

Abstract
Whether interleukin (IL)-17 promotes a diabetogenic response remains unclear. Here we examined the effects of neutralization of IL-17 on the progress of adoptively transferred diabetes. IL-17-producing cells in non-obese diabetic (NOD) mice were identified and their role in the pathogenesis of diabetes examined using transfer and co-transfer assays. Unexpectedly, we found that in vivo neutralization of IL-17 did not protect NOD–severe combined immunodeficiency (SCID) mice against diabetes transferred by diabetic splenocytes. In NOD mice, γδ+ T cells were dominated by IL-17-producing cells and were found to be the major source of IL-17. Interestingly, these IL-17-producing γδ T cells did not exacerbate diabetes in an adoptive transfer model, but had a regulatory effect, protecting NOD mice from diabetes by up-regulating transforming growth factor (TGF)-β production. Our data suggest that the presence of IL-17 did not increase the chance of the development of diabetes; γδ T cells protected NOD mice from diabetes in a TGF-β-dependent manner, irrespective of their role as major IL-17 producers.
Keywords: autoimmune diabetes, interleukin-17, non-obese diabetic (NOD), transforming growth factor-β, γδ T cells
Introduction
The non-obese diabetic (NOD) mouse spontaneously develops autoimmune diabetes and represents a good model for human type 1 diabetes.1 The pathogenic roles of T helper type 1 (Th1) cells in autoimmune diabetes have been well defined. Th1 cell cytokines, such as interleukin (IL)-12 and interferon (IFN)-γ, have been implicated in promoting diabetogenesis, although there have been recent reports that IFN-γ is non-pathogenic in the later phase of diabetes.2 Therapies designed to balance the Th1/Th2 response in NOD mice have been tested and found to work well in preventing diabetes in this model, which finally defined the pathogenic roles of Th1 cells in type 1 diabetes in these mice.3,4 However, the aetiology of autoimmune diabetes is complex and the mechanisms involved need to be explored to find more effective intervention strategies.
Recently, a new CD4+ T-cell lineage, the Th17 cell lineage, was identified and has been found to be involved in many autoimmune and hypersensitivity diseases, such as multiple sclerosis, rheumatoid arthritis and asthma.5–8 IL-17 knockout or neutralization significantly delays or prevents the disease process in collagen-induced arthritis and experimental autoimmune encephalomyelitis (EAE).6,7 However, the pathogenesis of the prevalent autoimmune disease, type 1 diabetes, and the role of Th17 cells in it remain largely unclear. Recently, using polarized Th17 and Th1 cells from NOD.BDC transgenic mice as cell models, some groups reported that Th17 cells play a less important role in the pathogenesis of diabetes than Th1 cells.9,10 However, under natural conditions, the production and role of IL-17 in the progress of diabetes in NOD mice remain largely unclear.
In addition to Th17 cells, γδ T-cell receptor (TCR)-bearing cells (γδ T cells) are an important source of IL-17.11,12 In contrast to conventional T cells, γδ T cells constitute only a small proportion (1–5%) of the lymphocytes that circulate in the blood and peripheral organs. The use of mice genetically engineered to be deficient in γδ T cells in experimental models of infectious and autoimmune diseases, including autoimmune diabetes, has focused attention on the immunoregulatory role of these cells.13–19 However, the mechanisms used by γδ T cells and the identity and molecular nature of the cells with which they interact have not been fully determined.20 As γδ T cells have recently been shown to express high levels of IL-17,11 it is of interest to evaluate the relationship between IL-17-producing γδ T cells and autoimmune diseases.
To investigate the roles of Th17 cells and the IL-17 they produce in the pathogenesis of autoimmune diabetes, we first measured IL-17 production in diabetic and non-diabetic NOD mice. We then performed in vivo IL-17 neutralization to evaluate the effects on the pathogenesis of diabetes. The kinetics of IL-17 and IFN-γ production during the disease process in NOD mice were also followed and compared. Importantly, we found that γδ T cells from NOD mice were dominated by IL-17+ T cells, and therefore studied the roles of IL-17-producing γδ T cells in the pathogenesis of autoimmune diabetes and the possible mechanisms involved.
Materials and methods
Mice
Diabetes-prone NOD mice and immunodeficient NOD-SCID mice (used for all transfer experiments except one in which γδ T cells were transferred into NOD mice to examine their possible regulatory effect) were originally obtained from the Jackson Laboratory (Jersey, NJ), and then bred in our facilities under specific pathogen-free conditions. The care, use and treatment of mice in this study were in strict agreement with the guidelines for the care and use of laboratory animals of the Institute of Basic Medical Sciences. In our institute, the incidence of spontaneous diabetes in NOD mice is 80–90% at 30 weeks of age. Blood glucose levels were measured biweekly or every other day (for NOD-SCID recipients) and the mice were considered to have type 1 diabetes when glucose levels exceeded 11·3 mm, as, in our facilities, spontaneous decreases after this level has been reached are rarely observed.
