

Abstract
Vasculopathies caused by varicella zoster virus (VZV) are indicative of a productive virus infection in cerebral arteries after either reactivation of VZV (shingles) or primary infection (chickenpox). VZV vasculopathy can cause ischaemic infarction of the brain and spinal cord, as well as aneurysm, subarachnoid and cerebral haemorrhage, carotid dissection, and, rarely, peripheral arterial disease. VZV vasculopathy in immunocompetent or immunocompromised individuals can be unifocal or multifocal with deep-seated and superficial infarctions. Lesions at the grey–white matter junction on brain imaging are a clue to diagnosis. Involvement of both large and small arteries is more common than that of either alone. Most patients have a mononuclear cerebrospinal fluid pleocytosis, often with red blood cells. Cerebrospinal fluid pleocytosis and rash are absent in about a third of cases. Anti-VZV IgG antibody in the cerebrospinal fluid is found more frequently than VZV DNA. In recent years, the number of recognised VZV vasculopathies has grown, and accurate diagnosis is important for the effective treatment of these disorders.
Introduction
Primary VZV infection, which usually occurs in children, results in chickenpox (varicella), after which the virus becomes latent in ganglionic neurons along the entire neuraxis. Years later, as cell-mediated immunity to VZV declines with age or from immunosuppression (such as in organ-transplant recipients or patients with cancer or AIDS), VZV can reactivate to cause zoster (shingles). Zoster is often followed by chronic pain (postherpetic neuralgia), as well as vasculopathy, myelopathy, retinal necrosis, and cerebellitis (figure 1). VZV reactivation can also cause pain without rash (zoster sine herpete); infact, all neurological complications of VZV reactivation can occur without rash.
Figure 1. Neurological disease caused by reactivation of varicella zoster virus.

*Can occur after varicella and can also occur without a rash.
Over the past few decades there has been an increasing number of reports of vascular disease after VZV reactivation. Unlike early cases of acute hemiplegia after contralateral zoster caused by large-artery disease, the recognised clinical range of this disease has expanded to include transient ischaemic attacks and protracted illness involving both small and large arteries. In addition to ischaemic infarction, VZV can cause aneurysm, cerebral and subarachnoid haemorrhage, and arterial ectasia, and might be a co-factor, along with trauma, in the pathogenesis of cerebral arterial dissection. Furthermore, VZV can also cause peripheral arterial disease. In adults, the exact incidence of VZV vasculopathy is difficult to estimate, although it is more common in immunocompromised individuals. In children, VZV vasculopathy has been proposed to account for 31% of all arterial ischaemic strokes;1 moreover, stroke was preceded by chickenpox in 44% of children with transient cerebral arteriopathy.2
In this Review, we outline the ever-widening spectrum of vascular disease after VZV reactivation (zoster), as well as after primary infection (varicella), and discuss the underlying mechanisms of the disease. We also emphasise the importance of accurate diagnosis to enable appropriate treatment of VZV vasculopathies.
History
The earliest recorded description of VZV vasculopathy was about 50 years ago when Cravioto and Feigin3 described what they believed was “a non-infectious granulomatous angiitis with a predilection for the nervous system, characterized by thrombosis in cerebral arteries and distinguished from other vasculitides by the nature of the inflammatory response, which consisted predominantly of histiocytes, mononuclear cells and multinucleated giant cells”. Years later, Rosenblum and Hadfield4 described granulomatous angiitis of the nervous system in patients with herpes zoster and lymphosarcoma, characterised by infiltrates of mononuclear cells and multinucleated giant cells in cerebral arteries. The first angiographic studies of the large arteries in the neck and intracranial arteries in a patient with herpes zoster ophthalmicus and delayed contralateral hemiparesis revealed segmental arteritis in the area of the carotid siphon.5 Until recently, these older cases of VZV vasculopathy were described as granulomatous angiitis, VZV vasculitis, or zoster ophthalmicus and delayed contralateral hemiparesis, although VZV vasculopathy can occur after zoster anywhere in the body.
