

Abstract
The evidence for the promising potential for derivatives of Vitamin D (deltanoids) in the treatment of myeloid leukemias is increasing, but currently is not matched by the understanding of the precise mechanisms by which these anti-neoplastic effects are achieved. Unlike solid tumors in which growth retardation by deltanoids appears to result from inhibition of cell proliferation and the promotion of cell death by apoptosis, control of myeloid leukemia proliferation by deltanoids results from the induction of differentiation of the immature myelo-monocytic cells towards functional monocytic cells. We present here the accumulating evidence that a pathway that is initiated by deltanoid activation of Vitamin D receptor (VDR) and leads to monocytic differentiation of human myeloblastic HL60 cells, includes the MEK-ERK and JNK mitogen-activated protein kinases (MAPKs), their positive and negative regulators and a downstream effector C/EBPβ. As in other cells, the abundance of VDR protein increases shortly after an exposure of HL60 cells to 1α,25-dihydroxyvitamin D3 (1α,25(OH)2 D3). Other early events include a parallel upregulation of kinase suppressor of Ras (KSR-1) and the activation of the ERK MAPK pathway and data suggest that KSR-1 acts to amplify the signal provided by low concentrations of 1α,25(OH)2 D3. Maintenance of monocytic differentiation may be enhanced by JNK, but diminished by p38, MAPK signaling. Downstream, one of the targets of these pathways is C/EBPβ, which can directly interact with the promoter for CD14, a gene characteristically expressed in monocytes. Importantly, in freshly obtained acute myeloid leukemia (AML)-M2 cells exposed to PRI-2191, a novel deltanoid with a modified side chain, upregulation of C/EBPβ paralleled the induction of monocytic differentiation. These data provide a basis for the hypothesis that deltanoid-induced upregulation of C/EBPβ bypasses the block to granulocytic differentiation in myeloid leukemia cells by redirecting the cells to monocytic differentiation.
Keywords: Vitamin D, Myeloid leukemia, Differentiation, KSR-1, MAPKs, C/EBP
1. Introduction
Conversion of malignant stem cell-like cancer cells to mature non-proliferating cells may be an ideal therapeutic strategy. Remarkably, 1α,25(OH)2 D3 and analogs, i.e. the deltanoids, have the ability to induce such conversion reviewed in Ref. [1], but the biological rationale and the sequence of steps required to achieve the non-malignant phenotype have not been clear. In this report, we present for the first time the hypothesis that, in at least one system, there is a simple explanation for the ability of deltanoids to reverse the malignant transformation. This concept is that in myeloid hematopoietic cells blocked from granulocytic differentiation by mutations affecting genes required for this lineage of differentiation, deltanoids redirect the blocked, immature leukemic cells from the original, developmentally determined differentiation lineage to an alternative, still intact, pathway to monocytic terminal differentiation. We also suggest a potential mechanism that may contribute to the maturation of leukemic cells towards this alternative form of differentiation.
2. Materials and methods
2.1. Deltanoids
The PRI Vitamin D analogs were synthesized in the Vitamin D Laboratory of the Pharmaceutical Research Institute, Warsaw, Poland by the previously described methods [2–4]. 1α,25(OH)2 D3 was a kind gift from Dr. Milan Uskokovic (Bioxell, Nutley, NJ).
2.2. Chemicals and antibodies
MEK1 inhibitor PD98059 was purchased from Cell Signaling Technology (Beverly, MA) and the p38 kinase inhibitor SB202190 from Calbiochem-Novabiochem Corp. (San Diego, CA). JNK inhibitor SP600125 was a generous gift from Signal Research Division, Celgene Corporation and ZK1590222 was a gift from Dr. A Steinmyer, Schering AG (Berlin, Germany). The antibodies to KSR-1 (N-19, goat polyclonal), Raf-1 (C-12, rabbit polyclonal), MEK1 (C-18, rabbit polyclonal), p90RSK (C-21, rabbit polyclonal), VDR (C-20), C/EBPα (14AA, rabbit polyclonal), C/EBPβ (C19, rabbit polyclonal), pRb (IF-8, mouse polyclonal) and Crk-L (C-20, rabbit polyclonal) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The antibodies used to detect phospho-Raf (Ser 259), phospho-MEK (Ser 217/221) and phospho-p90RSK (Ser 381), all rabbit polyclonal, were purchased from Cell Signaling Technology.