Isolation of γδ T cells
γδ T cells were isolated from splenocytes using Microbeads (Miltenyi Biotec Inc., Bergish Gladbach, Germany) according to the manufacturer’s protocol. Seven- to eight-week-old non-diabetic female NOD mice were used as the source of γδ T cells. Briefly, splenocytes were collected and lymphocytes prepared by Ficoll centrifugation and washed twice with phosphate-buffered saline (PBS), pH 7·2, containing 0·5% bovine serum albumin (BSA) and 2 mm ethylenediaminetetraacetic acid (EDTA) (PBS-BSA-EDTA). An aliquot containing 107 cells was then spun down and the cells re-suspended in 50 μl of PBS-BSA-EDTA, and then 5 μl of phycoerythrin (PE)-conjugated anti-γδ antibody (GL3; eBiosciences, San Diego, CA) was added and the mixture was incubated at 4° for 15 min. After two PBS-BSA-EDTA washes, the cells were re-suspended in 40 μl of PBS-BSA-EDTA and 10 μl of anti-PE Microbeads (Miltenyi Biotec Inc.) was added and the mixture was incubated at 4° for 15 min. Finally, after two PBS-BSA-EDTA washes, the cells were applied to MACS Separator columns (Miltenyi Biotec Inc.) and separated into bound and non-bound fractions. The bound fraction was eluted and the purity of the isolated cell fraction determined by flow cytometry analysis (95% pure γδ T cells). Data collection and analysis were performed by FACScaliber flow cytometry (BD Biosciences, San Jose, CA) using cellquest software (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA) measurement of cytokine production
Cytokine secretion by splenocytes or pancreatic draining lymph node cells was determined by ELISA as recommended by the kit manufacturer (eBioscience). Briefly, cells from female NOD mice (5 × 105) were incubated in 96-well flat-bottomed microtitre plates with 0·1 μg/ml of anti-CD3 antibody (1452C11) in the presence or absence of 10 ng/ml of recombinant mouse IL-23 (eBioscience) and the supernatants were harvested after 48 hr. Levels of IL-17, IFN-γ, TGF-β and IL-10 were determined in triplicate in 0·1 ml of supernatant using a sandwich ELISA method. For TGF-β analysis, samples were first activated with acid (1N HCL), followed by neutralization with 1N NaOH as described in the manufacturer's protocol to determine active TGF-β levels.
Adoptive transfer experiments
For disease transfer using splenocytes from diabetic NOD mice (‘diabetic splenocytes’) and for the IL-17 neutralization experiment, 5 × 106 diabetic splenocytes were injected intravenously (i.v.) into female NOD-SCID mice (4 to 6 weeks old). Anti-IL-17 antibody (50104·11; R&D Systems, Minneapolis, MN; 200 μg/mouse) was injected i.v. on the day of transfer, and two additional i.v. injections were given on days 4 and 16 after transfer.21 Mice receiving diabetic splenocytes plus immunoglobulin G (IgG) isotype antibody or splenocytes from 6- to 8-week-old non-diabetic mice (‘naïve splenocytes’) were used as controls.
To examine whether γδ T cells protected against the transfer of diabetes, (i) 1·5 × 106 purified γδ T cells from 8-week-old non-diabetic NOD mice were injected i.v. into 7-week-old female NOD mice or (ii) purified γδ T cells from 8-week-old non-diabetic NOD mice were co-transferred with diabetic splenocytes at different ratios into female NOD-SCID mice (4 to 6 weeks old).
Blood glucose levels were then monitored.
Administration of anti-TGF-β antibody
Splenocytes from 15-week-old diabetic NOD mice (5 × 106) and γδ T cells from 8-week-old non-diabetic NOD mice (1·5 × 106) were co-transferred into NOD-SCID mice. One group of recipients was injected i.v. with 0·5 mg of anti-TGF-β antibody (1D11; mouse IgG1; R&D Systems) every 4–5 days and the other group received the same amount of control mouse IgG.22 NOD-SCID recipients receiving 5 × 106 diabetic splenocytes were used as positive controls.
Flow cytometry
For staining for CD4, CD8 or γδ TCR, splenocytes or cells from the pancreatic draining lymph nodes were incubated for 30 min at 4° with PE-conjugated anti-CD4 or anti-CD8 antibodies or fluorescein isothiocyanate (FITC)-conjugated anti-γδ TCR antibodies (all from Biolegend, San Diego, USA), and then washed and analysed. For intracellular IL-17 staining, cells were stimulated in vitro for 4 hr with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA), 1 μg/ml of ionomycin, and 1 μg/ml of brefeldin A (Sigma, St Louis, MO), and then stained with FITC-conjugated anti-γδ TCR antibodies for 30 min at 4°. They were then washed, fixed overnight with Fix/Perm buffer (eBioscience), washed with permeabilization buffer (eBioscience), and stained with PE-conjugated anti-IL-17 antibodies (clone FJK-16s; eBioscience) and analysed on a FACScalibur flow cytometer.