Clinical features and diagnosis
Clinical presentation
Although early case reports emphasised that patients present with acute stroke, many patients have transient ischaemic attacks with protracted neurological symptoms and signs. Common clinical features are not limited to acute hemiplegia and include headache, changes in mental status, aphasia, ataxia, hemisensory loss, and both hemianopia and monocular visual loss. Less frequently, patients with VZV vasculopathy present with aneurysm, subarachnoid or cerebral haemorrhage, carotid dissection, and, rarely, peripheral arterial disease. Rare presentations of monocular visual loss exemplify the capacity for VZV to infect small arteries. The first reported case of monocular loss of vision was that of a patient who developed occlusion of the ipsilateral central retinal artery 2 weeks after trigeminal-distribution zoster.6 The second case was that of a patient who developed sudden monocular visual loss 5 months after ipsilateral ophthalmic-distribution zoster.7 The patient had a pale optic nerve without retinal oedema or a cherry-red spot, indicating involvement of the posterior ciliary artery. At the time of visual loss, there was a reduced serum to cerebrospinal fluid (CSF) ratio of anti-VZV IgG antibody. The serum also contained anti-VZV IgM antibody, indicating active infection. Prompt antiviral treatment resulted in complete resolution of the neurological deficit. The 5-month interval between zoster and visual loss is consistent with reports of VZV vasculopathy occurring up to 6 months after zoster.7,8 We have also encountered patients in whom the only symptom of VZV vasculopathy was headache that was prolonged for months.9
VZV vasculopathy affects both immunocompetent and immunocompromised patients, occurs after zoster or varicella, and can be either unifocal or multifocal. Clinically, the notion of a pure unifocal vasculopathy has been replaced by the realisation that more than one artery is often involved, resulting in a multifocal vasculopathy. This involvement of several arteries is exemplified by the onset of posterior circulation strokes caused by involvement of large and small arteries in an HIV-positive patient who had a Ramsay Hunt syndrome 2 weeks earlier.10 Most often, both large and small arteries are involved, followed by small arteries alone, and, least often, by large arteries alone.9 In 37% of cases, VZV vasculopathy develops without rash.9 Finally, VZV vasculopathy often coexists with VZV meningitis, radiculitis, and myelitis.11,12
Brain imaging
Brain imaging reveals abnormalities in most cases. An analysis of 30 cases of virologically confirmed VZV vasculopathy revealed changes in every patient, except in one individual with exclusively posterior ciliary artery involvement.7,9 To our knowledge, the only case of VZV vasculopathy with a normal MRI was the patient described above who developed ipsilateral posterior ciliary artery disease after ophthalmic-distribution zoster.7
Abnormalities are cortical and deep, and occur in both the grey and white matter and at grey–white matter junctions13 in particular (figure 2A)—a clue to the cause of disease. Although lesions of the grey and white matter are commonly seen in patients with other disorders such as metastatic carcinoma and embolic disease, VZV vasculopathy should be included in the differential diagnosis of patients with lesions at grey–white matter junctions. Most lesions are ischaemic, but haemorrhagic lesions also occur; some lesions enhance on MRI with contrast, indicating breakdown of the blood–brain barrier.
Figure 2. MRI scan and cerebral angiogram of patients with VZV vasculopathies.
