

Abstract
The serine/threonine kinase Akt – also known as protein kinase B (PKB) – has emerged as one of the most frequently activated protein kinases in human cancer. In fact, most, if not all, tumors ultimately find a way to activate this important kinase. As such, Akt activation constitutes a hallmark of most cancer cells, and such ubiquity presumably connotes important roles in tumor genesis and/or progression. Likewise, the hypermetabolic nature of cancer cells and their increased reliance on “aerobic glycolysis”, as originally described by Otto Warburg and colleagues, are considered metabolic hallmarks of cancer cells. In this review, we address the specific contributions of Akt activation to the signature metabolic features of cancer cells, including the so-called “Warburg effect”.
Keywords: Energy metabolism, Glycolysis, Oxygen consumption, Oxidative phosphorylation, Cancer
1. Background
1.1. The serine/threonine kinase Akt/PKB
The evolutionarily conserved serine/threonine kinase Akt – also known as protein kinase B (PKB) – was identified nearly two decades ago as the cellular homologue of the v-Akt oncogene [1,2] and a novel structural relative of both protein kinase A (PKA) and protein kinase C (PKC) [3,4]. The major cellular functions of this putative kinase, however, remained obscure for several years until it was recognized as both a major downstream effector of phosphatidylinositol 3-kinase (PI3K) signaling [5,6] and a transducer of growth factor effects on cell survival [7,8]. Mammalian cells express three highly homologous Akt isoforms (Akt1, Akt2, and Akt3) encoded by separate genes, but sharing over 80% amino acid sequence identity. The pathway leading to Akt activation is also highly conserved across species. Growth factor receptor–ligand interaction activates the catalytic p110 subunit of PI3K via p85 regulatory subunit recruitment or by activating Ras, which directly activates p110. Following activation, p110 phosphorylates phosphoinositides (PI) at the D3-position of the inositol ring to yield PI(3,4,5)P3 (PIP3). The binding of PIP3 to the pleckstrin homology (PH) domain of Akt promotes its translocation to the plasma membrane, which is the rate-limiting step in Akt activation. Akt is then phosphorylated by PI3K-dependent kinase-1 (PDK1) at a threonine residue within the catalytic domain and at a serine residue in the carboxy-terminal hydrophobic motif by mammalian target of rapamycin complex 2 (mTORC2) (Fig. 1). Phosphorylation at both sites is required for full Akt activity (reviewed in [9]). Conversely, PI3K antagonism negatively regulates Akt activity. This is best exemplified by the action of the tumor suppressor PTEN (for phosphatase and tensin homolog deleted on chromosome ten) [10], a phospholipid phosphatase that directly antagonizes PI3K activity by dephosphorylating PIP3. These signaling effectors, which tightly regulate Akt activity, collectively comprise a major metabolic regulatory pathway in nematodes and flies, and this function is conserved in mammals at both the organismal and cellular levels. At the organismal level, activators of PI3K–Akt signaling play a central role in glucose (Glc) homeostasis, and this can be attributed, in large part, to major roles in the regulation of cellular energy metabolism via mechanisms involving, but not fully restricted to, the cellular uptake and utilization of Glc. Mice with defects in the signaling pathway linking the insulin receptor to Akt – including insulin receptor substrate (IRS)-, PI3K-, and Akt-deficient mice – develop insulin resistance and diabetes (for review see [11,12]). Mammalian cells with hyperactive Akt accumulate ATP, whereas cells with reduced Akt abundance or activity exhibit markedly reduced ATP content [13,14]. Thus, the most conserved function of Akt seems to involve the preservation of energy metabolism at both the organismal and cellular levels.
Fig. 1.