Histology
The pancreas was removed from NOD-SCID mice and fixed in 10% buffered formalin, embedded in paraffin, sectioned at 4·5 μm, and stained with haematoxylin and eosin (H&E). Insulitis grade was determined as follows: 0, normal islet; 1, mononuclear infiltration, largely in the periphery, in < 25% of the islet; 2, 25–50% of the islet showing mononuclear infiltration; 3, > 50% of the islet showing mononuclear infiltration; and 4, small, completely destroyed islet with few mononuclear cells.
Statistical analysis
The log-rank test was used for analysis of the incidence of diabetes in the experimental and control groups. For the remainder of the experiments, P-values were calculated with the two-tailed Student’s unpaired t-test. A P-value of ≤ 0·05 was considered to be statistically significant.
Results
In vivo neutralization of IL-17 does not prevent the development of adoptively transferred diabetes
To examine the role of IL-17 in the pathogenesis of diabetes in diabetes-prone NOD mice, we first compared IL-17 production in diabetic and non-diabetic NOD mice at about 15 weeks of age. As serum IL-17 was undetectable (data not shown), in vitro IL-17 production by lymphocytes was measured after 48 hr of stimulation with anti-mouse CD3 antibody (0·1 μg/ml). Our data showed that splenocytes from diabetic mice (‘diabetic splenocytes’) produced significantly higher levels of IL-17 than those from non-diabetic age-matched NOD mice (Fig. 1a). To directly test whether IL-17 played an important role in the pathogenesis of autoimmune diabetes, a diabetes cell transfer study in immunodeficient NOD-SCID mice was performed in which neutralizing antibody was given simultaneously to block IL-17 function. We found that mice co-injected with diabetic splenocytes and anti-IL-17 antibody or isotype control showed a comparable incidence of diabetes with no significant difference in onset (Fig. 1b), indicating that neutralization of IL-17 did not prevent transfer of diabetes by diabetic splenocytes. Proof that the antibody was effective in vivo was obtained by examining the effect of neutralization on the IL-17-producing ability of recipient T cells when the mice were killed at 4 weeks after cell transfer. In response to stimulation with anti-CD3 antibody, T cells from anti-IL-17 antibody-injected NOD-SCID mice showed significantly decreased IL-17 production compared with isotype control-injected mice (data not shown). These results show that antibody administration efficiently blocked IL-17 production, but did not prevent diabetes in NOD mice.
Figure 1.
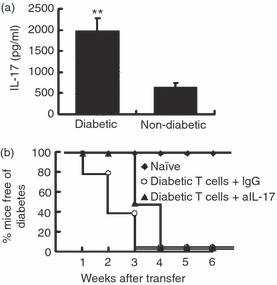
In vivo neutralization of interleukin (IL)-17 does not protect against diabetes transferred by diabetic splenocytes. (a) Splenocytes from 15-week-old female diabetic or age- and sex-matched non-diabetic non-obese diabetic (NOD) mice were stimulated in vitro for 2 days with anti-CD3 antibody (0·1 μg/ml) and IL-23 (10 ng/ml), and then the supernatants were collected and tested for IL-17 production by enzyme-linked immunosorbent assay (ELISA) (n = 4 per group). Cells cultured with medium alone were used as a negative control. IL-17 was undetectable in the control well (data not shown). **P < 0·01 by Student’s t-test. (b) One group of NOD–severe combined immunodeficiency (SCID) mice were injected intravenously (i.v.) with non-diabetic splenocytes (from 6- to 8-week-old mice) and left untreated, while two groups were injected with diabetic splenocytes (5 × 106 per mouse), and then, on days 0, 4 and 16, with 200 μg of either rat anti-mouse IL-17 antibody (50104·11; R&D Systems) or control rat immunoglobulin (IgG), and blood glucose was measured weekly. Compared with IgG-treated controls, anti-IL-17 antibody administration did not reduce the incidence of diabetes in NOD-SCID recipients (P > 0·05 by log-rank test). The data shown are representative of those for three independent experiments. In each experiment, 10 mice were used in each group.
IL-23-expanded diabetic splenocytes produce higher levels of IL-17 than non-treated cells, but induce comparable levels of diabetes
To test the role of IL-17 in the pathogenesis of diabetes in another way, diabetic splenocytes were expanded by incubation for 24 hr with anti-mouse CD3 antibody with or without recombinant mouse IL-23 (10 ng/ml). Compared with the non-IL-23-treated cells, the diabetic splenocytes expanded in the presence of IL-23 expressed higher levels of IL-17 (Fig. 2a) and comparable levels of IFN-γ (Fig. 2b). When the two different preparations of diabetic splenocytes were transferred into NOD-SCID mice, they also induced a comparable incidence of diabetes (Fig. 2c). This again suggests that the presence of IL-17 did not increase the incidence of diabetes development. As IFN-γ is a signature cytokine of Th1 cells, which might be involved in the onset of diabetes, we compared the kinetics of IL-17 and IFN-γ production by splenocytes in response to stimulation with anti-mouse CD3 antibody (0·1 μg/ml) in NOD mice at different ages. We found that production of IFN-γ peaked at 7 weeks of age, the age of insulitis onset in NOD mice, and then gradually decreased, while IL-17 production remained at a similar level or increased slightly with age (Fig. 2d), which, to some extent, suggests that Th1 cells, and not Th17 cells, are involved in the pathogenesis of type 1 diabetes in NOD mice. Similar results were obtained using pancreatic draining lymph nodes (data not shown).