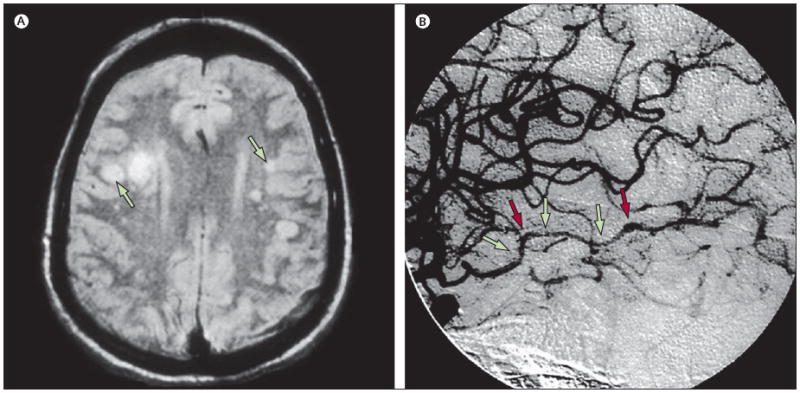
(A) MRI scan of a patient with VZV multifocal vasculopathy. Proton-density brain MRI scan shows multiple areas of infarction in both hemispheres, particularly involving the white matter. Arrows point to lesions at the grey–white matter junctions. Reproduced from Gilden and co-workers,13 with permission from Taylor & Francis. (B) Cerebral angiogram from a patient with VZV vasculopathy. Focal areas of stenosis (green arrows) and poststenotic dilatation (red arrows) involving the right posterior cerebral artery can be seen (left side of picture, right side of patient). Reproduced from Russman and co-workers,14 with permission from the American Medical Association. VZV=varicella zoster virus.
Angiographic features
Typical angiographic changes include segmental constriction, often with poststenotic dilatation (figure 2B).14 In the largest series reported so far of 30 patients with virologically verified VZV vasculopathy, 23 (77%) patients underwent vascular studies (conventional angiography or magnetic resonance angiography) and 16 of these patients (70%) had vascular abnormalities. Brain imaging and vascular studies indicated involvement of both large and small arteries in 15 (50%), pure small-artery involvement in 11 (37%), and pure large-artery disease in four (13%) patients.9 Although the presence of stenosis or occlusion is helpful in diagnosing VZV vasculopathy, a negative angiogram does not exclude the diagnosis, most probably because disease in small arteries is not detected as readily as in large arteries. Overall, disease of both large and small arteries is encountered more often than pure small-artery disease, and pure large-artery disease is reported least often.
Abnormalities in the CSF
Abnormalities in the CSF are common. A modest pleocytosis, generally of fewer than 100 cells and predominantly mononuclear, is seen in two-thirds of patients with VZV vasculopathy.9 Many patients also have red blood cells in their CSF. Concentrations of CSF protein are commonly increased, whereas those of glucose are normal. Oligoclonal bands are commonly present. As in other chronic infectious CNS diseases (such as neurosyphilis, subacute sclerosing pan encephalitis, and cryptococcal meningitis), in which the oligoclonal IgG is directed against the causative agent (reviewed elsewhere15), the oligoclonal IgG in VZV vasculopathy is directed against VZV.16
Virological confirmation
When a clinical diagnosis of VZV vasculopathy is suspected and is corroborated by single or multiple characteristic lesions on MRI or CT, virological confirmation is required. In 14 patients, the diagnostic value of detecting anti-VZV IgG antibody in the CSF was greater than that of detecting VZV DNA.17 Additional analysis of a further 16 patients from several institutions in Europe and Japan revealed that 28 of 30 patients with VZV vasculopathy had anti-VZV IgG antibody in the CSF, compared with only nine with VZV DNA in the CSF.9 Although a positive PCR for VZV DNA in CSF is helpful, a negative PCR does not exclude the diagnosis; only negative results in both VZV PCR and anti-VZV IgG antibody tests in the CSF can reliably exclude the diagnosis of VZV vasculopathy.
