

Abstract
Healing of the burn wound is a critical component of the burn patient's successful recovery. While inflammation is a critical component of the healing process, it is unknown whether the inflammatory response differs between non-burn and burn wounds. To study this, mice were subjected to major burn injury or sham procedure. Wound cells were collected by implantation of polyvinyl alcohol sponges beneath the burn site in injured mice or beneath uninjured skin in sham mice (i.e., non-burn wound). Three days thereafter, skin, wound fluid and infiltrating cells were collected for analysis. Significant levels of tumor necrosis factor (TNF)-α, interleukin (IL-6), monocyte chemoattractant protein (MCP)-1 and keratinocyte-derived chemokine (KC) were observed in burn wound tissue and the wound fluid from both non-burn and burn wounds. Burn injury induced 3-fold higher levels of KC and 50-fold higher levels of IL-6 in the wound fluid as compared to non-burn injury. Significant numbers of the cells from both burn and non-burn wounds were CD11b+, GR1+ and F4/80+, suggestive of a myeloid suppressor cell phenotype, whereas CD3+ T-cells were negligible under both conditions. LPS induced TNF-α, IL-6, IL-10, MCP-1, KC and nitric oxide production in both cell populations, however; IL-6, IL-10, MCP-1 and KC levels were suppressed in burn wound cell cultures. These findings indicate that significant differences in the wound inflammatory response exist between burn and non-burn cutaneous wounds and that the unique characteristics of the inflammatory response at the burn site may be an important contributing factor to post-burn wound healing complications.
Keywords: T-cells, trauma, cytokines, chemokines, nitric oxide
Introduction
Thermal injury remains a significant health problem, with approximately 50,000 people in the United States sustaining burn injuries that require hospitalization each year [1]. Despite advances in patient care; immunosuppression, increased susceptibility to sepsis, wound healing complications, and multiple organ failure remain major concerns in this severely compromised and unique patient population. This has been studied extensively in experimental models [2;3]. The proper healing of the burn wound site is critical to the burn patient's successful recovery [4]. The process of wound healing is complex and involves a series of overlapping phases that include: hemostasis, inflammation, proliferation and resolution. The inflammatory phase of wound healing is instrumental in supplying growth factors, cytokines, and chemokines that orchestrate the cellular movement and infiltration necessary for tissue repair [5]. The immune cell subsets (i.e., the cellular components of inflammation) are not only effector cells against invading microbes, but are also important in the catabolic phase of tissue degradation via production of proteases and reactive oxygen intermediates and also in the anabolic phase of tissue formation via production of growth factors [6].
In cutaneous wounds, neutrophils are the first immune cells to arrive and their primary role is wound debridement and protection against microbial infection. Nonetheless, activated neutrophils release proteases and reactive oxygen intermediates, which can lead to significant tissue damage [7]. Macrophages migrate to the wound within 48 hr and become the predominant immune cell type. They are essential to successful wound healing, as they produce large amounts of cytokines, chemokines and growth factors such as platelet derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which initiate the formation of granulation tissue [5]. Macrophage derived growth factors are central to the initiation and propagation of new tissue growth in the wound bed. Studies have shown that macrophage depletion leads to impaired wound debridement and fibroplasia, that isolated wound macrophages induce angiogenesis and that macrophages contribute to increased collagen synthesis and wound breaking strength [8]. Thus, macrophages appear to play a pivotal role in the transition from the inflammatory to the proliferative phase of wound healing. While these previous experimental studies have examined the inflammatory infiltrate in cutaneous wounds, independent of burn injury, systematic analysis of the inflammatory response and infiltrate at the burn wound site has been more limited [9-11]. Based upon the markedly different host responses to traumatic tissue injury (non-burn) and burn injury, it is reasonable to suspect that the cellular and inflammatory responses between these two insults might differ and contribute to differences in healing of the wound site.
Materials and Methods
Animals
C57BL/6 male mice (18-22 g; 8-10 weeks of age, Charles River Laboratories, Wilmington, MA) were used for all experiments. The mice were allowed to acclimatize in the animal facility for at least one week prior to experimentation. Animals were randomly assigned into either a thermal injury group or a sham treatment group. The experiments in this study were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham, and were performed in accordance with the National Institutes of Health guidelines for the care and handling of laboratory animals.
Thermal injury procedure
Mice received a scald burn as described previously [12]. Briefly, the mice were anesthetized by intra-peritoneal injection of ketamine/xylazine and the dorsal surface was shaved. The anesthetized mouse was placed in a custom insulated mold exposing 12.5% of their total body surface area (TBSA) along the right dorsum. The mold was immersed in 70° C water for 10 sec to produce a major burn injury. The mice were then resuscitated with 1 ml of Ringer's lactate solution administered by intra-peritoneal injection and returned to their cages. The cages were placed on a heating pad for 2 hr until the mice were fully awake, at which time they were returned to the animal facility. Sham treatment consisted of resuscitation with Ringer's lactate solution only.