Figure 2.

Interleukin (IL)-23-expanded diabetic splenocytes produced higher levels of IL-17, but induced comparable diabetes in non-obese diabetic–severe combined immunodeficiency (NOD-SCID) mice compared with non-treated diabetic splenocytes. (a, b) Diabetic splenocytes were expanded in vitro for 24 hr using anti-mouse CD3 antibody with or without IL-23, and then the supernatants were collected and analysed for IL-17 (a) and interferon (IFN)-γ (b) production (n = 4 per group). **P < 0·01 by Student’s t-test. (c) Diabetic splenocytes expanded with anti-mouse CD3 antibody plus recombinant IL-23 (rIL-23) or medium were transferred into NOD-SCID mice, and blood glucose levels were followed (n = 10 per group). Compared with the medium-cultured controls, IL-23-expanded diabetic splenocytes did not increase the incidence of diabetes in NOD-SCID recipients (P > 0·05 by log-rank test). (d) Female mice of different ages were killed and their splenocytes stimulated in vitro for 48 hr with anti-CD3 antibody, and then the supernatants were tested for IL-17 and IFN-γ by enzyme-linked immunosorbent assay (ELISA) (n = 4 per group). The data are representative of those for two independent assays.
IL-17 is mainly expressed by CD4− CD8−γδ T cells in NOD mice
Next, to test the potential role of IL-17 in the pathogenesis of type 1 diabetes in another way, IL-17-producing cells and their effects in NOD mice were examined. IL-17 was found to be mainly expressed by γδ T cells, and not CD4+ or CD8+ T cells (Fig. 3a), and this was more evident when T cells were in vitro expanded with recombinant mouse IL-23 (Fig. 3b). Finally, purified γδ T cells were tested for IL-17 expression and production and the results showed that purified γδ T cells were dominated by IL-17-expressing cells (Fig. 3c) and, in response to IL-23 stimulation, produced 10-fold more IL-17 than γδ TCR-negative splenocytes (Fig. 3d).
Figure 3.

Interleukin (IL)-17 expression in non-obese diabetic (NOD) mice is dominated by CD4− CD8−γδ+ T cells. (a, b) Freshly isolated (a) or IL-23-expanded (b) splenocytes from female NOD mice were double-stained with fluorescein isothiocyanate (FITC)-conjugated antibodies against CD4, CD8 or γδ T-cell receptor (TCR) and phycoerythrin (PE)-conjugated anti-IL-17 antibody (n = 3–6 per group). (c) γδ T cells were purified as described in the Materials and methods, and then the purified γδ T cells (> 95% purity) were double-stained with FITC-conjugated antibody against the γδ TCR and PE-conjugated anti-IL-17 antibody. (d) Purified γδ+ T cells and splenocytes lacking γδ TCR (γδ–) were stimulated for 48 hr with anti-mouse CD3 antibody and IL-23, and then the supernatants were collected and assayed for IL-17. The data are representative of those for three independent assays. **P < 0·01 by Student’s t-test.
IL-17-producing γδ T cells protect NOD mice from the induction of diabetes
As γδ T cells produce high levels of IL-17, it was of interest to examine their role in autoimmune diabetes. To investigate this, we first examined the percentage of γδ T cells in NOD mice and found that the percentage of γδ T cells in the peripheral lymphocytes was higher in non-diabetic than in age-matched diabetic NOD mice (Fig. 4a), and this difference was even more marked after the T cells had been expanded with recombinant IL-23 in vitro (Fig. 4b). Similar results were obtained with lymphocytes from the pancreatic draining lymph nodes (data not shown). This suggested that γδ T cells might have a protective effect in NOD mice. To examine whether there were differences between γδ T cells derived from non-diabetic and diabetic NOD mice, IL-17 production by γδ T cells were analysed, but the two populations produced comparable levels of IL-17 (Fig. 4c). To directly investigate the effects of IL-17-producing γδ T cells in NOD mice, γδ T cells from non-diabetic NOD mice were purified and injected into female 7-week-old NOD mice and different ratios of purified γδ T cells from non-diabetic NOD mice and diabetic splenocytes were co-transferred into NOD-SCID mice. Unexpectedly, we found that IL-17-producing γδ T cells did not induce diabetes in NOD mice, but actually decreased its incidence (P = 0·0046 by log-rank test) (Fig. 5a). In addition, the co-transfer study showed that γδ T cells had a marked effect in protecting NOD-SCID mice from the insulitis and diabetes induced by diabetic splenocytes (Fig. 5b–d) (P < 0·001).