2.3. Cell culture
HL60-G cell, a subclone of human promyelocytic leukemia cells HL60 [5] and mononuclear cells isolated from patient's peripheral blood were cultured in RPMI1640 medium (Mediatech, Washington, DC) supplemented with 10% bovine calf serum (Hyclone, Logan, UT) at 37 °C in 5% CO2. Cells in all experimental groups were seeded in fresh tissue culture medium at 3 × 105 cells/ml for HL60 cells and 1 × 106 cells/ml for primary cells in 25 cm2 flasks and incubated with different concentrations of 1α,25(OH)2 D3, analogs or combinations of these deltanoids with other compounds for the indicated times. Each experiment with HL60 cells was repeated at least three times. The specimen of peripheral blood from a patient with acute myeloid leukemia (AML) FAB classification M2 was obtained following the patient's informed consent according to the Institutional IRB protocol.
2.4. Isolation of the mononuclear cells from patient's peripheral blood
Peripheral blood obtained from the patient was prediluted with phosphate buffered saline (PBS) by 1:1 ratio. Volume of Ficoll (Histopaque-1077, Sigma, St. Louis, MO) equal to that of the diluted blood was added to a sterilized conical centrifuge tube, then the blood was carefully layered onto the top of Ficoll. The blood on Ficoll was then centrifuged at 400 × g for 30 min at room temperature. After centrifugation, the opaque interface (mononuclear cells) was carefully transferred into a clean conical centrifuge tube and washed for three times with PBS. The cells were then placed into cell culture as described above.
2.5. Assessment of differentiation
To analyze expression of cell surface markers, aliquots of 1 × 106 cells were harvested, washed and stained with MY4-RD-1 (anti-CD14) and M01-FITC (anti-CD11b) antibodies (Coulter, Miami, FL). Two-parameter analysis was performed using a Coulter flow cytometer instrument and Cell Quest Software (Becton Dickinson, Franklin Lakes, NJ). Isotypic mouse IgG1 was used to set threshold parameters. Slides were made to monitor the monocytic phenotype by cytochemical determination of the monocyte specific esterase (MSE) activity, also known to as non-specific esterase (NSE), as described previously [6].
2.6. Immunoblotting
Whole cell extracts were prepared for immunoblotting. The cells were harvested and washed twice with ice-cold 1× PBS. The washed cell pellets were solubilized with a lysis buffer containing 20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, followed by centrifugation at 12,000 × g for 5 min. The protein concentrations of the extracts were determined by using Bio-Rad protein assay kit. Equal amounts of 3 × SDS sample buffer containing 150 mM Tris–HCl, pH 6.8, 30% glycerol, 3% SDS, 1.5 mg/ml bromophenol blue dye and 100 mM dithiothreitol were then added to each sample. Equal amounts of whole cell extracts (40 μg of protein) were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech Piscataway, NJ). The membranes were blocked with 5% milk in TBS/0.1% Tween-20 for 1 h, subsequently blotted with primary antibodies and then the membranes were blotted with a horseradish-linked secondary antibody for 1 h The protein bands were visualized with a chemiluminescence assay system (Amersham). The protein loading of the gel and efficiency of the transfer were controlled by stripping the membrane and reprobing for Crk-L, a constitutively expressed protein in HL60 cells. The optical density of each band was quantitated using an image quantitator (Molecular Dynamics).
2.7. RT-PCR
Total RNA was extracted using Tri-zol reagent (Invitrogene, Carlsbad, CA). GeneAmp RNA PCR Core Kits (Perkin-Elmer, Boston, MA) were used. The primers were as follows: (1) RB upstream primer (5′-TACCTAGCTCAA-GGGTTAAT-3′) and RB downstream primer (5′-TAGCCA-TATGCACATGAATG-3′), (2) C/EPBβ upstream primer (5′-GTTCTTGACGTTCTTCGGCCG-3′) and C/EPBβ downstream primer (5′-TGGACAAGCACAGCGACGAGT-3′), (3) β-actin upstream primer (5′-TGACGGGGTCACCC-ACACTGTGCCCAGCTA-3′) and β-actin downstream primer (5′-CTAGAAGCATTTGCCGGTGGACGATGGA-GGG-3′). RT-PCR was performed according to the manufacturer's recommended procedure. For reverse transcription, samples were incubated in a Perkin-Elmer GeneAmp PCR system 9600 at 42 °C for 15 min, then 99 °C for 5 min and 5 °C for 5 min. For PCR, samples were incubated in GeneAmpPCR System9600 as follows: 95 °C for 105 s, 35 cycles of 95 °C for 15 s and 60 °C for 30 s and 72 °C for 7 min. The RT-PCR products were separated in 1.2% agarose gels and stained with ethidium bromide. The intensities of the bands were measured using Image QuaNT program (Molecular Dynamics).
2.8. Antisense oligonucleotides
Phosphorothioate antisense (5′-GCTTCTTCTTGGGCA-TCT-3′, corresponding to the start codon of MEK1) and sense (5′-AGATGCCCAAGAAGAAGC-3′) oligonucleotides were synthesized by the Molecular Resource Facility of the New Jersey Medical School. Cells were incubated with the oligonucleotides (5 μM) described above for 24 h, then supplemented with different concentrations of 1,25D3 for the indicated time.