Elicitation/collection of wound immune cell infiltrates
Modification of the methods of Albina and co-workers was employed to elicit and collect wound infiltrates [13]. In brief, immediately following thermal injury, one burn site (right dorsal surface) was prepped with 70% ethanol solution. A 1-cm full-thickness skin incision was made close to the burn edge and a bloodless subcutaneous pocket developed under the burn site. Four sterile polyvinyl alcohol (PVA) sponges (∼6-mm dia. × 3-mm thick; PVA Unlimited, Warsaw, IN) were implanted into the pockets through the skin incision and under the burn site, which was subsequently closed with wound clips. PVA sponges were similarly implanted under uninjured skin in sham animals, creating a non-burn wound. Sponge implantation was done using sterile techniques and no evidence of wound infection was observed. Three days after injury the animals were sacrificed, the incisional wounds reopened, and all of the sponges retrieved. Cell suspensions were obtained from the sponges by compression and resuspended in complete media (RPMI 1640 containing 10% heat inactivated FBS, 5 μg/ml gentimycin and 100 μg/ml of streptomycin and penicillin GibcoBRL, Grand Island, NY). The initial wash in 1 ml PBS was defined as the “wound fluid”.
Collection and processing of skin samples
At 3 days after thermal injury or sham procedure, skin samples from the burn wound and normal uninjured skin were excised down to the level of the musculofascia by sharp dissection and were immediately snap frozen in liquid nitrogen. Burn skin was collected from the left dorsal injury site not used to elicit wound exudate cells. Uninjured skin from burn and sham mice was collected from sites distal to the dorsal surface where the PVA sponges were implanted. All skin samples were stored at −80° C until analysis. Tissue samples were homogenized in protease inhibitor cocktail (Calbiochem, San Diego, CA) as described elsewhere [14]. Cytokine levels in the skin samples were assessed as described below and normalized to μg of protein as determined using the BioRad Protein Assay (Bio-Rad Laboratories, Hercules, CA).
Determination of wound cell phenotype
Wound cells were collected, fixed, stained with hemotoxylin-eosin and visualized by light microscopy using standard techniques. Cellular phenotype was determined by staining with a combination of antibodies (CD3, GR1, CD11b, F4/80) conjugated to either FITC or PE. The manufacturer's suggested methodology was employed (BD Biosciences, Mountain View, CA). Antibodies against CD3, GR1 and CD11b and appropriate isotype controls were obtained from BD Pharmingen. Antibodies against F4/80 and an appropriate isotype control were obtained from Serotec (Raleigh, NC). FITC and PE were analyzed with a LSRII flow cytometer (BD Biosciences). The entire lymphocyte and monocyte populations (as determined for forward and side scatter) were gated in the analysis. A minimum of 10,000 events was collected and WinMIDI 2.8 software was used to analyze the results.
In vitro culture of wound cells
Wound exudate cells (4 × 106/ml) were cultured in complete media for 48 hr with media alone (no stimulation; NS) or LPS (1 μg/ml) and cell-free supernatants collected and stored at −80° C until analysis for cytokine and nitrite/nitrate levels.
Analysis of cytokine and nitrite levels
The concentrations of interleukin (IL)-6, IL-10, IL-12p70, interferon (IFN)-γ, monocyte chemoattractant protein (MCP)-1 and tumor necrosis factor (TNF)-α in the skin homogenates, wound fluids and culture supernatants were measured by commercially available cytometric bead array (CBA) Mouse Inflammation Kits (BD Biosciences), according to the manufacturer's instructions, with some modification. Briefly, 25 μl of mixed capture beads were incubated with 25 μl of supernatant and 25 μl of PE detection reagent for 2 hr at room temperature. The immunocomplexes were then washed and analyzed using the LSRII flow cytometer (BD Biosciences). The data was processed with the accompanying FACSDiva and BD CBA software. Similarly, keratinocyte-derived chemokine (KC) and granulocyte/monocyte- colony stimulating factor (GM-CSF) were measured by CBA Mouse Flex Sets (BD Biosciences) according to the manufacturer's instructions with some modification. In brief, 25 μl of mixed capture beads were incubated with 25 μl of skin homogenates, wound fluid or culture supernatant for 1 hr at room temperature in the dark. Twenty-five μl of PE detection reagent was then added and incubated for 1 hr. The immunocomplexes were then washed twice with wash buffer and centrifuged at 200 × g for 5 min. Analysis was then carried out processed as described above. Cytokine concentrations in the skin homogenates were normalized against protein content. Nitrite/nitrate levels were determined using the Greiss reaction using a kit from Cayman Chemical (Ann Arbor, MI).
Statistical analysis
Data are expressed as mean ± SE. Comparisons were analyzed using ANOVA and Tukey's test for multiple comparisons. A P value of < 0.05 was considered to be statistically significant for all analyses.