Figure 4.

Non-diabetic non-obese diabetic (NOD) mice have a higher percentage of γδ+ T cells than diabetic mice. (a, b) The percentage of γδ T cells in (a) freshly isolated splenocytes and (b) interleukin (IL)-23-expanded splenocytes from diabetic and non-diabetic NOD mice of the same age was determined by flow cytometry. (c) γδ T cells isolated from 8-week-old non-diabetic and diabetic NOD mice were stimulated for 48 hr with anti-mouse CD3 antibody and IL-23, and then the supernatants were collected and assayed for IL-17. Six mice were used in each group. The data are representative of those for three independent assays.
Figure 5.

Purified γδ+ T cells protect non-obese diabetic (NOD) mice from diabetes. (a) γδ T cells (1·5 × 106) were injected intravenously (i.v.) into 7-week-old female NOD mice, untreated age- and sex-matched NOD mice being used as controls, and blood glucose was measured biweekly. γδ T cells protected NOD mice from diabetes (P= 0·0046 by the log-rank test). The data are representative of those for two independent experiments. More than nine mice were used in each group. (b) Diabetic splenocytes (5 × 106) or purified γδ T cells (5 × 106) alone or diabetic splenocytes (5 × 106) plus 1·5 × 106 (30%γδ T cells) or 7·5 × 105 (15%γδ T cells) purified γδ T cells were injected i.v. into NOD–severe combined immunodeficiency (SCID) mice and blood glucose was measured weekly. Both 30%γδ T cells (P < 0·001) and 15%γδ T cells (P < 0·01) protected NOD-SCID recipients from diabetes (by the log-rank test). Ten mice were included in each experimental group. The data are representative of those for three independent assays. (c) Photomicrographs of representative islets from NOD-SCID mice 6 weeks after transfer of diabetic splenocytes, diabetic splenocytes plus 30%γδ T cells, or diabetic splenocytes plus 15%γδ T cells. Animals (n = 6 per group) were killed and a histological examination of the pancreatic islets (at least 60 islets per mouse) was performed on sections stained with haematoxylin and eosin (H&E). Transfer of γδ T cells alone does not cause insulitis (data not shown). The photomicrographs shown are representative of those from more than five animals per group. (d) The insulitis score in the animals in (c) was determined as described in the Materials and methods.
γδ T cells protect NOD mice from diabetes by up-regulating TGF-β
The mechanism by which γδ T cells protect against diabetes was then examined. As IL-17 can inhibit the production of IFN-γ,23–25 we first examined whether γδ T cells protected against diabetes by suppressing IFN-γ production. To investigate this, splenocytes from NOD-SCID mice at 4 weeks after transfer of diabetic splenocytes or co-transfer of γδ T cells and diabetic splenocytes were stimulated with anti-mouse CD3 antibody, and then, 48 hr later, the supernatants were collected and IL-17 and IFN-γ production measured by ELISA. As shown in Fig. 6a, similar amounts of IFN-γ were produced by splenocytes from NOD-SCID mice that had received both γδ T cells and diabetic splenocytes or only diabetic splenocytes, indicating that the protective effect of γδ T cells was not mediated by suppression of IFN-γ production. Co-transfer of γδ T cells with splenocytes significantly up-regulated IL-17 production (Fig. 6b). To investigate the effect of γδ T cells on IFN-γ production in another way, γδ T cells were isolated and co-cultured with diabetic T cells and anti-mouse CD3 antibody, and then, 48 hr later, the supernatants were collected and IFN-γ and IL-17 production measured by ELISA. Again, the γδ T cells did not suppress the production of IFN-γ by diabetic splenocytes (data not shown), suggesting that γδ T cells protected NOD mice from diabetes via a mechanism not involving IFN-γ.
Figure 6.

Protection by γδ T cells does not involve suppression of interferon (IFN)-γ production. (a, b) Splenocytes from non-obese diabetic–severe combined immunodeficiency (NOD-SCID) mice that had undergone transfer of γδ T cells or diabetic splenocytes or diabetic splenocytes plus 30%γδ T cells were isolated and stimulated for 48 hr with anti-CD3 antibody and interleukin (IL)-23, and then the supernatants were assayed for IFN-γ (a) or IL-17 (b) (n = 4 per group). The data are representative of those for three independent assays. *P < 0·05 by Student’s t-test.
As many reports have shown that γδ T cells exert regulatory effects by producing TGF-β and/or IL-10,13,15,17 we measured levels of TGF-β and IL-10 in NOD-SCID mice that had undergone transfer of γδ T cells or diabetic splenocytes or co-transfer of γδ T cells plus diabetic splenocytes. TGF-β levels were significantly increased in the γδ T cell-protected NOD mice (Fig. 7a), but up-regulation of IL-10 was not seen (data not shown). When cells were also tested for TGF-β expression after in vitro stimulation with anti-CD3 antibody and IL-23, purified γδ T cells were found to produce higher levels of TGF-β than γδ-negative T cells (Fig. 7b), suggesting that TGF-β is the critical cytokine in the protection against diabetes provided by γδ T cells.