2.9. Statistical methods
All experiments on HL60 cells were repeated a minimum of three times. Significance of differences between mean values was assessed by ANOVA analysis followed by the Bonferroni post-test. All computations were performed using an IBM personal computer using Microsoft EXCEL + ANALYSE, IT Program.
3. Results and discussion
3.1. Initial signals for induction of monocytic differentiation
The well established initial event through which deltanoids exert their biological effects is an interaction with Vitamin D receptor (VDR), which acts as a transcription factor and upregulates the classical Vitamin D-dependent genes [7]. As in many other cells which already express VDR [8], its abundance rapidly increases in HL60 cells after exposure to 1α,25(OH)2 D3, even at 1 nM concentration (Fig. 1). In addition to the 50 kDa VDR protein, a more slowly migrating band was also seen in this immunoblot and may correspond to the VDR isoform B1, described by Gardiner et al. [9]. However, the upregulation of the putative VDR B1 by 1α,25(OH)2 D3 was less marked (Fig. 1).
Fig. 1.
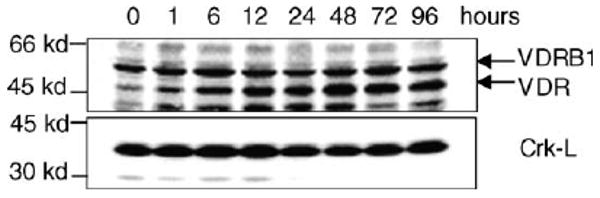
Exposure of HL60 cells to 1 nM 1α,25(OH)2 D3 for the stated times results in a rapid increase in the 50 kDa VDR protein levels shown by immunoblotting. The 56 kDa Vitamin D receptor B1 (VDR B1) is a putative isoform of VDR. The 36 kDa Crk-L protein which is constitutively expressed in HL60 cells and not regulated by 1α,25(OH)2 D3 is shown as a loading control.
While the liganding and the upregulation of VDR followed by transcriptional activation of genes that have VDR-responsive elements (VDREs) in their promoters may be sufficient to explain some biological consequences of exposure of mammalian cells to deltanoids, not all phenomena associated with differentiation can be explained by this mechanism. The observed events include rapid membrane effects of deltanoids [10,11] and more sustained effects on MAP kinase signaling induced in leukemia cells [12–19]. Thus, the presence of auxiliary pathways for differentiation, that include mitogen-activated protein kinase (MAPK) cascades, seems a likely postulate.
The link between the differentiation-signaling deltanoid and MAPK activation remains to be firmly established, but our data suggest that kinase suppressor of Ras-1 (KSR-1) plays a part in such linkage. Fig. 2 shows that the increase in KSR-1 protein abundance (panel A) parallels both the upregulation of VDR illustrated in Fig. 1 and the appearance of the monocytic phenotype demonstrated by three markers, CD14, CD11b and monocyte-specific esterase (MSE) shown in panel B. Raf-1 phosphorylation is also increased during 1α,25(OH)2 D3-induced monocytic differentiation in parallel with the increase inKSR-1 abundance (Fig. 2, panel A) and this phosphorylation is at least in part VDR-dependent, as it is inhibited by compound ZK159222, an antagonist of VDR-mediated actions of 1α,25(OH)2 D3 (Fig. 3). It was previously reported that KSR-1 is principally membrane-associated and can be activated by ceramide, a compound that is one of the products of membrane phospholipids that are cleaved by deltanoid-activated sphingomyelinases in leukemic cells [20]. Thus, KSR-1 is a good candidate for the intersection of VDR (the upregulation of KSR-1 expression) and membrane-associated (KSR-1 activation by ceramide) signaling pathways.
Fig. 2.

Parallel increases in KSR-1 protein abundance, Raf-1 (74 kDa) activation by phosphorylation and monocytic differentiation induced by 1 nM 1α,25(OH)2 D3 in HL60 cells. (A) Demonstration of increased protein expression of 97 kDa KSR-1 and 74 kDa phospho-Raf-1 Ser 259 after 1α,25(OH)2 D3 treatment. The protein Crk-L is shown as the loading control for the immunoblots. (B) Summary of changes in differentiation markers determined by flow cytometry (CD11b and CD14) and cytochemistry (MSE), as described in Section 2. The bars represent mean values of four experiments and show S.E.; MSE, monocyte-specific esterase.
Fig. 3.

Exposure of HL60 cells to 10 nM 1α,25(OH)2 D3 (1,25D) for 24 or 48 h increases the abundance and phosphorylation of the 74 kDa Raf-1 protein and is VDR-dependent, as shown by its inhibition by the 1α,25(OH)2 D3 antagonist ZK159222. Crk-L is a loading control.