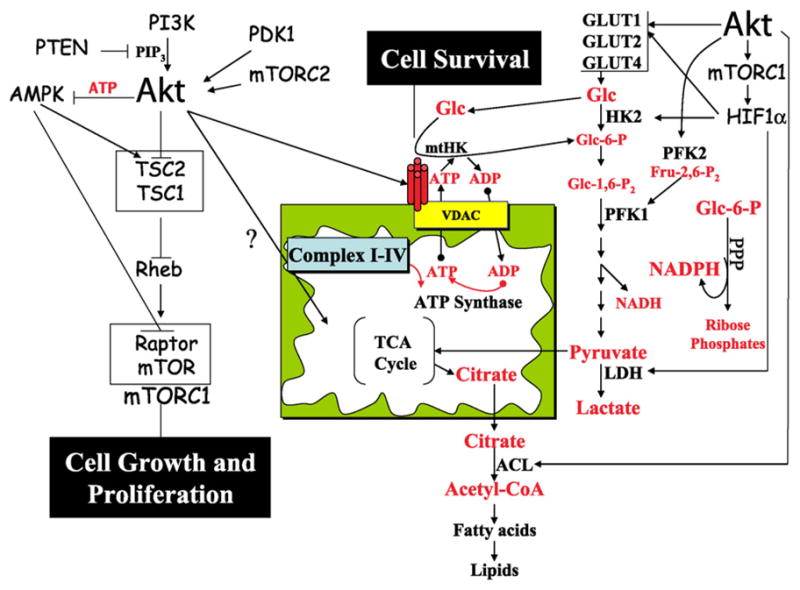
Coupling between Akt-mediated cellular energy metabolism, cell survival and proliferation. Following activation by PI3K, PDK1, and mTORC2, Akt increases cellular ATP production by accelerating both glycolytic and oxidative metabolism. Akt may increase oxidative phosphorylation by enhancing metabolic coupling between glycolysis and oxidative phosphorylation through facilitation of mitochondrial hexokinase (mtHK; i.e. HKI and HKII) association with VDAC and mitochondria, and by as yet unknown mechanisms. By facilitating mtHK association with mitochondria, Akt increases cell survival. Akt also enhances glycolytic flux by multiple mechanisms. First, it increases glucose (Glc) uptake by increasing the expression of Glc transporters (GLUT1, GLUT2, and GLUT4), and, in some cases, by promoting translocation to the plasma membrane. Second, enhanced coupling between oxidative phosphorylation and glycolysis may also kinetically favor enhanced glycolytic flux. Third, hyperactive Akt activates mTORC1, which promotes HIF1α accumulation under normoxic conditions and increases GLUT1, HKII, and lactate dehydrogenase (LDH) abundance. The increased capacity for both Glc transport and phosphorylation results in increased glucose-6-phosphate (Glc-6-P) availability for utilization in both glycolysis and the pentose phosphate pathway (PPP). Fourth, Akt phosphorylates and activates phosphofructokinase-2 (PFK2), which leads to allosteric activation of phosphofructokinase-1 (PFK1) by the PFK2 product fructose-2,6-bisphosphate (Fru-2,6-P2 ). Citrate generated in the mitochondrial TCA cycle is exported to the cytoplasm, where it is utilized for acetyl-coA generation by ATP-citrate lyase (ACL), which is directly phosphorylated and activated by Akt. By increasing citrate utilization, Akt may help drive TCA cycle flux, in addition to providing precursors for lipid biosynthesis for new membrane generation. Finally, Akt-increased cellular ATP levels serve to maintain low AMPK activity, which is required for full mTORC1 activation. As the most critical downstream effector of Akt, mTORC1 is largely responsible for the contributions of Akt to cell growth, cell proliferation, and susceptibility to oncogenic transformation.
Akt is perhaps the most frequently activated oncoprotein in human cancers, and it is activated by multiple mechanisms (reviewed in [9]). The predominant mechanisms by which Akt is activated in human cancer are through mutational inactivation of PTEN and mutational activation of the catalytic subunit of PI3K. Hyperactivation of Akt in cancer cells also occurs via oncogenic mutations in Ras genes and growth factor receptor activation, particularly involving heterodimeric ErbB2/ErbB3 receptors which possess six docking sites for PI3K. Additionally, amplifications of the genes encoding both Akt and p110 have been observed in a subset of human cancers. More recently, an activating mutation in the PH domain of Akt1 that increases affinity for PIP3 has been identified in another subset of human cancers (reviewed in [15]).
Akt hyperactivation can contribute to the genesis of cancer by inhibiting apoptosis, by increasing cell proliferation, and/or by accelerating oncogenic mutation rates. As discussed below, these potential contributions are coupled to Akt’s role in energy metabolism, and Akt hyperactivation is closely associated with cellular processes that contribute to the classical biochemical phenotype of cancer cells, including the so-called “Warburg effect”.
1.2. Warburg’s observations on the nature and development of cancer
In the 1920s and 1930s, Warburg and colleagues rigorously interrogated cancer metabolism in search of clues to the essential nature of tumorigenesis and cancer progression. In the process, they described what is arguably the most consistent cancer phenotype, namely the aberrant shift to glycolytic energy dependence in the presence, as well as absence, of molecular oxygen—or so-called “aerobic glycolysis”. This ubiquitous metabolic phenotype presumably confers competitive survival or growth advantages to cancer cells over their normal counterparts by favorably adapting them to growth in hypoxic tumor microenvironments [16]. This classic biochemical signature of cancer cells also serves as the mechanistic basis for using FDG-PET to clinically identify and stage metastatic tumors [17,18].