The detection of VZV antibody is better than that for VZV DNA and might depend on the often protracted clinical course of VZV vasculopathy, which generally lasts for weeks to months. This longer course contrasts with the acute encephalitis caused by herpes simplex virus-1 (HSV-1), in which the CSF is positive for HSV-1 DNA by PCR and negative for antibody to HSV-1 during the first week of disease, whereas viral DNA begins to disappear from the CSF as anti-HSV-1 antibody becomes detectable during the second week.18 In a case reported by Nagel and co-workers9 (case 16), serial CSF analysis revealed VZV DNA on the day of admission and after 7 days; VZV DNA then became undetectable on days 14–50 when anti-VZV IgG antibody was found. In 30 patients with virologically verified VZV vasculopathy, the average time from onset of neurological symptoms to virological analysis was 4·2 months; thus, the fact that most patients did not have detectable VZV DNA but did have anti-VZV IgG antibody in their CSF is not surprising. Therefore, although testing for VZV DNA in the CSF only is common in clinical practice, testing for anti-VZV IgG antibody will identify more cases of VZV vasculopathy. The diagnostic value of PCR in HSV encephalitis was shown by comparison with HSV-positive brain biopsy samples, which has served as the gold standard for diagnosis; however, a gold standard that shows VZV in the arterial wall of patients with VZV vasculopathy has not been established.
Many of the same symptoms and signs, as well as CSF, imaging, and arteriographic abnormalities that occur in VZV vasculopathy, are seen in other CNS disorders (such as primary angiitis of the nervous system) and in the granulomatous angiitides (such as CNS sarcoidosis, neurosyphilis, tuberculous, and fungal infections of the nervous system). Thus, the assessment of all patients with granulomatous or other CNS angiitides should include CSF analysis for both VZV DNA and anti-VZV IgG antibody.
A prototypical case of VZV vasculopathy
An important case has been described19 that shows many of the clinical, imaging, and CSF features seen in VZV vasculopathy. The patient was a 73-year-old immunocompetent man who developed headache, fever, progressive changes in mental status, and focal deficit that progressed over 20 days. There was no history of zoster. Repeated CSF examinations revealed a persistent pleocytosis with both red and white blood cells (predominantly mononuclear). Brain MRI scans revealed multiple superficial and deep infarcts in the central grey matter, white matter, and brainstem. Angiography revealed focal narrowing in the internal carotid, anterior, and middle cerebral arteries. Anti-VZV antibody was present in the CSF. The patient was hospitalised for many months and treated with steroids and cyclophosphamide, but deteriorated and died. At autopsy, multiple benign infarcts were seen throughout the brain. The cerebral arteries showed disruption of the internal elastic lamina and multinucleated giant cells. Virological analysis indicated both VZV DNA and VZV antigen in the posterior cerebral and basilar arteries.20
This case shows many previously unrecognised features of VZV vasculopathy, including the presence of headache and focal changes that were progressive in the absence of rash, with red cells in the CSF on several occasions, involvement of brain structures supplied by large and small arteries, and a productive virus infection in cerebral arteries but not in the brain.
Less typical complications of VZV vasculopathy
Spinal-cord infarction
Rare cases of putative spinal-cord infarction produced by VZV vasculopathy have been diagnosed on the basis of acute-onset myelopathy associated with zoster or on the basis of virological evidence of VZV infection in acute myelopathy cases without rash. Only a few cases of VZV spinal-cord infarction have been verified pathologically.21 Before diffusion-weighted MRI, postmortem spinal-cord necrosis secondary to VZV vasculitis with productive VZV infection was the only way to establish the diagnosis of VZV vasculopathy in the spinal cord.21,22