Results
Effect of burn injury on inflammatory cytokine content of the skin
Samples of uninjured skin and burn skin were obtained from mice at 3 days post-burn or sham procedure and cytokine content assessed (Fig 1). Burn injury caused a significant (P<0.05) elevation in the levels of TNF-α, IL-6, MCP-1 and KC in samples collected from the burn injury site. In contrast, normal or uninjured skin from burned animals had levels of these mediators that were not significantly different from skin collected from sham mice. While IL-12, IL-10, IFN-γ and GM-CSF were also assessed, significant levels of these mediators were not observed in the samples (data not shown).
Figure 1. Skin cytokine and chemokine levels.

Samples of skin were collected from sham mice and from 2 sites on thermally-injured mice (normal [i.e., uninjured] and burn site) at 3 days after injury. Tissue cytokine and chemokine levels were determined by BD™ Cytometric Bead Array (CBA) analysis as described in the Materials and Methods section. Tissue levels for TNF-α [A], IL-6, [B], MCP-1 [C], and KC [D] were determined. Data are mean ± SEM; n = 4-5 mice/group. * P<0.05 as compared with sham group.
Comparison of the dermal inflammatory response to burn and non-burn injury
Wound fluid was collected from non-burn and burn wound sponges and assessed for inflammatory mediators as shown in Fig. 2. Significant levels of TNF-α, IL-6, MCP-1 and KC were observed. As with the skin samples, IL-12, IL-10, IFN-γ and GM-CSF levels were undetectable. Wound fluid collected from burn sponges had significantly higher levels of IL-6 and KC as compared with non-burn wounds. TNF-α and MCP-1 levels were comparable between the burn and non-burn groups. Protein content of the wound fluid from non-burn and burn wound sponges was similar (2.34 ± 0.33 and 2.19 ± 0.10 mg/ml, respectively; mean ± SE, n=4/group).
Figure 2. Wound fluid cytokine and chemokine levels.

Wound fluid was collected from non-burn and burn wounds at 3 days after injury and cytokine and chemokine levels were determined by BD™ Cytometric Bead Array (CBA) analysis as described in the Materials and Methods section. Wound fluid levels for TNF-α [A], IL-6, [B], MCP-1 [C], and KC [D] were determined. Data are mean ± SEM; n = 5-6 mice/group. * P<0.05 as compared with non-burn group.
Phenotypic analysis of cells from burn and non-burn wounds
Cell yields from the wound sponges were comparable for non-burn and burn wounds (1.25 × 106 and 1.30 × 106, respectively; n=4 sponges/animal). The majority of cells collected displayed morphological characteristics of the monocyte/macrophage lineage (Fig. 3A). The cells were irregular in shape and vacuolated. Another characteristic in many of the cells was a horseshoe-shaped nucleus. Very few of the cells displayed a segmented nucleus characteristic of neutrophils. Morphological characteristics of wound cells were qualitatively similar in non-burn and burn wounds. The scatter pattern (i.e., side scatter for granularity/complexity and forward scatter for size) as determined by FACs analysis showed that the majority of the cells were positioned in the monocyte/macrophage gate and devoid of lymphocytes in both cell populations (Fig. 3B). Cells collected from the wound sponges were also analyzed for lymphoid, myeloid and granulocyte markers. Negligible numbers of CD3+ T-cells (1.5-4.9%) were found in either wound cell population (data not shown). Dual staining of the wound cells revealed that significant numbers of the cells from both populations were positive for both CD11b and GR1 (Fig. 4A). The percentage of this double-positive cell population was comparable in non-burn and burn wounds. In contrast, dual staining for the myeloid markers CD11b and F4/80 revealed that a significantly greater percentage of cells in the burn population were positive for CD11b and F4/80 as compared with non-burn cells (43% and 22%, respectively; Fig. 4B).
Figure 3. Wound cell morphology.
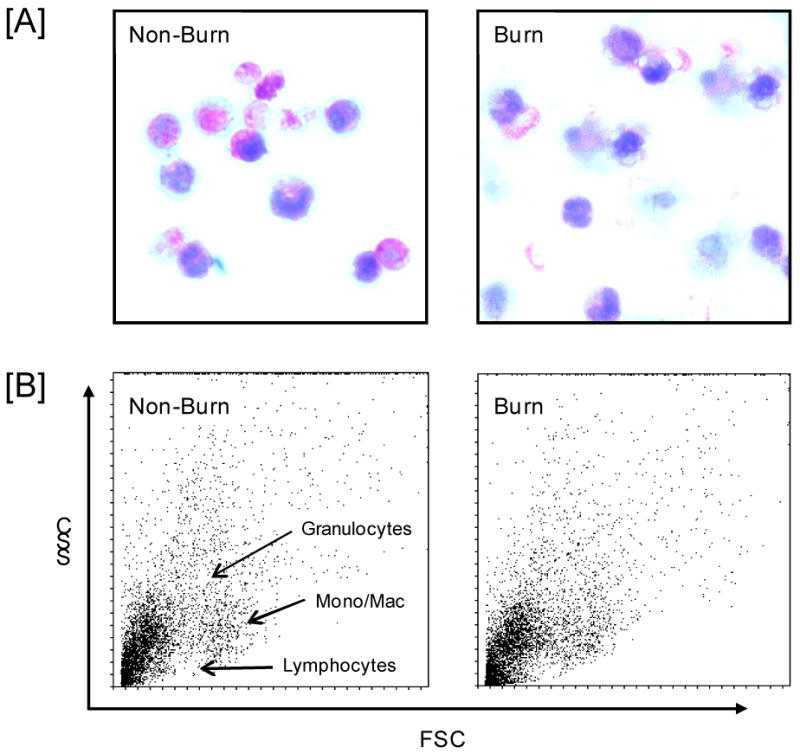
Wound cells were collected from mice at 3 days after injury as described in the Materials and Methods section. In panel [A] cells were stained with hemotoxylin-eosin and evaluated by light microscopy (× 400). Panel [B] shows the forward (FSC) and side scatter (SSC) of the wound cells as determined by FACs. Regions were granulocytes, lymphocytes and monocyte/macrophages are located are labeled and similar for both cell populations. Representative samples of 3-5 separate experiments are shown.