In normal cells, the presence of oxygen indirectly suppresses glycolysis, a process commonly referred to as the “Pasteur effect” in recognition of Pasteur’s original observations of this phenomenon in yeast [19]. Although the mechanistic underpinning of this effect remain incompletely understood, there is general agreement that these changes reflect intramitochondrial–extramitochondrial communication, likely via direct metabolic coupling mechanisms such as those facilitated by hexokinase (HK)–mitochondria interaction [18]. The corresponding existence of the “Crabtree effect”, wherein elevated Glc concentrations reversibly suppress the TCA cycle and oxidative phosphorylation, suggests that these lines of communication must be bidirectional – although not necessarily identical – in normal cells. By gating the exchange of limiting metabolites such as adenine nucleotides into and out of mitochondria, we have previously proposed that phosphohexose accumulation-associated changes in HK–mitochondria interaction may directly contribute to the Crabtree effect [18,20]. In principle, the persistence of tumor cell glycolysis in the presence of oxygen may thus reflect altered intramitochondrial–extramitochondrial coupling, rather than a primary impairment of oxidative phosphorylation. In keeping with this notion, Warburg observed in his preface to the 1930 English edition of The Metabolism of Tumours that “Aerobic glycolysis results if the respiration of growing cells is injured, whether by diminishing its extent or by interfering with the relationship which holds between respiration and fermentation (glycolysis)” [21]. In these and subsequent works from the Kaiser–Wilhelm Institute, Warburg and colleagues established that the relative contributions of glycolysis (lactate accumulation) and respiration (oxygen consumption) are altered in most tumors. The observed metabolic shift to increased cellular glycolysis and relative dependence upon Glc as an energy substrate is not, however, mutually exclusive with substantive contributions by oxidative phosphorylation, as some interpretations of this work have suggested. This notion is affirmed by Warburg’s contention that, “In order to destroy tumor cells in the living animal by lack of energy, it is necessary, as with the experiments in vitro, to inhibit the respiration as well as the fermentation” [22]. Persistent – or even increased – oxidative metabolism in many cancers is also compatible with this contention. In this review, we address a number of coordinated and sometimes competing mechanisms whereby Akt may contribute to altered respiratory function and/or coupling with extramitochondrial metabolism and thereby account for many of the phenotypic features of cancer cells. As a master regulator of many of these features, we further suggest that Akt may constitute a “Warburg kinase” that can be directly targeted to alter cancer cell susceptibility to therapeutic intervention.
2. Coupling between individual Akt functions and cellular energy metabolism
2.1. Akt regulates energy metabolism
The activation of Akt can increase total cellular ATP content by as much as two- to threefold, whereas deletion of Akt1 alone or in combination with Akt2 markedly decreases intracellular ATP levels in knockout mouse embryonic fibroblasts [13,14]. These Akt-dependent changes in cellular ATP content involve both glycolysis and oxidative phosphorylation [13]. Also, oxygen consumption is elevated in cells expressing activated Akt and is reduced in Akt-deficient cells [23]. Taken together, these findings suggest major roles for Akt in the coordinated regulation of both glycolytic and oxidative metabolism.
Akt-induced glycolysis may be mediated by multiple non-exclusive mechanisms (Fig. 1), including the expression and membrane translocation of glucose transporters and effects on HK expression, activity, and mitochondrial interaction. Akt may also indirectly activate the important rate-controlling enzyme phosphofructokinase-1 (PFK1) by directly phosphorylating and activating phosphofructokinase-2 (PFK2) [24], whose principal reaction product, fructose-2,6-bisphosphate (Fru-1,6-P2), is the most potent known allosteric activator of PFK1. In addition, suppression of glycolytic gene expression by the transcription factor FoxO could be reversed by its phosphorylation and inactivation by hyperactive Akt [25]. Finally, Akt hyperactivity can increase mTORC1 activity, thereby increasing HIF1α abundance [26] and HIF1α-associated glycolytic enzyme and Glc transporter expression.
As indicated above, Akt affects not only glycolysis, but also oxidative phosphorylation. By analogy with glycolysis, Akt may affect respiration both directly and indirectly. For example, Akt may indirectly promote oxidative phosphorylation via increased formation of and access to glycolysis-derived substrates essential for TCA cycle activity and oxidative phosphorylation (e.g. pyruvate, ADP, and NADH). The increased coupling efficiency between glycolysis and respiration afforded by Akt-stimulated mitochondrial HK association [13] may also provide kinetic advantages that serve to simultaneously increase the effective rates of both glycolysis and respiration. Additional direct and indirect effects of Akt on respiration are also possible (Fig. 1).