Figure 7.

Involvement of transforming growth factor (TGF)-β in γδ T cell-mediated protection against diabetes. (a) Serum was collected from non-obese diabetic–severe combined immunodeficiency (NOD-SCID) mice that had undergone transfer of non-diabetic naïve splenocytes (from 6- to 8-week-old mice), γδ T cells, diabetic splenocytes or diabetic splenocytes plus 30%γδ T cells and assayed for TGF-β (n = 5 per group). (b) Purified γδ+ and γδ− T cells were stimulated for 48 hr with anti-CD3 antibody and interleukin (IL)-23, and then the supernatants were assayed for TGF-β. The data are representative of those for three independent experiments. *P < 0·05; **P < 0.01 by Student’s t-test.
The protective action of γδ T cells is inhibited by anti-TGF-β antibody
To determine whether this up-regulation of TGF-β in NOD-SCID mice that had received γδ T cells was functionally relevant to the inhibition of diabetes, diabetic splenocytes and purified γδ T cells were co-transferred into NOD-SCID mice, half of which were injected with anti-TGF-β antibody every 4–5 days and half of which were injected with control mouse IgG. All the mice given control IgG were diabetes-free at 6 weeks after transfer, while 50% of those given anti-TGF-β antibody developed diabetes by 3 weeks after transfer (P < 0·001 by the log-rank test) (Fig. 8), showing that TGF-β is important in the protection against diabetes afforded by γδ T cells.
Figure 8.

The protective effect of γδ T cells against diabetes is inhibited by anti-transforming growth factor (TGF)-β antibodies. Diabetic splenocytes (5 × 106) plus 1·5 × 106γδ T cells were injected intravenously (i.v.) into non-obese diabetic–severe combined immunodeficiency (NOD-SCID) mice, and then one group was left untreated, while two others were injected i.v. with either 0·5 mg of anti-TGF-β antibody [1D11; mouse immunoglobulin G1 (IgG1)] or the same amount of normal mouse IgG on the day of transfer, and then every 4–5 days. NOD-SCID mice that only received diabetic splenocytes (5 × 106) were used as a positive control. Blood glucose was measured weekly. Anti-TGF-β antibodies significantly inhibited the protective effect of γδ T cells in vivo (P < 0·001 by the log-rank test). The data are representative of those for three independent assays. In each experiment, 10 mice were used in each group.
Discussion
The roles of Th17 cells in autoimmune disease remain largely unclear. In the present study, we found that, although diabetic T cells produced higher levels of IL-17 than non-diabetic cells, in vivo neutralization of IL-17 did not prevent or delay the development of adoptively transferred diabetes. This suggests that the role of IL-17 in autoimmune diabetes is not as critical as in multiple sclerosis or rheumatoid arthritis, in which the disease can be delayed or prevented by neutralization or knock-out of IL-17.6–8 Although some groups reported that Th17 cells play a less important role in the pathogenesis of diabetes than Th1 cells,9,10 the precise roles of IL-17 and IL-17-producing cells in natural conditions in autoimmune diabetes remain to be investigated.
It is well known that IL-23 is the most important cytokine for expanding Th17 cells and promoting IL-17 production, but is less effective in promoting IFN-γ production.23,25 Thus, IL-23 significantly enhanced IL-17 production in diabetic T cells, while IFN-γ secretion was not affected compared with the non-IL-23-treated cells (Fig. 2a,b). However, transfer of diabetic splenocytes expanded by IL-23 did not result in an increased incidence of diabetes in recipients (Fig. 2c), again indicating that the presence of IL-17 did not increase the incidence of diabetes development in NOD mice.
IFN-γ was found to be involved in the pathogenesis of autoimmune diabetes in NOD mice in early reports.2 When the kinetics of IL-17 and IFN-γ production in NOD mice were examined, we found that IFN-γ production peaked at the age of onset of insulitis in NOD mice, while IL-17 production was virtually unchanged with age. This result contrasts with the pattern seen in EAE, in which the IL-17 peak appears earlier than the IFN-γ peak.26 These data again suggest that IFN-γ and not IL-17 is involved in the progression of diabetes.
Many types of cell, including CD4+, CD8+ and γδ T cells, are known to produce IL-17.11 What are the IL-17-producing cells in NOD mice and do they affect the process of diabetes? IL-17-producing cells in NOD mice were identified and their function investigated. Our data showed that (i) γδ T cells were the major source of IL-17 in NOD mice, (ii) γδ T cells were dominated by IL-17-expressing cells, and (iii) purified γδ T cells produced 10 times more IL-17 than splenocytes lacking γδ TCR in response to stimulation by anti-mouse CD3 antibody and IL-23. These data suggested that γδ T cells might dominate the IL-17 response in some circumstances, which is in accordance with the findings of other studies.11,27.
