates the subcellular localization of hKSR-2, whereas CA-5 is involved in augmenting mitogen-activated protein kinase signaling either by a scaffolding function or by phosphorylation of c-Raf-1. These functions of hKSR-2 have not been established yet, and this is likely to be a major undertaking, given the controversy regarding the source of kinase activity associated with KSR-1 (e.g., refs. 37, 38). In either case, because in 1,25-(OH)2D3-treated HL60 cells, up-regulation of hKSR-2 is accompanied by an early increase in c-Raf-1 and extracellular signal-regulated kinase 1/2 activity (13, 15), the CA-5 function of hKSR-2 may be responsible for this increase and transmit the signal down the mitogen-activated protein kinase pathway to transcription factors such as ATF-1 and CREB, reported to be required for the up-regulation of Bcl-2 levels (39).
It has been shown previously that down-regulation of Bcl-2 increases the cytotoxic action of AraC (40–42), probably because AraC generates ROS, the toxicity of which Bcl-2 has the capacity to diminish (25, 43, 44). In this context, an important mechanistic insight established by this study is that Bcl-2 is up-regulated in 1,25-(OH)2D3-treated HL60 cells and that this up-regulation requires the hKSR-2 protein. Interestingly, depletion of hKSR-2 followed by exposure to 1,25-(OH)2D3 reduced Bcl-2 levels below their basal abundance in proliferating cells, resulting in a dramatic decrease in Bcl-2/Bax and Bcl-2/Bad ratios (Fig. 6B; data not shown). This correlated with the activation of caspase-3 and increased apoptosis.
In addition to promoting apoptosis, reduction of the levels of hKSR-2 by electroporation of siRNA into HL60 cells resulted in the appearance of some necrotic cells. It is likely that electroporation, which affects the cell membrane, can predispose to necrosis in cells with low levels of hKSR-2. This form of cell death is difficult to study mechanistically, although there is increasing interest in intracellular oxidative and glycolytic events that lead to necrosis (45). Necrotic cells are also difficult to enumerate precisely, as on one hand apoptotic cells in suspension culture usually undergo secondary necrosis, whereas on the other hand necrotic cells disintegrate and disappear. Thus, we have focused on the study of apoptosis at this time, although it is acknowledged that hKSR-2 may also play a role in overall cell survival, preventing apoptosis and perhaps necrosis as well.
There is at this point of time only very scant literature on human KSR-2, so it is not surprising that this is the first report documenting that physiologic functions of hKSR-2 in myeloid leukemia cells HL60 include increased survival in the presence of a ROS-producing and DNA-damaging agent. Physiologically, such function could serve to protect a phagocytic cell from endogenous DNA damage coincident with the destruction and digestion of phagocytosed material, and this may have important carryover implications for the chemotherapy of acute myeloid leukemia, where AraC is a usual component of the treatment regimens (e.g., ref. 25). Thus, the potential of 1,25-(OH)2D3 for supplementing chemotherapy in the treatment of neoplastic diseases may require the presence of compounds or agents which down-regulate hKSR-2 expression, and these need to be identified. Our findings also have potential implications for the control of the vitamin D status of patients undergoing cytotoxic therapy that includes AraC.
Acknowledgments
We thank Dr. Milan Uskokovic (BioXell) for the gift of 1α,25-dihydroxyvitamin D3; Dr. Sharer for help with photography of slides; Drs. Jonathan Harrison, Michael Danilenko, and Dariusz Galkowski for comments on the article, and Vivienne Lowe for secretarial assistance in the preparation of this article.