2.2. The anti-apoptotic role of Akt is coupled to its role in energy metabolism
The classical apoptotic cascade in mammalian cells is strongly antagonized by growth factor signaling involving PI3K and its principal downstream effector Akt. Although these phylogenetically conserved opposing signal transduction pathways are observed in C. elegans as well as mammals, no compelling evidence exists to support an anti-apoptotic role for Akt in nematodes [18,20]. The more highly conserved role of trophic factor-stimulated PI3K–Akt signaling in regulating cellular metabolism suggests that this function was evolutionarily recruited to the regulation of apoptosis in parallel with the recruitment of mitochondria to the apoptotic cascade [18]. There is considerable experimental support for this hypothesis, although alternate explanations can also be put forth. Both Akt and Bcl-2 promote mitochondrial integrity and inhibit cytochrome c release in mammalian cells, but Akt, unlike Bcl-2, requires the presence of Glc to achieve these effects. This distinction originally attracted our attention to mitochondrial HK (mtHK), which directly interacts with voltage-dependent anion channels (VDAC) and the outer mitochondrial membrane (OMM) and catalyze the first committed step of Glc metabolism—namely, the ATP-dependent phosphorylation of Glc to yield glucose 6-phosphate (Glc-6-P). The physical and functional interactions between HK and mitochondria have been recognized for over a half century and can be largely attributed to specific binding between high affinity HKI and HKII isoforms and the OMM at contact sites with the inner mitochondrial membrane (IMM). HK binding is both dynamic and regulated and is mediated, in part, by specific interactions between VDAC in the OMM and hydrophobic alpha-helical mitochondrial binding domains found in the N-terminus of HKI and HKII, but not HKIII or GK(HKIV) [18,20]. A fifth expressed HK isoform, HKDC1, has recently been identified [27], and inspection of its N-terminal sequence suggests a similar alpha-helical hydrophobic domain. Although presently speculative, this raises the theoretical possibility that HKDC1 may also interact with mitochondria. This would have clear functional implications insofar as the ability to specifically bind mitochondria presently distinguishes HKI and HKII from all other glycolytic enzymes, including other HK isoforms, and this property plays important roles in both HK regulation and functions relevant to cancer pathobiology. The dynamic binding of HKs to VDAC and mitochondria is mediated primarily by a feedback mechanism involving the principal downstream reaction products, Glc-6-P and Glc-1,6-P2, which are thought to induce conformational changes in mtHK that inhibit enzymatic activity and promote dissociation from mitochondria (reviewed in [30]). Thus, the net rate of phosphohexose accumulation, which reflects the rates of both Glc-6-P formation and consumption, could largely determine both the activity and the duration of HK association with mitochondria. By providing “privileged access” to intramitochondrial ATP, mitochondrial localization is thought to confer functional advantages to mtHK. Consistent with this view, mtHK exhibit increased specific activity and decreased allosteric inhibition relative to their cytosolic counterparts. The ability of mtHK to directly couple the first committed step of Glc utilization to oxidative phosphorylation also makes it uniquely suited to contribute to both the Pasteur and Crabtree effects. In fact, during active oxidative phosphorylation, mtHK preferentially, if not exclusively, utilize ATP of intramitochondrial origin to phosphorylate Glc, even if extramitochondrial ATP is freely available [28,29]. ADP generated through this reaction can then be redirected back into the mitochondrion to support oxidative phosphorylation. This relationship between ADP-generating mtHK and ADP-consuming mitochondria is critical to the proposed model [18,20] insofar as ADP is normally limiting for oxidative phosphorylation in fully coupled mitochondria.
By binding to mitochondrial contact sites and modulating anionic metabolite exchange across the OMM via VDAC, mtHK physically act at a site where competing survival and apoptotic signals converge to modulate mitochondrial integrity and cytochrome c release. HKI and HKII compete with each other [30] and with the binding and the oligomerization of Bax and Bak at the OMM [31,32]. As Bax and Bak oligomers mediate the release of cytochrome c from mitochondria, this could explain, in part, the ability of mtHK to maintain mitochondrial integrity and inhibit apoptosis [31,32]. Growth factors and Akt facilitate the association of HK with mitochondria, which is required for Akt to inhibit cytochrome c release and apoptosis (reviewed in [18]). Mechanistically, the association of HK with mitochondria interferes with cytochrome c release induced by Bax and Bak, as well as the ensuing activation of caspases and apoptosis. However, it appears that mtHK association is important for mitochondrial integrity even in the absence of Bax and Bak [18,33]. Although still unproven, there is a wealth of evidence to support the notion that mtHK–VDAC interaction regulates the VDAC open state and directly influences mitochondrial integrity through effects on transmembrane anionic metabolite exchange. Octameric mitochondrial creatine kinase (mtCK) also associates with the intramitochondrial aspect of VDAC, but only when mtHK is not associated with its cytosolic aspect (reviewed in [20]). VDAC and the IMM adenine nucleotide translocator (ANT) directly interact when complexed with mtHK [34], whereas mtCK interposition mediates VDAC–ANT interaction when mtHK is absent. As such, mtCK, like mtHK, may function to modulate VDAC activity and VDAC–ANT interaction. Both kinases have been proposed to influence mitochondrial contact site formation through complexes with VDAC (reviewed in [20]). Although the exact mechanisms are incompletely defined, it is widely accepted that VDAC participates in the formation of the mitochondrial permeability transition pore (PTP) [35]. With this in mind, recent work has suggested a role for mtHK in preventing PTP formation, albeit via a VDAC-independent mechanism [36]. Although difficult to mechanistically reconcile with our findings and those of others, we take these findings as generally supportive of the notion that Akt-directed mtHK association favorably influences mitochondrial energetics and intercompartmental communication between mitochondria and the cytosol and also prevents pro-apoptotic Bak and Bax oligomerization and binding at the level of the mitochondrion. The importance of coupling between mitochondrial and extramitochondrial metabolism and the anabolic, as well as catabolic, roles of the TCA cycle in cancer has received increased recent attention [18,37], and the Akt–HK axis seems uniquely well-suited to regulating these processes.