As the γδ T cells were dominated by IL-17-producing cells, the function of γδ T cells in diabetes is of interest. Different roles of γδ T cells have been reported in inflammatory and autoimmune diseases. Some reports showed that γδ T cells in type 1 diabetes and other autoimmune diseases are protective or regulatory,13–19 while others showed that they could either act as antigen-presenting cells to initiate immunity or provide a first line of defence through the recognition of self-antigens expressed on damaged or stressed cells.28,29 These results show that γδ T cells perform different functions according to their tissue distribution, antigen-receptor structure, and local microenvironment.
The roles of IL-17-producing γδ T cells in autoimmune diabetes were subsequently examined. When purified γδ T cells were co-transferred with diabetic T cells at different ratios, γδ T cells showed regulatory effects in NOD mice. Consistent with the above results, the percentage of γδ T cells was found to be lower in the diabetic than in the non-diabetic NOD mice.
We then examined the mechanism by which IL-17-producing γδ T cells prevented diabetes in NOD-SCID mice. Because some reports have shown that expression of IL-17 and IFN-γ is mutually suppressive,23,24 we first tested whether the higher level of IL-17 production by γδ T cells suppressed IFN-γ production. Our data showed that IFN-γ production was not suppressed in the γδ T cell-protected mice and IL-17 production was positively correlated with the number of transferred γδ T cells (Fig. 6). Why IFN-γ production was not suppressed might be explained as follows: (i) γδ T cells might function as antigen-presenting cells and promote the production of IFN-γ by splenocytes;28 and (ii) as shown by Peng,18 regulatory γδ T cells might secrete IFN-γ. Our data suggested that γδ T cells protect NOD mice from diabetes through a mechanism not involving IFN-γ.
As γδ T cells have been reported to have regulatory effects by producing TGF-β and/or IL-10,13,15,17 we examined whether γδ T cells protected NOD mice via a mechanism involving TGF-β and/or IL-10. To investigate this, we first measured the production of TGF-β and IL-10 by purified γδ T cells in response to anti-mouse CD3 antibody and IL-23. Purified γδ+ T cells secreted significantly more TGF-β than γδ− T cells. However, no significant difference in IL-10 production was seen between the two groups (data not shown). Our data indicate that γδ T cells might exert regulatory effects by producing TGF-β. To further determine whether transferred γδ T cells protected NOD mice from diabetes by up-regulating TGF-β production in vivo, TGF-β levels were measured in serum collected from recipient NOD-SCID mice that had undergone transfer of γδ T cells, diabetic splenocytes, or non-diabetic splenocytes alone or γδ T cells plus diabetic splenocytes. As expected, γδ T cell-protected NOD mice produced significantly higher levels of TGF-β than control mice (Fig. 7). To confirm this, an in vivo TGF-β neutralization study was performed and, as shown in Fig. 8, administration of anti-TGF-β antibody blocked the γδ T cell-mediated protection against diabetes (P < 0·001), indicating that TGF-β is involved in the mechanism underlying the γδ T cell-mediated protection.
Recently, there have been reports that the function of γδ T cells is subset-dependent.20,30 However, on the question of whether one γδ T cell subset produces IL-17 while another produces TGF-β, the results are not consistent. For example, Roark30 showed that the vγ4 subset is the major source of IL-17 and that the vγ1 subset produces virtually no IL-17. However, Romani et al.31 found that the vγ1 subset mainly produces IL-17 and the vγ4 subset mainly produces IL-10. Again, this suggests that γδ T cells perform different functions depending on their tissue distribution and local microenvironment.
Here we showed that IL-17-producing γδ T cells were suppressive. However, the roles of IL-17-producing CD4+ Th17 cells in NOD mice remain to be determined. Recent findings obtained by McGeachy,32 who showed that CD4+ Th17 cells that co-produce IL-10 have regulatory properties, suggest that the presence of IL-17 may not increase the chance of disease development, but the ability of IL-17-producing cells to secrete other cytokines may be a major determinant in their function as pathogenic or tolerogenic cells.
Overall, our data suggest that IL-17 is not the critical pro-inflammatory cytokine in the pathogenesis of autoimmune diabetes in NOD mice. γδ T cells, the major source of IL-17 in NOD mice, protect NOD mice from diabetes in a TGF-β-dependent way.
Acknowledgments
This work was supported by the National Natural Sciences Foundation of China (Grant No. 30801029) and a National ‘973’ Fund Grant of China (No. 2007CB512406).