Figure 4. Wound cell phenotype.
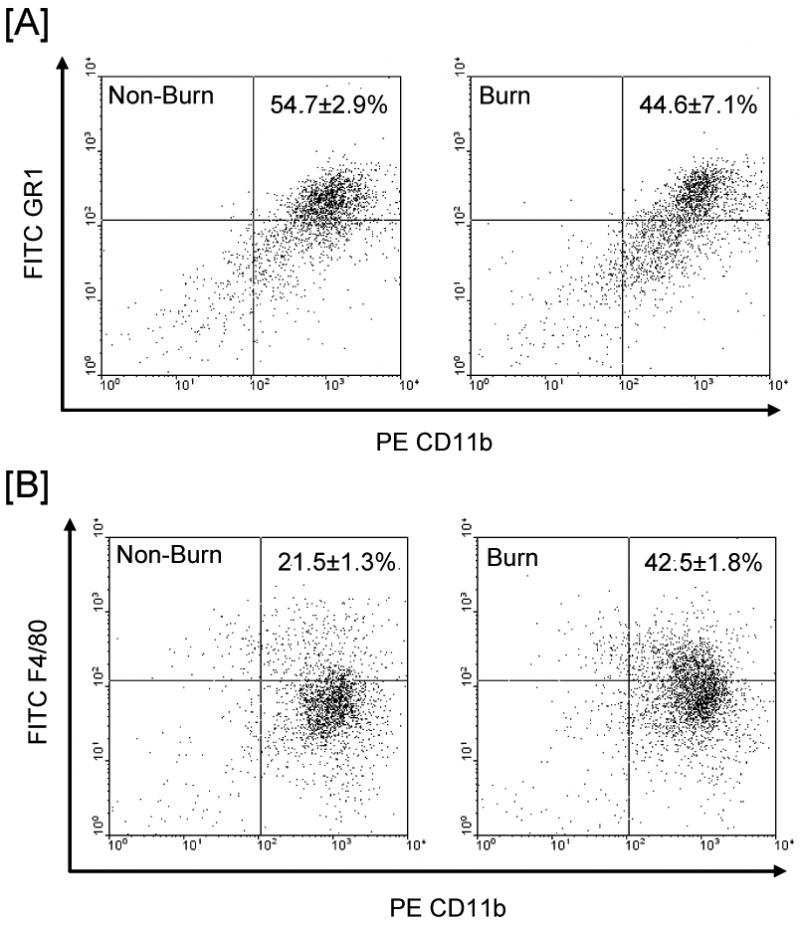
Cell phenotyping was determined by FACs analysis at 3 days after injury as described in the Materials and Methods section. A representative experiment for wound cell CD11b and GR1 expression by cells from non-burn and burn wounds are shown in panels A and panel B shows a representative experiment for F4/80 and CD11b expression by such cells. The values in each panel represent the percentage of the gated population positive for both phenotypic markers (mean ± SE for 5 mice/group).
To compare the relative expression of the phenotypic immune cell markers the mean fluorescent intensity (MFI) was determined (Table 1). The MFI for GR1 and F4/80 expressions were comparable between the two groups, indicating that the relative expression of these cell markers was similar. In contrast, the MFI for CD11b was slightly reduced by approximately 6% in the burn group as compared with the non-burn group (p<0.05).
Table 1.
Relative expression of phenotypic markers by cells from non-burn and burn woundsa.
| F4/80 | GR-1 | CD11b | |
|---|---|---|---|
| Non-Burn | 38.0 ± 9.7 | 73.5 ± 6.3 | 782.3 ± 42.8 |
| Burn | 39.5 ± 9.9 | 69.3 ± 9.3 | 603.3 ± 44.1b |
Wound cells were collected from mice at 3 days after injury and phenotyped by FACs analysis and the mean florescence intensity (MFI; an indicator of relative cell expression) determined as described in the Materials and Methods section. Data are mean ± SEM; n = 5 mice/group.