The specific mechanism(s) whereby Akt promotes mitochondrial HK association remain(s) obscure, but both metabolic and classical signaling effectors have been implicated in this process. One possibility is that by facilitating glycolytic flux downstream of Glc-6-P, Akt activation maintains low levels of intracellular Glc-6-P, and thereby prolongs HK–mitochondria association. As noted earlier, the rate-controlling enzyme PFK1, which acts distal to both Glc-6-P and Glc-1,6-P2, can be indirectly stimulated by Akt through direct PFK2 activation to generate the principal positive allosteric effector of PFK1, Fru-1,6-P2 [24]. In principle, the coordinated induction and/or activation of downstream glycolytic enzymes could serve to enhance downstream glycolytic flux and thereby ensure that mtHK-dissociating phosphohexoses do not accumulate.
Another mechanism whereby Akt may indirectly contribute to increased HK–VDAC interaction involves the phosphorylation and inhibition of glycogen synthase kinase 3β (GSK3β), which, in the absence of the inhibitory influence of Akt, has been reported to directly phosphorylate VDAC and thereby inhibit mtHK binding [38]. Interestingly, overexpression of HK together with GLUT1 has been reported to increase the phosphorylation and inhibition of GSK3 through an increase in glycolysis [39], suggesting the possibility that HK can facilitate its own interaction with VDAC.
It is also possible that Akt may directly influence HK-mitochondria association or function. There is limited evidence for regulation of HK activity or function by post-translational modifications, including phosphorylation, but it is pertinent to note that HKII contains a conserved consensus Akt phosphorylation site (RxRxxS/T) at T473 [13,40]. It has recently been reported that Akt can directly phosphorylate HKII at this site in leukemia inhibitory factor-stimulated cardiomyocytes and that HKII phosphorylation correlates with mitochondrial localization and anti-apoptotic function [40]. If confirmed, these findings could suggest a specific mechanism whereby Akt recruits HKII to mitochondria in this cell type, although they do not clearly distinguish between phospho-HKII targeting to mitochondria and in situ phosphorylation of mtHKII already resident on the OMM [40]. Considering the latter possibility, it remains to be seen whether HKII phosphorylation has kinetic consequences or is critical for mitochondrial protein complex assembly in this model. These findings also do not address the established interaction between mitochondria and the other mtHK isoform, HKI, which is also prominently expressed in cardiomyocytes [41]. As such, the specific biological relevance of direct HKII phosphorylation by Akt remains to be fully established. Akt also increases HKII expression (R.B.R., unpublished data), but the relationship between HK pool size and mitochondrial HK association is still incompletely defined.
In addition to its effects on HKII expression and HK-mitochondria interaction, Akt could promote cell survival through enhanced expression and translocation of Glc transporters [42,43], which share major control over Glc uptake and utilization with HK [18]. A link between HK, Glc metabolism, and stabilization of Mcl-1, an anti-apoptotic member of the Bcl-2 family, has also recently been suggested, with co-expression of HKII and GLUT1 facilitating the phosphorylation and inactivation of GSK3 [39]. This antagonizes the ability of GSK3 to phosphorylate and destabilize Mcl-1 [44]. Finally, inhibitory coupling between PPP flux and the apoptotic cascade has also been reported [45]. Exogenous Glc-6-P addition to non-glycolytic Xenopus egg extracts inhibits mitochondrial cytochrome c release and caspase activation via a mechanism involving Glc-6-P utilization in the PPP and the subsequent generation of NADPH, which in turn activates CaMKII. CaMKII then phosphorylates a serine residue in the pro-domain of caspase-2 and thereby inhibits its activity [45]. When activated by cellular stress, DNA damage, or trophic factor deprivation, caspase-2 acts upstream of mitochondria through the activation of “BH3-only” proteins [46–49]. It has been shown that caspase-2 can cleave and activate Bid [46,50–52], and that its pro-apoptotic activity is dependent on Bid [53]. However, it remains to be seen whether PPP flux exerts such effects on caspase-2 activation in somatic cells and whether mtHK also maintain mitochondrial integrity via upstream caspase-2 inhibition.