Evidence has been presented that KSR-1 can function as a scaffold that facilitates Ras-activated Raf–MEK–ERK pathway by bringing some of these proteins together [21,22]. Others, and our laboratory, have found that KSR-1 can also phosphorylate Raf-1 in intact cells [15,18,23,24]. Irrespective of whether KSR-1 functions exclusively as a scaffold, or a scaffold and a Raf-1 kinase, KSR-1 can upregulate signaling downstream from Raf-1 [18,24].
Additional evidence that KSR-1 has a role in 1α,25(OH)2 D3 -induced differentiation of HL60 cells has been recently published [18]. It includes inhibition of monocytic differentiation induced by relatively low concentrations of 1α,25(OH)2 D3 by oligonucleotides antisense to the KSR-1 coding sequence, while ectopic expression of wild-type, but not kinase-inactive, KSR-1 potentiates 1α,25(OH)2 D3-induced monocytic differentiation. Importantly, the changes in differentiation modulated by KSR-1 are associated with corresponding changes in phosphorylation of Raf-1 and of at least one of the downstream effectors of MAPK pathway, p90RSK [18]. Collectively, experimental data strongly suggest that KSR-1 participates in signaling of monocytic differentiation induced by low concentrations of 1,25D, and presumably by other deltanoids, through a Raf-1 regulated MAPK pathway, in a VDR-dependent manner.
3.2. MAPKs as auxiliary pathways in deltanoid-induced monocytic differentiation
To confirm that analogs of 1α,25(OH)2 D3 modulate the activities of MAPK pathways, we used three compounds synthesized in the Pharmacological Research Institute (PRI) in Warsaw, Poland. The calcemic activities of these compounds have been extensively evaluated in mice by Verstuyf and co-workers and the deltanoids were shown to be approximately 10 times less calcemic than 1α,25(OH)2 D3 [25,26]. As recently demonstrated, the relative differentiation potencies of 1α,25(OH)2 D3 and the three analogs parallel the activation of all three branches of the principal MAPK pathways, i.e. the ERK, JNK and p38MAPK cascades [25]. Interestingly, however, while the inhibition of ERK and JNK pathways also inhibits differentiation, inhibition of the p38MAPK pathway actually potentiates differentiation, suggesting that p38MAPK signaling provides a negative feedback in deltanoid-induced monocytic differentiation (top panels of Figs. 4, 6 and 7).
Fig. 4.
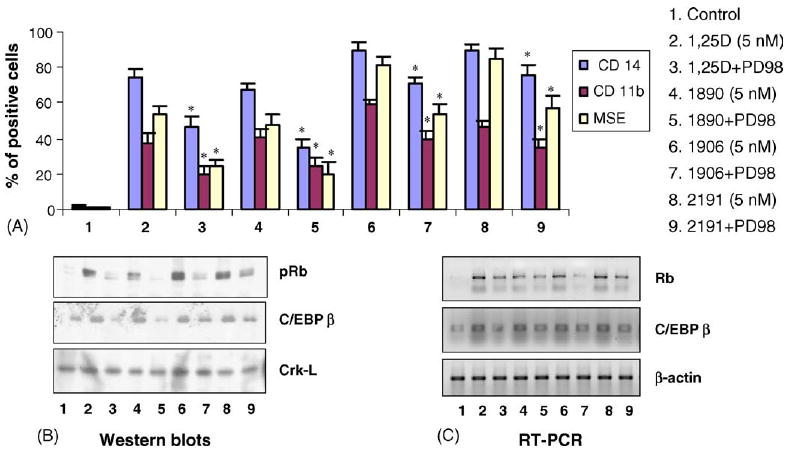
Deltanoids with varying differentiation—inducing potencies induce parallel increases in the expression of pRb and C/EBPβ (41 and 38kDa), which are inhibited by the inhibitor of MEK-ERK MAPK pathway, PD98059 (PD). (A) HL60 cells were treated for 48 h with 5 nM 1,25D or analog, with and without 10 μM of PD98059, then differentiation markers were determined as described in Fig. 2. (B) The protein expression of Rb (110 kDa) and C/EBPβ were determined by immunoblotting. (C) The corresponding mRNA levels were determined by RT-PCR. Each experiment was repeated for at least three times. The MEK inhibitor reduced Rb and C/EBPβ protein and mRNA expression; * p < 0.05, compared to treatment with the same concentration of 1,25D3 or analog.
Fig. 6.