P<0.05 as compared with respective non-burn group.
Comparative analysis of in vitro functional capacity of non-burn and burn wound cells
The capacity of wound cells to produce a range of inflammatory mediators following LPS stimulation was assessed. LPS stimulation induced significant production of TNF-α, IL-6, IL-10, MCP-1, KC and NO2-/NO3- in both cell populations as compared to unstimulated cells (Fig. 5). Significant levels of IL-12, IFN-γ or GM-CSF were not observed in the cell supernatants (data not shown). LPS-stimulated TNF-α (Fig. 5A) and NO2-/NO3- (Fig. 5F) were comparable between the non-burn and burn wound cell cultures, whereas the production of IL-6 (Fig. 5B), IL-10, (Fig 5C), MCP-1 (Fig. 5D) and KC (Fig. 5E) was significantly less in the cells obtained from burned mice. The suppressed productive capacity ranged between 50-70% of non-burn cell levels.
Figure 5. Wound cell inflammatory mediator production.

Wound cells were isolated from non-burn and burn wounds at 3 days after injury and cultured with media (no stimulation; NS) or LPS (1 μg/ml) for 48 h and cell-free supernatants collected. Cytokine and chemokine levels were determined by BD™ Cytometric Bead Array (CBA) analysis and nitrite/nitrate levels determined by the Greiss reaction as described in the Materials and Methods section. Supernatant levels of TNF-α [A], IL-6, [B], IL-10 [C], MCP-1 [D], and KC [E] and NO2-/NO3- [F] were determined. Data are mean ± SEM; n = 4-5 mice/group. *P<0.05 as compared with NS; †P<0.05 as compared with respective non-burn group.
Discussion
Complications induced by major burn injury include; immunosuppression, increased incidence of infections, and delayed wound healing. The experimental model employed was developed to compare the infiltrating inflammatory response in a non-burn dermal wound to that of a burn wound. The implantation of sub-cutaneous sponges to create a wound site is a well-established model [15;16]. The non-burn wound model is associated with pronounced inflammation and the release of a wide range of inflammatory mediators. The implantation of the sponges beneath the burn site allows for the “trapping” of the infiltrating inflammatory cells and resultant inflammatory mediators that are responding to the burn injury. While our burn model is a full-thickness injury, (i.e., injury to the epidermal, dermal and sub-dermal layers), the findings with regard to inflammatory response indicate that viable tissue remains at the wound site at 3 days post-injury. Thus, it could be suggested that the burn injury model that we have employed is more consistent with a deep partial-thickness burn [17].
We observed a significant inflammatory response at the burn site at 3 days post-injury. This inflammatory response was associated with increased levels of the pro-inflammatory cytokines TNF-α and IL-6 and the chemokines MCP-1 and KC. These findings are consistent with our previous results showing an elevated level of various growth factors at the burn site at 3 days post-injury [14] and recent clinical findings regarding the systemic inflammatory response [18;19]. Others have also shown elevated levels of IL-6, TNF-α MCP-1 and KC at the burn site [20-22]. With regard to the skin, the inflammatory response to burn injury appears to be somewhat localized to the injury site. In contrast, our previous findings and others have shown elevated levels of nitrite/nitrate and growth factors in uninjured skin from burn injured mice [14;23]. In contrast, Rodriguez et al. [24] showed that in burn patients IL-6 levels in normal skin were elevated. These apparent discrepancies may be related to the time post-injury examined or the degree of injury. The wound fluid collected from burn and non-burn wounds contained significant levels of TNF-α, IL-6, MCP-1 and KC; however, differences between non-burn and burn wounds were only observed with regard to IL-6 and KC. Thus, the inflammatory response induced by wound infiltrating cells differs significantly between non-burn and burn wounds. This difference may be related to the greater degree of tissue injury in the burn group, as IL-6 is considered a marker of tissue injury [25].
A critical aspect of chemokine function is the trafficking of immune cells. KC is a potent neutrophil chemotactic factor and a key mediator of neutrophil recruitment in response to tissue injury and infection [22;26]. Consistent with the findings of Faunce et al. [22] we observed a profound elevation in the levels of KC in both the burn skin and the wound fluid. Since cytokines such as IL-1, TNF-α and PDGF have been shown to induce KC expression [27-29], the elevated KC levels in the burn skin and wound fluid may be related to increased TNF-α at the injury site [14]. MCP-1 is important to the healing process as it acts to stimulate endothelial and smooth muscle cell proliferation and monocytes associated with angiogenesis and correlates with macrophage infiltration [21;30;31]. MCP-1 can be induced by TNF-α and PDGF [32;33].