2.3. The role of Akt in cell growth and proliferation is coupled to its role in energy metabolism
In addition to its pivotal role in cell survival, Akt plays important roles in normal cell proliferation and is a determinant of cellular susceptibility to oncogenic transformation [54]. Of the numerous Akt target proteins reported to-date, mTORC1 appears to be the most critical downstream effector of Akt-dependent cell proliferation and altered susceptibility to oncogenic transformation [54]. Akt is both required and sufficient for mTORC1 activation by growth factors [55]. mTORC1 also regulates cell mass (cell growth) by increasing protein synthesis and this could explain the effect of Akt on cell growth in mammals both at the cellular and organismal levels (reviewed in [9,56]). In recent years, the mechanisms by which Akt regulates mTORC1 have been delineated. The principal barrier to mTORC1 activation is the heterodimeric tuberous sclerosis complex (TSC1/TSC2). This complex acts as a GTPase activating protein (GAP) to negatively regulate the small GTPase Rheb, which is required for mTORC1 activation. Akt phosphorylates and inactivates TSC2, thereby activating both Rheb and subsequently mTORC1 (reviewed in [9,56]; see Fig. 1). However, the role of Akt in energy metabolism is required for full activation of mTORC1 (Fig. 1). Heterodimeric TSC1/TSC2 is sensitive to cellular energy status. When cellular energy stores are low, increasing AMP levels and decreasing ATP levels activate AMP-activated kinase (AMPK), which then phosphorylates and activates TSC2 [57]. It appears that, by regulating cellular ATP content, Akt indirectly controls AMPK activity, which is required for full mTORC1 activation [14]. Furthermore, it has been recently shown that mTORC1 itself is sensitive to cellular ATP content, as Raptor, which is the defining component of mTORC1, is phosphorylated and inactivated by AMPK [58]. Finally, Akt-mediated increases in respiration [23] have anabolic, as well as catabolic, consequences that are needed to support cell proliferation. For example, increased TCA cycle flux serves as an ongoing source of citrate needed for sequential acetyl-coA generation, lipid biosynthesis, and membrane renewal in rapidly proliferating cells such as cancer cells. Interestingly, ATP citrate lyase (ACL), is also an Akt target [59] and is required for Akt-mediated tumorigenesis [60]. It is also possible that Akt-stimulated increases in citrate utilization per se could help drive TCA cycle flux and respiration. Collectively, these observations suggest that the major impact of Akt on cell growth and proliferation is tightly coupled to its roles in energy metabolism.
Akt-stimulated increases in oxidative metabolism can contribute to reactive oxygen species (ROS) accumulation [23]. Cells exhibiting Akt hyperactivity display high levels of ROS, not only as a byproduct of accelerated oxidative phosphorylation, but also as a consequence of ROS scavenger inhibition by Akt [23]. The elevated ROS induced by Akt could contribute to tumorigenesis by activating signaling pathways via direct oxidation of catalytic cysteine residues within the active sites of certain phosphatases [61]. However, the principal effect is probably mediated by increased mutation rates after oxidation of both genomic and mitochondrial DNA. The propensity of Akt to increase intracellular ROS generation also constitutes its Achilles’ heel by leading to premature senescence and/or increased susceptibility to ROS-induced apoptosis. Surprisingly, despite the ability of Akt to inhibit apoptosis induced by multiple apoptotic stimuli it cannot inhibit apoptosis induced by ROS. In fact, the combination of Akt’s abilities to elevate intracellular ROS and to impair ROS scavengering, together with its inability to inhibit ROS-induced cell death, hypersensitizes cells to ROS-mediated cell death [23]. The hypersensitivity of cells displaying hyperactive Akt could be used to selectively eradicate cancer cells that display hyperactive Akt by exposing them to ROS inducers [23].
3. Akt contributions to the Warburg effect and the biochemical phenotype of cancer cells
As indicated earlier, most tumors cells share two common features: (i) the activation of Akt, which is a major regulator of cellular energy metabolism, and (ii) increased glycolysis with excess lactate accumulation in both the presence and absence of oxygen, commonly referred to as the Warburg effect. However, activation of Akt increases ATP generated by both glycloysis and oxidative phosphorylation with a concomitant increase in oxygen consumption [13,23]. How can these observations be reconciled with the classical notion that respiration is impaired in highly glycolytic tumor cells? Increased lactate accumulation indicates that the increased aerobic glycolytic flux is not strictly coupled to changes in oxidative metabolism. Warburg deduced that tumor cells acquire specific lesions that impair mitochondrial respiration in these cells. However, careful reexamination of the available data suggests that respiration may remain intact or even increase in a large number of tumor cells despite high levels of aerobic glycolysis [62]. As such, the subset of tumors cells categorized as having both increased aerobic glycolysis and diminished respiration may be smaller than conventionally held and would be predicted to include cells harboring heritable mutations in nuclear genes encoding electron transport chain components or TCA cycle enzymes such as succinate dehydrogenase (SDH) or fumarate hydratase (FH) [63]. Thus, there is a good general agreement with Warburg’s observation that tumor cells have elevated glycolysis but it does not follow that they must also have impaired oxidative phosphorylation. Increased Glc uptake and glycolysis not only adapt cells to hypoxic microenvironments, but are also required to meet the anabolic and catabolic demands for glycolysis-derived intermediates to support rapid tumor cell proliferation. For instance, pentose phosphates required for nucleic acid biosynthesis are generated by the PPP, and NADPH generated by the same pathway contributes to anabolic cellular processes such as fatty acid biosynthesis and is redox coupled to antioxidant metabolisms involving glutathione peroxidase [18]. Glycolysis, in addition to providing ATP, provides metabolic intermediates that participate in anabolic reactions, particularly in hypoxic conditions. Finally, excess lactate production and proton-coupled lactate extrusion via monocarboxylate transporters may contribute to local extracellular acidification and support tumor invasion, particularly in hypoxic tumor microenvironments. Extruded lactate can also be taken up by adjacent stromal cells or neighboring non-hypoxic tumor cells and utilized as an energy substrate to support their growth. The ability of Akt to increase tumor cell Glc uptake and glycolysis may also be shared with other oncoproteins such as Myc [16]. In addition, hypoxia inducible factor 1α (HIF1α) abundance increases in hypoxic cells and induces adaptive gene expression programs involving both Glc transporters and glycolytic enzymes, including GLUT1 and HKII [64]. Activated Akt may also contribute to increased HIF1α abundance under aerobic conditions via mTORC1-mediated effects on HIF1α mRNA translation [26]. Lastly, a subset of tumor cells exhibit increased HIF1α abundance as a consequence of von Hippel Lindau protein (pVHL) deficiency, which normally targets HIF1α for proteasomal degradation under aerobic conditions following oxygen-dependent prolyl hydroxylation [65].