Deltanoids with varying differentiation—inducing potencies induce parallel increases in the expression of pRb and C/EBPβ (41 and 38 kDa), which are inhibited by the JNK inhibitor SP600125 (10 μM). (A) HL60 cells were treated for 48 h with 5 nM 1,25D3 or analog, with and without 10 μM SP600125 (SP) and the differentiation markers were determined as described in Fig. 2. The JNK inhibitor reduces differentiation. (B) The protein expression of Rb (110 kDa) and C/EBPβ were determined by immunoblotting. (C) The corresponding mRNA levels were determined by RT-PCR. The JNK inhibitor reduced Rb and C/EBPβ protein and mRNA expression; n = 3, * p < 0.05, compared to treatment with the same concentration of 1,25D3 or analog.
Fig. 7.

Deltanoids with varying differentiation—inducing potencies induce parallel increases in the expression of pRb and C/EBPβ (41 and 38kDa), that are potentiated by the inhibitor of p38MAPK pathway, SB202190. (A) HL60 cells were treated for 48h with 5 nM 1,25D3 or analog, with and without 10 μM SB202190 (SB) and the differentiation markers were determined as described in Fig. 2. The p38 inhibitor increased differentiation. (B) The protein expression of Rb (110 kDa) and C/EBPβ were determined by immunoblotting. (C) The corresponding mRNA levels were determined by RT-PCR. The p38 inhibitor also increased Rb and C/EBPβ protein and mRNA expression; n = 3, * p < 0.05, compared to treatment with the same concentration of 1,25D3 or analog.
3.3. C/EBP β transcription factor and retinoblastoma protein (pRb) as effectors of deltanoid-induced monocytic differentiation
To provide evidence that would help to validate our hypothesis that deltanoids can shift the balance of transcription factors from those that favor granulocytic differentiation to those that favor the monocytic lineage, we focused on deltanoid-induced changes in the expression of C/EBPβ and pRb [25]. In addition to the well-known importance of pRb in cell cycle control [27], the rationale for this approach includes the accumulating data that C/EBPα has an important role in granulocytic differentiation [28,29]. Given that several subtypes of acute myeloid leukemia have mutations that abrogate the expression of C/EBPα [30], that functional cooperation between C/EBPβ and VDR has recently been reported [31] and C/EBPβ is required for 1,25D-induced monocytic differentiation [32], possibly due to the presence of C/EBP binding site in the CD14 promoter [33], we asked if C/EBPβ expression could be linked to deltanoid-induced changes in the pathways described above.
The dependence of C/EBPβ and pRb expression on the MEK–ERK pathway was first examined using the pharmacological inhibitor of MEK activity, PD98059 [34]. Fig. 4 shows that, as mentioned above, the MEK inhibitor reduces monocytic differentiation induced by each deltanoid and that the expression of pRb and C/EBPβ is reduced in parallel. This is evident at both the protein and the mRNA level, suggesting transcriptional regulation (Fig. 4). These results were also obtained by alternative approaches (Fig. 5), which included the use of antisense oligonucleotide to MEK and the anthrax toxin that destroys the MEK protein [35]. In these experiments, the efficacy of inhibition of MEK activity was demonstrated by the reduced phosphorylation of a downstream target, p90RSK (Fig. 5).
Fig. 5.

C/EBPβ is under control of the MEK1 pathway in HL60 cells. Inhibition of MEK activity by several strategies inhibited phosphorylation of its downstream target p90RSK and of the 1α,25(OH)2 D3-induced increase in the expression of C/EBPβ. In these experiments, MEK1 protein was knocked out by antisense oligo, pharmacological inhibitor PD98059 (20 μM) or anthrax toxin (PA = 100 ng/mL, LF = 10 ng/mL), then treated with 1,25D for 24 h. Protein levels of MEK1, phospho-p90RSK and C/EBPβ were determined by Western blotting. Knock out of the MEK1 protein inhibited C/EBPβ protein expression. Crk-L protein was used as internal control; AS, antisense; MM, mismatch; PA, anthrax protective antigen; LF, anthrax lethal factor.
Inhibition of JNK pathway by SP600125 and of p38MAPK pathway by SB202190 showed that that expression of C/EPBβ and pRb are modulated by deltanoids in parallel with the modulation of deltanoid-induced monocytic differentiation (Figs. 6 and 7). Taking together all data presented and discussed above, we suggest a circuit that may be apart of still not completely elucidated network of pathways required for deltanoid-induced differentiation (Fig. 8).
Fig. 8.

Schematic representation of VDR-dependent and auxiliary pathways that lead to deltanoid-induced monocytic differentiation of myeloid leukemia cells. Deltanoids entering the cell activate p50VDR, as indicated by an asterisk. This results in modulation of the activity of genes containing VDREs by the VDR/RXR heterodimers and by the activation of differentiation–auxiliary pathways that include the three principle MAPK signaling cascades. KSR-1 may amplify the mitogen-activated Raf–MEK–ERK pathway, the MAPK cascade upregulated in the initial phase of deltanoid-induced monocytic differentiation [13,16] and may result in phosphorylation of C/EBPβ. The source of the activation of JNK and p38 pathways is currently unclear, but it appears that the p38 pathway has a negative effect on differentiation. The suggested involvement of the transcription factor AP-1 [17] and of the cell cycle controlling protein pRb [25] have not been discussed in this paper. The C/EBPβ appears to be an effector of monocytic differentiation, perhaps through a C/EBP element in the promoter of the gene encoding CD14, a monocyte-specific protein [33]. Another monocyte-specific protein, MSE, is also upregulated by deltanoids, but the mechanism is still unknown.