Based on the expression of GR1, CD11b and F4/80 by the wound cells, our findings suggest that the wound infiltrating cells, irrespective of burn injury, are likely to be myeloid suppressor cells (MSC). Makarenkova et al. [34] have recently characterized a MSC population in the spleen following traumatic stress that was heterogenesis expressing a range of phenotypic markers that included MHC 1, MHC II, CD80, CD86, CD40 and CD34, in addition to GR1 and CD11b. In the trauma group MHC 1, CD40 and CD34 were expressed, whereas these molecules were not expressed in the control cell group. Others have shown that MSC can also express CD11c and CD115 [35;36]. While the present study did not examine these phenotypic markers, it is likely that they are in part expressed by wound cells in our system. Due to the limited number of cells collected from the sponges functional analysis of MSC activity (i.e., suppression T-cell proliferation) was not possible. Future studies will need to address the functional characteristics of this wound cell population to confirm the MSC phenotype. Somewhat surprisingly, the wound infiltrate was devoid of T-cells. We have previously shown that T-cells of the γδ T-cell lineage are important to cellular infiltration of the wound site and distal organs [37;38], and the induction of growth factors at the burn wound site at 3 days post-injury [14]. The present findings suggest that T-cells resident in the skin are important in the orchestrating the inflammatory influx, but are not the actual cell type infiltrating the wound at 3 days post-injury.
GR-1, while generally considered a neutrophil marker, is expressed by immature macrophages thought to function in immunosuppression via an NO-mediated process [39;40]. The cells isolated from the wound sponges in our model are likely to be immunosuppressive to T-cells through an NO-dependent mechanism and may in part explain the lack of CD3+ cells found in the wound infiltrates. Nonetheless, expression and activity of all three NOS isoforms are important to the wound healing process [41]. We have previously shown that a hyperactive splenic macrophage phenotype post-burn is associated increased CD11b expression [42]. In contrast in the current study, a decrease in the relative CD11b expression was observed in the burn wound group. This observation suggests that the cells from the burn wound are “less-activated” than cells from the non-burn group. The cells isolated from the burn wounds hyporesponsive and produced lower levels of a number of important inflammatory mediators (i.e., IL-6, IL-10, MCP-1 and KC) in response to LPS stimulation. Since inflammation is a critical component of the wound healing process, the suppressed inflammatory response in the burn wound may contribute to the clinical observations of wound healing complications [4].
Activation of a pro-inflammatory cascade after burn injury is important in the development of subsequent immune dysfunction. In this regard, previous findings have shown that macrophage productive capacity for these mediators is markedly enhanced following thermal injury and thereby contributing to immune dysfunction [2;43]. Moreover, since both macrophages and inflammation contribute to the wound healing process [7;8], dysregulation of the macrophage inflammatory response, as seen with burn injury, likely contributes to wound healing complications. Nonetheless, the current study observed a hypoactive macrophage phenotype (i.e., suppressed productive capacity for inflammatory mediators) at the wound site following burn injury. Our previous findings have shown that the expression of macrophage hyperactivity (i.e., increased productive capacity for inflammatory mediators) post-burn was in part limited to the fixed-tissue compartment [44]. Splenic macrophages displayed enhanced IL-6 and TNF-α production post-burn, whereas such activity was suppressed in peripheral blood mononuclear cells (PBMC). In the current study it can be speculated that the cells infiltrating the wound sponges might have originated from the circulating PBMC population, since aspects of their inflammatory phenotype were similarly suppressed post-burn. The inability of infiltrating burn wound cells to mount an appropriate inflammatory response may contribute to wound healing complications. In particular, the suppressed ability of burn wound cells to be produce the chemokines KC and MCP-1 upon activation may contribute to reduced cellular infiltration and resultant complications, such as delayed healing or infection. Nonetheless, this concept is speculative and additional studies are warranted to verify such a concept.
In conclusion, the skin contains a network of various immune cells that function in concert to maintain organ function and respond to cellular stress. The migration, activation and clearance of these diverse cells are likely to be temporally dependent after burn injury. Due to previously observed differences in responses at the burn site, we focused the current studies on 3 days post-injury [14]. It remains to be determined how burn injury influences the inflammatory wound response at other times and during the entire healing process. These findings show that, while the infiltrating cells at the non-burn and burn wound site were phenotypically similar, the burn wound inflammatory response was exaggerated as compared to non-burn wounds, but the subsequent responsiveness of the burn wound cells to appropriate activation was attenuated. Thus, the burn wound inflammatory response and subsequent immune cell responsiveness differs significantly from that of non-burn cutaneous wounds. These burn specific differences in the wound inflammatory response may be important contributing factors to delayed wound healing, increased wound infections and similar complications in burn patients. An improved understanding of the unique characteristics of the burn wound inflammatory response and how it differs from that of non-burn cutaneous wounds may contribute to the development of improved therapeutic regimes specific for burn patients and warrants further investigation.