The highly glycolytic phenotype of cancer cells is widely appreciated and provides obvious advantages for tumor cell growth and proliferation, particularly in hypoxic microenvironments. But what possible advantage could tumor cells derive from irreversibly impaired respiration? This would be a seemingly disadvantagous strategy for a tumor cell to adopt given the dual catabolic and anabolic demands of proliferating tumor cells. This would seem especially true following angiogenic neovascularization or during metastasis when physiologic levels of oxygen tension may be encountered. Rapidly proliferating tumor cells demand high levels of ATP for growth and proliferation, largely due to protein synthesis, which is a major consumer of cellular energy. Likewise, rapidly dividing cells require increased lipid generation for new membrane synthesis. Acetyl-coA, which is a required precursor for lipid biosynthesis, is generated from citrate produced by the TCA cycle. Thus, diminishing respiration would be disadvantageous for tumor cells from an anabolic standpoint. Therefore, Akt hyperactivation provides selective advantages to tumor cells by increasing both glycolysis and oxidative phosphorylation. If Akt hyperactivation occurs early in tumorigenesis, then increased oxygen consumption combined with diminished anti-oxidant capacity could lead to elevated intracellular ROS and subsequent premature senescence [23] unless a lesion capable of overriding premature senescence occurs. If Akt hyperactivation occurs after an immortalizing event, such as p53 inactivation, or if such an event follows Akt activation, then Akt activation may directly or indirectly contribute to tumorigenesis via multiple non-exclusive mechanisms. These include increased glycolysis, increased respiration, altered coupling between glycolytic and oxidative metabolism, and decreased apoptotic susceptibility due to increased HK–mitochondria interaction. Although speculative, the increased oxidative phosphorylating capacity of tumor cells exhibiting hyperactive Akt may not be fully realized, in part, due to the inhibitory influence of increased aerobic glycolysis on oxidative phosphorylation (Crabtree effect). ROS generated as a consequence of Akt activation may also contribute to tumorigenesis by facilitating tumor-promoting signaling pathways and by increasing mutation rates. Mitochondrial DNA is particularly susceptible to accelerated mutagenesis by intramitochondrial ROS due to its close physical proximity, the absence of histones, and the low efficiency of mitochondrial DNA repair [66]. In principle, mutations associated with Akt hyperactivity could lead to impairment of cellular respiration. In some cases, these mutations could augment ROS generation at levels supportive of metastasis [67]. It bears noting, however, that oxidant-induced somatic mutations occur randomly, and given the existence of both multiple mitochondria per cell and multiple DNA copies per mitochondrion, such mutations in mitochondrial DNA could easily be lost by dilution during ongoing DNA replication unless positive selective pressures exist to maintain them. For instance, respiration-damaging mitochondrial DNA mutations could be enriched in tumor cells restricted to a hypoxic microenvironment without compromising their viability and eventually lead to irreversibly impaired respiratory capacity. In contrast, exposure of the same tumor cells to increased oxygen tensions that might be anticipated during neovascularization or metastasis would ostensibly result in selection against such disadvantageous mutations. This mechanism would enable tumor cells to adapt to environmental cues with varying levels of oxygen tension and maximize their ability to thrive in both hypoxic and non-hypoxic environments. Implicit in this model, tumor cells are assumed to be heterogeneous with respect to respiratory capacity, and thus when tumor cells are isolated from primary tumors for in vitro culture under standard aerobic conditions, cells with intact or increased respiration will be selected for, unless a homogeneous population of hypoxic tumor cells was isolated. Similarly, tumor cells of metastatic origin would be predicted to possess intact or elevated respiration, especially if Akt is hyperactive. This may help explain inconsistencies regarding the observed magnitude of respiration in different tumors.
4. Conclusions
Akt hyperactivation directly or indirectly recapitulates many of the phenotypic features of cancer metabolism. As such, it is an excellent candidate master regulator responsible for the classical biochemical features of cancer cells. In this sense, Akt may constitute a “Warburg kinase” that can be specifically targeted to alter cancer cell energy metabolism for therapeutic benefit.
Acknowledgments
Dr. Robey is an Established Investigator of the American Heart Association. His laboratory is supported by the United States Department of Veterans Affairs, the American Heart Association, and the Hitchcock Foundation. Dr. Hay’s laboratory is supported by the National Institutes of Health.