3.4. The role of C/EBP β in deltanoid-induced bypass of block to granulocytic differentiation in leukemia cells ex vivo
The data presented above were obtained on the established myeloblastic leukemia cell line HL60. To determine if these findings can be reproduced in clinical cases of leukemia, we performed a pilot experiment using mononuclear leukemia cells isolated from the peripheral blood of a patient with AML, subtype M2. While 1α,25(OH)2 D3 had no significant differentiating effect on these cells, the deltanoid used here, PRI-2191, showed a modest differentiating effect, accompanied by modest upregulation of C/EBPβ (Fig. 9). It should also be noted, however, that the combination of the deltanoid with an antioxidant agent, carnosic acid and the p38 inhibitor SB202190, both of which were previously shown to potentiate differentiation activity of deltanoids on leukemia cells [14,36,37], markedly increased differentiation markers as well as C/EBPβ expression in these primary myeloid leukemic cells (Fig. 9).
Fig. 9.

Upregulation of C/EBPβ by deltanoid/antioxidant combination parallels the expression of monocyte-specific markers MSE and CD14. (A) Mononuclear leukemic cells isolated from peripheral blood obtained from a patient with AML M2 were treated for 72 h with the compounds at the concentrations indicated. C/EBPβ was determined by immunoblotting. Crk-L protein levels are displayed as a loading control; CA, carnosic acid; SB, SB 202190. (B) Optical densities (OD) of the protein levels were determined by Fluorimage with image Quant Software (Molecular Dynamics, Sunnyvale, CA) as described in Section 2. The OD ratios of C/EBPβ to Crk-L were plotted for the different groups, as indicated. Arithmetical means ± S.D. are shown, n = 3. The proportion of mononuclear cells positive for monocytic markers CD11b (C), MSE (D) and CD14 (E) were determined as described in Section 2.
In conclusion, our findings support the hypothesis that 1α,25(OH)2 D3 and low calcemic analogs induce differentiation of leukemic cells with intact potential for monocytic differentiation by changing the intracellular balance of transcription factor activity and this allows the cells to bypass the granulocytic differentiation block by diverting the cells to the monocytic lineage (Fig. 10). Auxiliary signaling pathways activated by the deltanoid may contribute to this shift in transcription factor activity.
Fig. 10.

The deltanoid-induced bypass of the block to differentiation in leukemia hypothesis. The upper panel postulates that in normal hematopoiesis, C/EBPα is critical to granulopoiesis, while C/EBPβ preponderance favors monocytic differentiation. The lower panel illustrates that when granulopoiesis is blocked by a mutation or a functional inactivation of C/EBPα, immature myeloid cell accumulate in the bone marrow and appear in the peripheral blood. However, when deltanoids are present, C/EBPβ levels increase and monocytic differentiation can take place, thus bypassing the block.
Acknowledgments
We thank Dr. Milan Uskokovic, BioXell, Nutley, NJ, for the gift of 1α,25-dihydroxyvitamin D3 and Dr. Steinmeyer, Schering AG, Berlin, Germany, for the 1α,25-dihydroxyvitamin D3 antagonist ZK 159222. We also thank Dr. Michael Danilenko, Ben-Gurion University of the Negev, Israel, Dr. Ewa Marcinkowska, University of Wroclaw, Poland and Edward Garay, UMDNJ, for comments on the manuscript. This study was supported by NIH Grant RO1-CA 44722 from the National Cancer Institute.