Grant support: NIH/National Cancer Institute grants RO1-CA-44722-18 and RO1-CA-117942-01.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.de las Heras B, Hortelano S, Giron N, Bermejo P, Rodriguez B, Bosca L. Kaurane diterpenes protect against apoptosis and inhibition of phagocytosis in activated macrophages. Br J Pharmacol. 2007;152:249–55. doi: 10.1038/sj.bjp.0707382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang XJ, Wiernik PH, Klein RS, Gallagher RE. Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Med Oncol. 1999;16:58–64. doi: 10.1007/BF02787360. [DOI] [PubMed] [Google Scholar]
- 3.Brown G, Bunce CM, Rowlands DC, Williams GR. All-trans retinoic acid and 1α,25-dihydroxyvitamin D3 co-operate to promote differentiation of the human promyeloid leukemia cell line HL60 to monocytes. Leukemia. 1994;8:806–15. [PubMed] [Google Scholar]
- 4.Castaigne S, Chomienne C, Daniel MT, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704–9. [PubMed] [Google Scholar]
- 5.Koeffler HP. Induction of differentiation of human acute myelogenous leukemia cells: therapeutic implications. Blood. 1983;62:709–21. [PubMed] [Google Scholar]
- 6.Studzinski GP, Wang X, Ji Y, et al. The rationale for deltanoids in therapy for myeloid leukemia: role of KSR-MAPK-C/EBP pathway. J Steroid Biochem Mol Biol. 2005;97:47–55. doi: 10.1016/j.jsbmb.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe E, Miyaura C, Sakagami H, et al. Differentiation of mouse myeloid leukemia cells induced by 1α,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1981;78:4990–4. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Studzinski GP. Antiapoptotic action of 1,25-dihydroxyvitamin D3 is associated with increased mitochondrial MCL-1 and RAF-1 proteins and reduced release of cytochrome c. Exp Cell Res. 1997;235:210–7. doi: 10.1006/excr.1997.3667. [DOI] [PubMed] [Google Scholar]
- 9.Wang HG, Miyashita T, Takayama S, et al. Apoptosis regulation by interaction of Bcl-2 protein and Raf-1 kinase. Oncogene. 1994;9:2751–6. [PubMed] [Google Scholar]
- 10.Weissinger EM, Eissner G, Grammer C, et al. Inhibition of the Raf-1 kinase by cyclic AMP agonists causes apoptosis of v-abl-transformed cells. Mol Cell Biol. 1997;17:3229–41. doi: 10.1128/mcb.17.6.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 12.Stadheim TA, Xiao H, Eastman A. Inhibition of extracellular signal-regulated kinase (ERK) mediates cell cycle phase independent apoptosis in vinblastine-treated ML-1 cells. Cancer Res. 2001;61:1533–40. [PubMed] [Google Scholar]
- 13.Wang X, Studzinski GP. Activation of extracellular signal-regulated kinases (ERKs) defines the first phase of 1,25-dihydroxyvitamin D3-induced differentiation of HL60 cells. J Cell Biochem. 2001;80:471–82. doi: 10.1002/1097-4644(20010315)80:4<471::aid-jcb1001>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Marcinkowska E. Evidence that activation of MEK1,2/ERK1,2 signal transduction pathway is necessary for calcitriol-induced differentiation of HL-60 cells. Anticancer Res. 2001;21:499–504. [PubMed] [Google Scholar]
- 15.Wang X, Studzinski GP. Raf-1 signaling is required for the later stages of 1,25-dihydroxyvitamin D3-induced differentiation of HL60 cells but is not mediated by the MEK/ERK module. J Cell Physiol. 2006;209:253–60. doi: 10.1002/jcp.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Studzinski GP. Kinase suppressor of RAS (KSR) amplifies the differentiation signal provided by low concentrations 1,25-dihydroxyvitamin D3. J Cell Physiol. 2004;198:333–42. doi: 10.1002/jcp.10443. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Wang TT, White JH, Studzinski GP. Induction of kinase suppressor of RAS-1(KSR-1) gene by 1,α25-dihydroxyvitamin D3 in human leukemia HL60 cells through a vitamin D response element in the 5′-flanking region. Oncogene. 2006;25:7078–85. doi: 10.1038/sj.onc.1209697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohmachi M, Rocheleau CE, Church D, Lambie E, Schedl T, Sundaram MV. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr Biol. 2002;12:427–33. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- 19.Channavajhala PL, Wu L, Cuozzo JW, et al. Identification of a novel human kinase supporter of Ras (hKSR-2) that functions as a negative regulator of Cot (Tpl2) signaling. J Biol Chem. 2003;278:47089–97. doi: 10.1074/jbc.M306002200. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Wang TT, White JH, Studzinski GP. Expression of human kinase suppressor of Ras 2 (hKSR-2) gene in HL60 leukemia cells is directly upregulated by 1,25-dihydroxyvitamin D(3) and is required for optimal cell differentiation. Exp Cell Res. 2007;313:3034–45. doi: 10.1016/j.yexcr.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baran CP, Zeigler MM, Tridandapani S, Marsh CB. The role of ROS and RNS in regulating Life and death of blood monocytes. Curr Pharm Des. 2004;10:855–66. doi: 10.2174/1381612043452866. [DOI] [PubMed] [Google Scholar]