Acknowledgments
These studies were presented in part at the 26th Annual Meeting of the Surgical Infection Society, La Jolla CA and the 29th Annual Conference on Shock, Broomfield, CO. Support was provided by National Institutes of Health grant GM079122, AI049960 and Department of Defense grant PRO034212 and completed in part while MGS was a faculty member at the University of Alabama at Birmingham. BMT was a fellow in the laboratory of IH Chaudry at the University of Alabama at Birmingham when these studies were conducted and was supported in part by NIH grant GM37127.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brigham PA, McLoughlin E. Burn incidence and medical care use in the United States: Estimates, trends, and data sources. J Burn Care Rehabil. 1996;17:95–107. doi: 10.1097/00004630-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Schwacha MG. Macrophages and post-burn immune dysfunction. Burns. 2003;29:1–14. doi: 10.1016/s0305-4179(02)00187-0. [DOI] [PubMed] [Google Scholar]
- 3.Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock. 1999;11:153–159. doi: 10.1097/00024382-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Shakespeare P. Burn wound healing and skin substitutes. Burns. 2001;27:517–522. [PubMed] [Google Scholar]
- 5.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 6.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001;69:513–521. [PubMed] [Google Scholar]
- 7.Moore K. Cell biology of chronic wounds: the role of inflammation. J Wound Care. 1999;8:345–348. [PubMed] [Google Scholar]
- 8.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg. 2004;187:11S–16S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 9.Lyuksutova OI, Murphey ED, Toliver-Kinsky TE, Lin CY, Cui W, Williams DL, Sherwood ER. Glucan phosphate treatment attenuates burn-induced inflammation and improves resistance to Pseudomonas aeruginosa burn wound infection. Shock. 2005;23:224–232. [PubMed] [Google Scholar]
- 10.Gibran NS, Heimbach DM. Current status of burn wound pathophysiology. Clin Plast Surg. 2000;27:11–22. [PubMed] [Google Scholar]
- 11.Singer AJ, McClain SA. Persistent wound infection delays epidermal maturation and increases scarring in thermal burns. Wound Repair Regen. 2002;10:372–377. doi: 10.1046/j.1524-475x.2002.10606.x. [DOI] [PubMed] [Google Scholar]
- 12.Alexander M, Chaudry IH, Schwacha MG. Relationships between burn size, immunosuppression, and macrophage hyperactivity in a murine model of thermal injury. Cell Immunol. 2002;220:63–69. doi: 10.1016/s0008-8749(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 13.Reichner JS, Meszaros AJ, Louis CA, Henry WL, Jr, Mastrofrancesco B, Martin BA, Albina JE. Molecular and metabolic evidence for the restricted expression of inducible nitric oxide synthase in healing wounds. Am J Pathol. 1999;154:1097–1104. doi: 10.1016/S0002-9440(10)65362-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander M, Daniel T, Chaudry IH, Choudhry M, Schwacha MG. T-cells of the γδ T-cell receptor lineage play an important role in the postburn wound healing process. J Burn Care Res. 2006;27:18–25. doi: 10.1097/01.bcr.0000188325.71515.19. [DOI] [PubMed] [Google Scholar]
- 15.Albina JE, Mills CD, Henry WL, Jr, Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol. 1990;144:3877–3880. [PubMed] [Google Scholar]
- 16.Angele MK, Knoferl MW, Schwacha MG, Ayala A, Bland KI, Cioffi WG, Josephson SL, Chaudry IH. Hemorrhage decreases macrophage inflammatory protein 2 and interleukin-6 release: a possible mechanism for increased wound infection. Ann Surg. 1999;229:651–660. doi: 10.1097/00000658-199905000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams WG, Phipps RP. Pathophysiology of the Burn Wound. In: Herndon DN, editor. Total Burn Care. Philadelphia: W.B. Saunders Company LTD; 1996. pp. 63–70. [Google Scholar]
- 18.Finnerty CC, Jeschke MG, Herndon DN, Gamelli R, Gibran N, Klein M, Silver G, Arnoldo B, Remick D, Tompkins RG. Temporal cytokine profiles in severely burned patients: a comparision of adults and children. Mol Med. 2008 doi: 10.2119/2007-00132.Finnerty. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauglitz GG, Song J, Herndon DN, Finnerty CC, Boehning DF, Barral JM, Jeschke MG. Characterization of the inflammatory response during acute and postacute phases after severe burn. Shock. 2008 doi: 10.1097/SHK.0b013e31816e3373. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S. Topical p38MAPK inhibition reduces dermal inflammation and epithelial apoptosis in burn wounds. Shock. 2006;26:201–209. doi: 10.1097/01.shk.0000225739.13796.f2. [DOI] [PubMed] [Google Scholar]
- 21.Gibran NS, Ferguson M, Heimbach DM, Isik FF. Monocyte chemoattractant protein-1 mRNA expression in the human burn wound. J Surg Res. 1997;70:1–6. doi: 10.1006/jsre.1997.5017. [DOI] [PubMed] [Google Scholar]