Glossary
Abbreviation:
- NOD
non-obese diabetic
Disclosures
The authors have no financial conflict of interest.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 2.Savinov AY, Wong FS, Chervonsky AV. IFN-g affects the homing of diabetogenic T cells. J Immunol. 2001;167:6637–43. doi: 10.4049/jimmunol.167.11.6637. [DOI] [PubMed] [Google Scholar]
- 3.Han G, Li Y, Wang J, et al. Active tolerance induction and prevention of autoimmune diabetes by immunogene therapy using recombinant adenoassociated virus expressing glutamic acid decarboxylase 65 peptide GAD (500-585) J Immunol. 2005;174:4516–24. doi: 10.4049/jimmunol.174.8.4516. [DOI] [PubMed] [Google Scholar]
- 4.Cooke A, Phillips JM, Parish NM. Tolerogenic strategies to halt or prevent type 1 diabetes. Nat Immunol. 2001;2:810. doi: 10.1038/ni0901-810. [DOI] [PubMed] [Google Scholar]
- 5.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–7. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 7.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto T, Akiyama K, Kobayashi N, Mori A. Comparison of IL-17 production by helper T cells among atopic and nonatopic asthmatics and control subjects. Int Arch Allergy Immunol. 2005;137:51–4. doi: 10.1159/000085432. [DOI] [PubMed] [Google Scholar]
- 9.Bending D, De La Pea H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A. Highly purified Th17 cells from BDC2·5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009 doi: 10.1172/JCI37865. doi 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–24. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by γδT cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 12.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vγ1γδT cells control early infiltration of Neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–72. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- 13.Bhagat G, Naiyer AJ, Shah JG, Harper J, Jabri B, Wang TC, Green PH, Manavalan JS. Small intestinal CD8+TCRγδ+NKG2A+ intraepithelial lymphocytes have attributes of regulatory cells in patients with celiac disease. J Clin Invest. 2008;118:281–93. doi: 10.1172/JCI30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison LC, DempseyCollier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gd T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184:2167–74. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seo N, Tokura Y, Takigawa M, Egawa K. Depletion of IL-10- and TGF-b-producing regulatory gd T cells by administering a daunomycin -conjugated specific monoclonal antibody in early tumor lesions augments the activity of CTLs and NK cells. J Immunol. 1999;163:242–9. [PubMed] [Google Scholar]
- 16.Locke NR, Stankovic S, Funda DP, Harrison LC. TCR gd intraepithelial lymphocytes are required for self-tolerance. J Immunol. 2006;176:6553–9. doi: 10.4049/jimmunol.176.11.6553. [DOI] [PubMed] [Google Scholar]
- 17.Nagaeva O, Jonsson L, Mincheva-Nilsson L. Dominant IL-10 and TGF-b mRNA expression in gdT cells of human early pregnancy decidua suggests immunoregulatory potential. Am J Reprod Immunol. 2002;48:9–17. doi: 10.1034/j.1600-0897.2002.01131.x. [DOI] [PubMed] [Google Scholar]
- 18.Peng G, Wang H, Peng W, Kiniwa Y, Seo KH, Wang R. Tumor-infiltrating gammadelta T cells suppress T and dendritic cell function via mechanisms controlled by a unique Toll-like receptor signaling pathway. Immunity. 2007;27:334–48. doi: 10.1016/j.immuni.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Tschöp J, Martignoni A, Goetzman HS, et al. gdT cells mitigate the organ injury and mortality of sepsis. J Leukoc Biol. 2008;83:1–8. doi: 10.1189/jlb.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carding SR, Egan PJ. γδT cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–45. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 21.Kohyama S, Ohno S, Isoda A, et al. IL-23 enhances host defense against Vaccinia virus infection via a mechanism partly involving IL-17. J Immunol. 2007;179:3917–25. doi: 10.4049/jimmunol.179.6.3917. [DOI] [PubMed] [Google Scholar]
- 22.Yu S, Sharp GC, Braley-Mullen H. TGF-beta promotes thyroid epithelial cell hyperplasia and fibrosis in IFN-gamma deficient NOD.H-2h4 Mice. J Immunol. 2008;181:2238–45. doi: 10.4049/jimmunol.181.3.2238. [DOI] [PubMed] [Google Scholar]
- 23.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–68. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 24.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 25.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn T, Reddy J, Gao W, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:4. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roark CL, Simonian PL, Fontenot AP, Born WB, O'Brien RL. Gamma delta T cells: an important source of IL-17. Curr Opin Immunol. 2008;20:1–5. doi: 10.1016/j.coi.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng L, Cui Y, Shao H, et al. Mouse γδ T cells are capable of expressing MHC class II molecules, and of functioning as antigen-presenting cells. J Neuroimmunol. 2008;203:3–11. doi: 10.1016/j.jneuroim.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thedrez A, Sabourin C, Gertner J, Devilder MC, Allain-Maillet S, Fournié JJ, Scotet E, Bonneville M. Self/non-self discrimination by gammadelta T cells: simple solutions for a complex. Immunol Rev. 2007;215:123–35. doi: 10.1111/j.1600-065X.2006.00468.x. [DOI] [PubMed] [Google Scholar]
- 30.Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol. 2007;179:5576–83. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romani L, Fallarino F, De Luca A, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–5. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 32.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-b and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nat Immun. 2007;8:12. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]