References
- 1.Danilenko M, Studzinski GP. Enhancement by other compounds of the anti-cancer activity of Vitamin D3 and its analogs. Exp Cell Res. 2004;298(2):339–358. doi: 10.1016/j.yexcr.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 2.Chodynski M, Wietrzyk J, Marcinkowska E, Opolski A, Szelejewski W, Kutner A. Synthesis and antiproliferative activity of side-chain unsaturated and homologated analogs of 1,25-dihydroxyvitamin D2. Steroids. 2002;67(9):789–798. doi: 10.1016/s0039-128x(02)00038-7. [DOI] [PubMed] [Google Scholar]
- 3.Tetich M, Kutner A, Leskiewicz J, Budziszewska B, Lason W. Neuroprotective effects of (24R)-1,24-dihydroxycholecalciferol in human neuroblastoma SH-SY5Y cell line. J Steroid Biochem Mol Biol. 2004;89–90(11–5):365–370. doi: 10.1016/j.jsbmb.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 4.Wietrzyk J, Pelczynska M, Madej J, Dzimira S, Kusnierczyk H, Kutner A, Szelejewski W, Opolski A. Toxicity and antineoplastic effect of (24R)-1,24-dihydroxyvitamin D3 (PRI-2191) Steroids. 2004;69(10):629–635. doi: 10.1016/j.steroids.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. Characterization of the continuous, differentiating myeloid cell line (HL60) from a patient with acute promyelocytic leukemia. Blood. 1979;54(3):713–733. [PubMed] [Google Scholar]
- 6.Wang X, Gardner JP, Kheir A, Uskokovic MR, Studzinski GP. Synergistic induction of HL60 cell differentiation by ketoconazole and 1-desoxy analogs of vitamin D3. J Natl Cancer Inst. 1997;20(16):1199–1206. doi: 10.1093/jnci/89.16.1199. [DOI] [PubMed] [Google Scholar]
- 7.Christakos S, Dhawan P, Liu Y, Peng X, Porta A. New insights into the mechanisms of Vitamin D action. J Cell Biochem. 2003;88(4):695–705. doi: 10.1002/jcb.10423. [DOI] [PubMed] [Google Scholar]
- 8.Krishnan AV, Feldman D. Regulation of Vitamin D receptor abundance. In: Feldman D, editor. Vitamin D. Academic Press; San Diego, CA: 1997. pp. 179–200. [Google Scholar]
- 9.Gardiner EM, Esteban LM, Fong C, Allison SJ, Flanagan JL, Kouzmenko AP, Eisman JA. Vitamin D receptor B1 and exon 1d: functional and evolutionary analysis. J Steroid Biochem Mol Biol. 2004;89–90(1–5):233–238. doi: 10.1016/j.jsbmb.2004.03.078. [DOI] [PubMed] [Google Scholar]
- 10.Norman AW, Nemere I, Zhou LX, Bishop JE, Lowe KE, Maiyar AC, Collins ED, Taoka T, Sergeev I, Farach-Carson MC. 1,25(OH)2-Vitamin D3, a steroid hormone that produces biologic effects via both genomic and nongenomic pathways. J Steroid Biochem Mol Biol. 1992;41(3–8):231–240. doi: 10.1016/0960-0760(92)90349-n. [DOI] [PubMed] [Google Scholar]
- 11.Marcinkowska E. A run for a membrane Vitamin D receptor. Biol Signals Recept. 2001;10(6):341–349. doi: 10.1159/000046902. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Rao J, Studzinski GP. Inhibition of p38 MAP kinase activity up-regulates multiple MAP kinase pathways and potentiates 1,25-dihydroxyvitamin D3-induced differentiation of human leukemia HL60 cells. Exp Cell Res. 2000;258(2):425–437. doi: 10.1006/excr.2000.4939. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Studzinski GP. Activation of extracellular signal-regulated kinases (ERKs) defines the first phase of 1,25-dihydroxyvitamin D3-induced differentiation of HL60 cells. J Cell Biochem. 2001;80(4):471–482. doi: 10.1002/1097-4644(20010315)80:4<471::aid-jcb1001>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Studzinski GP. Inhibition of p38MAP kinase potentiates the JNK/SAPK pathway and AP-1 activity in monocytic but not in macrophage or granulocytic differentiation of HL60 cells. J Cell Biochem. 2001;82(1):68–77. doi: 10.1002/jcb.1141. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Studzinski GP. Phosphorylation of raf-1 by kinase suppressor of ras is inhibited by “MEK-specific” inhibitors PD 098059 and U0126 in differentiating HL60 cells. Exp Cell Res. 2001;268(2):294–300. doi: 10.1006/excr.2001.5292. [DOI] [PubMed] [Google Scholar]
- 16.Marcinkowska E, Kutner A. Side-chain modified Vitamin D analogs require activation of both PI 3-K and Erk 1,2 signal transduction pathways to induce differentiation of human promyelocytic leukemia cells. Acta Biochim Pol. 2002;49(2):393–406. [PubMed] [Google Scholar]
- 17.Wang Q, Wang X, Studzinski GP. Jun N-terminal kinase pathway enhances signaling of monocytic differentiation of human leukemia cells induced by 1,25-dihydroxyvitamin D3. J Cell Biochem. 2003;89(6):1087–1101. doi: 10.1002/jcb.10595. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Studzinski GP. Kinase suppressor of RAS (KSR) amplifies the differentiation signal provided by low concentrations 1,25-dihydroxyvitamin D3. J Cell Physiol. 2004;198(3):333–342. doi: 10.1002/jcp.10443. [DOI] [PubMed] [Google Scholar]