- 22.Gallagher R, Collins S, Trujillo J, et al. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979;54:713–33. [PubMed] [Google Scholar]
- 23.Ramachandra S, Studzinski GP. Cell growth and apoptosis: a practical approach. Oxford (NY): IRL Press at the Oxford University Press; 1995. Morphological and biochemical criteria of apoptosis (Chapter 7) [Google Scholar]
- 24.McGahon AJ, Martin SJ, Bissonnette RP, et al. The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol. 1995;46:153–85. doi: 10.1016/s0091-679x(08)61929-9. [DOI] [PubMed] [Google Scholar]
- 25.Hu ZB, Yang GS, Li M, Miyamoto N, Minden MD, McCulloch EA. Mechanism of cytosine arabinoside toxicity to the blast cells of acute myeloblastic leukemia: involvement of free radicals. Leukemia. 1995;9:789–98. [PubMed] [Google Scholar]
- 26.Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–4. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 27.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996;166:523–36. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 28.Channavajhala PL, Rao VR, Spaulding V, Lin LL, Zhang YG. hKSR-2 inhibits MEKK3-activated MAP kinase and NF-κB pathways in inflammation. Biochem Biophys Res Commun. 2005;334:1214–8. doi: 10.1016/j.bbrc.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Yan F, John SK, Wilson G, Jones DS, Washington MK, Polk DB. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004;114:1272–80. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szatmari E, Kalita KB, Kharebava G, Hetman M. Role of kinase suppressor of Ras-1 in neuronal survival signaling by extracellular signal-regulated kinase 1/2. J Neurosci. 2007;27:11389–400. doi: 10.1523/JNEUROSCI.3473-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim M, Yan Y, Kortum RL, et al. Expression of kinase suppressor of Ras1 enhances cisplatin-induced extracellular signal-regulated kinase activation and cisplatin sensitivity. Cancer Res. 2005;65:3986–92. doi: 10.1158/0008-5472.CAN-03-2334. [DOI] [PubMed] [Google Scholar]
- 32.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 33.Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002;16:427–38. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ory S, Morrison DK. Signal transduction: implications for Ras-dependent ERK signaling. Curr Biol. 2004;14:R277–8. doi: 10.1016/j.cub.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–88. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 36.Roy F, Therrien M. MAP kinase module: the Ksr connection. Curr Biol. 2002;12:R325–7. doi: 10.1016/s0960-9822(02)00831-x. [DOI] [PubMed] [Google Scholar]
- 37.Michaud NR, Therrien M, Cacace A, et al. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci U S A. 1997;94:12792–6. doi: 10.1073/pnas.94.24.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing HR, Campodonico L, Kolesnick R. The kinase activity of kinase suppressor of Ras1 (KSR1) is independent of bound MEK. J Biol Chem. 2004;279:26210–4. doi: 10.1074/jbc.M401323200. [DOI] [PubMed] [Google Scholar]
- 39.Belkhiri A, Dar AA, Zaika A, Kelley M, El-Rifai W. t-Darpp promotes cancer cell survival by up-regulation of Bcl2 through Akt-dependent mechanism. Cancer Res. 2008;68:395–403. doi: 10.1158/0008-5472.CAN-07-1580. [DOI] [PubMed] [Google Scholar]
- 40.Decaudin D, Geley S, Hirsch T, et al. Bcl-2 and Bcl-XL antagonize the mitochondrial dysfunction preceding nuclear apoptosis induced by chemotherapeutic agents. Cancer Res. 1997;57:62–7. [PubMed] [Google Scholar]
- 41.Avramis VI, Nandy P, Kwock R, et al. Increased p21/WAF-1 and p53 protein levels following sequential three drug combination regimen of fludarabine, cytarabine and docetaxel induces apoptosis in human leukemia cells. Anticancer Res. 1998;18:2327–38. [PubMed] [Google Scholar]
- 42.Dasmahapatra G, Almenara JA, Grant S. Flavopiridol and histone deacetylase inhibitors promote mitochondrial injury and cell death in human leukemia cells that overexpress Bcl-2. Mol Pharmacol. 2006;69:288–98. doi: 10.1124/mol.105.016154. [DOI] [PubMed] [Google Scholar]
- 43.Hedley DW, McCulloch EA. Generation of reactive oxygen intermediates after treatment of blasts of acute myeloblastic leukemia with cytosine arabinoside: role of bcl-2. Leukemia. 1996;10:1143–9. [PubMed] [Google Scholar]
- 44.Kanno S, Higurashi A, Watanabe Y, Shouji A, Asou K, Ishikawa M. Susceptibility to cytosine arabinoside (Ara-C)-induced cytotoxicity in human leukemia cell lines. Toxicol Lett. 2004;152:149–58. doi: 10.1016/j.toxlet.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Deberardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]