Footnotes
Conflict of interest statement
None.
References
- 1.Staal SP, Hartley JW, Rowe WP. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci USA. 1977;74:3065–7. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine/threonine kinase containing an SH2-like region. Science. 1991 October;254:274–7. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 3.Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci USA. 1991;88:4171–5. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;201:475–81. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 5.Franke TF, Yang S, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995 June;81:727–36. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 6.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 7.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–5. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Develop. 1997;11:701–13. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 9.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12(4):487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 10.Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol. 2004;15(2):171–6. doi: 10.1016/j.semcdb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 11.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 13.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15(11):1406–18. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280(37):32081–9. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 15.Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12(2):104–7. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–30. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 17.Smith TA. The rate-limiting step for tumor [18F]fluoro-2-deoxy-D-glucose (FDG) incorporation. Nucl Med Biol. 2001;28(1):1–4. doi: 10.1016/s0969-8051(00)00177-3. [DOI] [PubMed] [Google Scholar]
- 18.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25(34):4683–96. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 19.Pasteur M. Expériences et villes nouvelles sur la nature des fermentations. 1861:1260–4. [Google Scholar]
- 20.Robey RB, Hay N. Mitochondrial hexokinases: guardians of the mitochondria. Cell Cycle. 2005;4(5):654–8. doi: 10.4161/cc.4.5.1678. [DOI] [PubMed] [Google Scholar]
- 21.Warburg O, Wind F, Negelein E. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology. Berlin-Dahlen, London: Constable & Co., Ltd; 1930. On the Metabolism of Tumours in the Body. [Google Scholar]
- 22.Warburg O. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlen. In: Warburg O, editor. London: Constable. Preface to the English edition. London: Constable & Co. Ltd; 1930. [Google Scholar]
- 23.Nogueira V, Park Y, Chen C-C, Xu P-Z, Chen M-L, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14(6):458–70. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272:17269–75. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281(15):10105–17. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 26.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–58. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- 27.Irwin DM, Tan H. Molecular evolution of the vertebrate hexokinase gene family: identification of a conserved fifth vertebrate hexokinase gene. Comp Biochem Physiol D. 2008;3(1):96–107. doi: 10.1016/j.cbd.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Arora KK, Pedersen PL. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. J Biol Chem. 1988;263(33):17422–8. [PubMed] [Google Scholar]
- 29.BeltrandelRio H, Wilson JE. Interaction of mitochondrially bound rat brain hexokinase with intramitochondrial compartments of ATP generated by oxidative phosphorylation and creatine kinase. Arch Biochem Biophys. 1992;299(1):116–24. doi: 10.1016/0003-9861(92)90252-r. [DOI] [PubMed] [Google Scholar]
- 30.Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- 31.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277(9):7610–8. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 32.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24(2):730–40. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase–mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16(5):819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 34.Adams V, Bosch W, Schlegel J, Wallimann T, Brdiczka D. Further characterization of contact sites from mitochondria of different tissues: topology of peripheral kinases. Biochim Biophys Acta. 1989;981(2):213–25. doi: 10.1016/0005-2736(89)90031-x. [DOI] [PubMed] [Google Scholar]
- 35.Grimm S, Brdiczka D. The permeability transition pore in cell death. Apoptosis. 2007;12(5):841–55. doi: 10.1007/s10495-007-0747-3. [DOI] [PubMed] [Google Scholar]
- 36.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE. 2008;3(3):e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65(22):10545–54. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27(12):4328–39. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15(3):521–9. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 41.Aubert-Foucher E, Font B, Gautheron DC. Rabbit heart mitochondrial hexokinase: solubilization and general properties. Arch Biochem Biophys. 1984;232(1):391–9. doi: 10.1016/0003-9861(84)90554-x. [DOI] [PubMed] [Google Scholar]
- 42.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6(3):683–92. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 43.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21(17):5899–912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–60. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123(1):89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297(5585):1352–4. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 47.Tinel A, Tschopp J. The PIDDosome a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304(5672):843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 48.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun. 2005;331(3):859–67. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 49.Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8(1):72–7. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- 50.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277(33):29803–9. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- 51.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277(16):13430–7. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 52.Wagner KW, Engels IH, Deveraux QL. Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J Biol Chem. 2004;279(33):35047–52. doi: 10.1074/jbc.M400708200. [DOI] [PubMed] [Google Scholar]
- 53.Gao Z, Shao Y, Jiang X. Essential roles of the Bcl-2 family of proteins in caspase-2-induced apoptosis. J Biol Chem. 2005;280(46):38271–5. doi: 10.1074/jbc.M506488200. [DOI] [PubMed] [Google Scholar]
- 54.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10(4):269–80. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 57.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 58.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277(37):33895–900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 60.Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24(41):6314–22. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- 61.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7(11):833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 62.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393–418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 63.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5(11):857–66. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- 64.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007. 2007;(407):cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 65.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2(9):673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 66.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 67.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]