- 19.Chen F, Wang Q, Wang X, Studzinski GP. Up-regulation of Egr1 by 1,25-dihydroxyvitamin D3 contributes to increased expression of p35 activator of cyclin-dependent kinase 5 and consequent onset of the terminal phase of HL60 cell differentiation. Cancer Res. 2004;64(15):5425–5433. doi: 10.1158/0008-5472.CAN-04-0806. [DOI] [PubMed] [Google Scholar]
- 20.Okazaki T, Bielawska A, Bell RM, Hannun YA. Role of ceramide as a lipid mediator of 1 alpha, 25-dihydroxyvitamin D3-induced HL-60 cell differentiation. J Biol Chem. 1990;265(26):15823–15831. [PubMed] [Google Scholar]
- 21.Michaud NR, Therrien M, Cacace A, Edsall LC, Spiegel S, Rubin GM, Morrison DK. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci. 1997;94(24):12792–12796. doi: 10.1073/pnas.94.24.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison DK. KSR: a MAPK scaffold of the Ras pathway. J Cell Sci. 2001;114(9):1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 23.Xing HR, Lozano J, Kolesnick R. Epidermal growth factor treatment enhances the kinase activity of kinase suppressor of Ras. J Biol Chem. 2000;275(23):17276–17280. doi: 10.1074/jbc.C900989199. [DOI] [PubMed] [Google Scholar]
- 24.Yan F, John SK, Polk DB. Kinase suppressor of Ras is necessary for tumor necrosis factor alpha activation of extracellular signal-regulated kinase/mitogen-activated protein kinase in intestinal epithelial cells. Cancer Res. 2001;61(3):963–969. [PubMed] [Google Scholar]
- 25.Ji Y, Kutner A, Verstuyf A, Verlinden L, Studzinski GP. Derivatives of Vitamins D2 and D3 activate three MAPK pathways and upregulate pRb expression in differentiating HL60 cells. Cell Cycle. 2002;1(6):410–415. doi: 10.4161/cc.1.6.269. [DOI] [PubMed] [Google Scholar]
- 26.Perlman K, Kutner A, Prahl J, Smith C, Inaba M, Schnoes H, DeLuca HF. 24-homologated 1,25-dihydroxyvitamin D3 compounds: separation of calcium and cell differentiation activities. Biochemistry. 1990;29(1):190–196. doi: 10.1021/bi00453a026. [DOI] [PubMed] [Google Scholar]
- 27.Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1(2):103–110. [PubMed] [Google Scholar]
- 28.Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci. 1997;94(2):569–574. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavor S, Park DJ, Gery S, Vuong PT, Gombart AF, Koeffler HP. Restoration of C/EBP alpha expression in a BCR-ABL+ cell line induces terminal granulocytic differentiation. J Biol Chem. 2003;278(52):52651–52659. doi: 10.1074/jbc.M307077200. [DOI] [PubMed] [Google Scholar]
- 30.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27(3):263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 31.Dhawan P, Peng X, Sutton AL, Macdonald PN, Croniger CM, Trautwein C, Centrella M, McCarthy TL, Christakos S. Functional cooperation between CCAAT/enhancer-binding proteins and the Vitamin D receptor in regulation of 25-hydroxyvitamin D3 24-hydroxylase. Mol Cell Biol. 2005;25(1):472–487. doi: 10.1128/MCB.25.1.472-487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji Y, Studzinski GP. Retinoblastoma protein and CCAAT/enhancer-binding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res. 2004;64(1):370–377. doi: 10.1158/0008-5472.can-03-3029. [DOI] [PubMed] [Google Scholar]
- 33.Pan Z, Hetherington CJ, Zhang DE. CCAAT/enhancer-binding protein activates the CD14 promoter and mediates transforming growth factor beta signaling in monocyte development. J Biol Chem. 1999;274(33):23242–23248. doi: 10.1074/jbc.274.33.23242. [DOI] [PubMed] [Google Scholar]
- 34.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270(46):27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 35.Chopra AP, Boone SA, Liang X, Duesbery NS. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J Biol Chem. 2003;278(11):9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- 36.Danilenko M, Wang X, Studzinski GP. Carnosic acid and promotion of monocytic differentiation of HL60-G cells initiated by other agents. J Natl Cancer Inst. 2001;93(16):1224–1233. doi: 10.1093/jnci/93.16.1224. [DOI] [PubMed] [Google Scholar]
- 37.Danilenko M, Wang Q, Wang X, Levy J, Sharoni Y, Studzinski GP. Carnosic acid potentiates the antioxidant and prodifferentiation effects of 1alpha,25-dihydroxyvitamin D3 in leukemia cells but does not promote elevation of basal levels of intracellular calcium. Cancer Res. 2003;63(6):1325–1332. [PubMed] [Google Scholar]