- 22.Faunce DE, Llanas JN, Patel PJ, Gregory MS, Duffner LA, Kovacs EJ. Neutrophil chemokine production in the skin following scald injury. Burns. 1999;25:403–410. doi: 10.1016/s0305-4179(99)00014-5. [DOI] [PubMed] [Google Scholar]
- 23.Oliveira GV, Shimoda K, Enkhbaatar P, Jodoin J, Burke AS, Chinkes DL, Hawkins HK, Herndon DN, Traber L, Traber D, Murakami K. Skin nitric oxide and its metabolites are increased in nonburned skin after thermal injuries. Shock. 2004;22:278–282. doi: 10.1097/01.shk.0000135259.90311.33. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez JL, Miller CG, Garner WL, Till GO, Guerrero P, Moore NP, Corridore M, Normolle DP, Smith DJ, Remick DG. Correlation of the local and systemic cytokine response with clinical outcome following thermal injury. J Trauma. 1993;34:684–694. doi: 10.1097/00005373-199305000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17:463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Kondo T, Novick AC, Toma H, Fairchild RL. Induction of chemokine gene expression during allogeneic skin graft rejection. Transplantation. 1996;61:1750–1757. doi: 10.1097/00007890-199606270-00015. [DOI] [PubMed] [Google Scholar]
- 27.Marmur JD, Poon M, Rossikhina M, Taubman MB. Induction of PDGF-responsive genes in vascular smooth muscle. Implications for the early response to vessel injury. Circulation. 1992;86:III53–III60. [PubMed] [Google Scholar]
- 28.Ohmori Y, Hamilton TA. Cell type and stimulus specific regulation of chemokine gene expression. Biochem Biophys Res Commun. 1994;198:590–596. doi: 10.1006/bbrc.1994.1086. [DOI] [PubMed] [Google Scholar]
- 29.Ohmori Y, Hamilton TA. IFN-gamma selectively inhibits lipopolysaccharide-inducible JE/monocyte chemoattractant protein-1 and KC/GRO/melanoma growth-stimulating activity gene expression in mouse peritoneal macrophages. J Immunol. 1994;153:2204–2212. [PubMed] [Google Scholar]
- 30.Buschmann I, Heil M, Jost M, Schaper W. Influence of inflammatory cytokines on arteriogenesis. Microcirculation. 2003;10:371–379. doi: 10.1038/sj.mn.7800199. [DOI] [PubMed] [Google Scholar]
- 31.DiPietro LA, Polverini PJ, Rahbe SM, Kovacs EJ. Modulation of JE/MCP-1 expression in dermal wound repair. Am J Pathol. 1995;146:868–875. [PMC free article] [PubMed] [Google Scholar]
- 32.Van Damme J, Proost P, Put W, Arens S, Lenaerts JP, Conings R, Opdenakker G, Heremans H, Billiau A. Induction of monocyte chemotactic proteins MCP-1 and MCP-2 in human fibroblasts and leukocytes by cytokines and cytokine inducers. Chemical synthesis of MCP-2 and development of a specific RIA. J Immunol. 1994;152:5495–5502. [PubMed] [Google Scholar]
- 33.Colotta F, Borre A, Wang JM, Tattanelli M, Maddalena F, Polentarutti N, Peri G, Mantovani A. Expression of a monocyte chemotactic cytokine by human mononuclear phagocytes. J Immunol. 1992;148:760–765. [PubMed] [Google Scholar]
- 34.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176:2085–2094. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 35.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 36.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 37.Daniel T, Thobe BM, Chaudry IH, Choudhry MA, Hubbard WJ, Schwacha MG. Regulation of the postburn wound inflammatory response by gammadelta T-cells. Shock. 2007;28:278–283. doi: 10.1097/shk.0b013e318034264c. [DOI] [PubMed] [Google Scholar]
- 38.Toth B, Alexander M, Daniel T, Chaudry IH, Hubbard WG, Schwacha MG. The role of γδ T cells in the regulation of neutrophil-mediated tissue damage after thermal injury. J Leukoc Biol. 2004;76:545–552. doi: 10.1189/jlb.0404219. [DOI] [PubMed] [Google Scholar]
- 39.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 40.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 41.Curran JN, Winter DC, Bouchier-Hayes D. Biological fate and clinical implications of arginine metabolism in tissue healing. Wound Repair Regen. 2006;14:376–386. doi: 10.1111/j.1743-6109.2006.00151.x. [DOI] [PubMed] [Google Scholar]
- 42.Schwacha MG, Schneider CP, Bland KI, Chaudry IH. Resistance of macrophages to the suppressive effect of interleukin-10 following thermal injury. Am J Physiol- Cell Physiol. 2001;281:C1180–C1187. doi: 10.1152/ajpcell.2001.281.4.C1180. [DOI] [PubMed] [Google Scholar]
- 43.O'Riordain M, Collins KH, Pitz M, Saporoschetz IB, Mannick JA, Rodrick ML. Modulation of macrophage hyperactivity improves survival in a burn-sepsis model. Arch Surg. 1992;127:152–157. doi: 10.1001/archsurg.1992.01420020034005. [DOI] [PubMed] [Google Scholar]
- 44.Schwacha MG, Schneider CP, Chaudry IH. Differential expression and tissue compartmentalization of the inflammatory response following thermal injury. Cytokine. 2002;17:266–274. doi: 10.1006/cyto.2001.1003. [DOI] [PubMed] [Google Scholar]