Because diffusion-weighted MRI is better than conventional MRI for the detection of spinal-cord ischaemia and infarction,23 the technique was used in conjunction with virological analysis of CSF to diagnose VZV infarction of the spinal cord in a patient who presented with abrupt-onset myelopathy followed by zoster.24 A spinal-cord infarction was identified with diffusion-weighted MRI and virological analysis confirmed the presence of anti-VZV IgG antibody, but not VZV DNA, in the CSF. A decreased serum to CSF ratio of anti-VZV IgG antibody was consistent with intrathecal synthesis of VZV IgG. The virological findings were the same as described in most cases of VZV vasculopathy in the brain, with detection of anti-VZV IgG antibody in the CSF being more sensitive as an indicator of VZV vasculopathy than detection of VZV DNA.17
Aneurysm, subarachnoid and intracerebral haemorrhage, arterial ectasia, and dissection
The protean complications of VZV vasculopathy include cerebral aneurysm, subarachnoid and intracerebral haemorrhage, ectasia, and dissection. The first report of a CNS aneurysm produced by VZV infection was in a 24-year-old healthy woman who developed ophthalmic-distribution zoster with peripheral facial palsy and contralateral hemiplegia. Although an initial angiogram was normal, she developed decreased hearing and left-sided Horner's syndrome 4 weeks later, and repeat angiography revealed an aneurysm in the intrapetrosal region of the left internal carotid artery (figure 3A).25 Three further cases of aneurysm caused by VZV infection in patients with AIDS were recently reported.26 In another case of VZV vasculopathy, a 42-year-old renal transplant recipient with concurrent vertebral-artery aneurysm and dissection was successfully treated with embolisation and aciclovir therapy.27 Two cases of dissection after VZV infection have been described.28 The first was a 15-year-old boy who, 4 weeks after ophthalmic-distribution zoster, developed a left hemiparesis while jogging. An angiogram revealed carotid-artery dissection. His clinical symptoms resolved and the angiogram normalised in 3 months. The other case was a 4-year-old boy who, 2 weeks after recovery from varicella, developed left-sided weakness while playfully wrestling with another child of the same age. A cerebral angiogram revealed a right internal carotid-artery dissection. Clinical examination and radiological improvement revealed full recovery after 2 months. Although trauma and exercise are frequently associated with arterial dissection, VZV infection in the wall of an artery can predispose the artery to dissection if it is subjected to further trauma. VZV has also been associated with dolichoectasia of the anterior and posterior circulation with multiple small deep-seated infarcts.29 Finally, a comprehensive study of VZV vasculopathy revealed the occurrence of stenosis, ectasia, and aneurysm in several patients.26 Elble30 described intracerebral haemorrhage after ophthalmic-distribution zoster. Another patient with VZV vasculitis presented with intracranial haemorrhage with concomitant zoster on the back; cerebral angiography revealed vasculitic changes involving small-sized and medium-sized vessels (figure 3B).31
Figure 3. Aneurysm and subarachnoid haemorrhage in patients with VZV vasculopathy.

(A) Aneurysm in a patient with VZV vasculopathy. Angiogram 4 weeks after zoster reveals an aneurysm in the intrapetrosal portion of the left internal carotid artery (arrow). Reproduced from Gürsoy and co-workers,25 with permission from Springer-Verlag. (B) Subarachnoid haemorrhage in a patient with zoster and VZV vasculopathy. Axial CT shows subarachnoid haemorrhage and a left para-midline haematoma (left side of picture, right side of patient). Reproduced from Jain and co-workers,31 with permission from the American Society of Neuroradiology. VZV=varicella zoster virus.
As VZV infection is primarily in the media of arteries with disruption of the internal elastic membrane, it is surprising that arterial ectasia and aneurysm with subarachnoid and intracerbral haemorrhage do not occur more frequently.
Peripheral artery disease
Recently, multifocal vasculopathy with peripheral thrombotic disease was reported in an adult with varicella,32 indicating that VZV vasculopathy is not always restricted to the brain and spinal cord. The patient was an adult man who developed varicella followed by stroke and multiple peripheral thrombotic events with stenosis and occlusion of the femoral and iliac arteries (figure 4A), as well as ischaemic lesions in the striatocapsular region and fronto-temporal cortex (figure 4B). Haemorrhagic foci were also seen. The patient had other antibody abnormalities, such as positive anti-cardiolipin IgM and IgM anti-α2-glycoprotein 1, along with anti-VZV IgM antibody.
Figure 4. VZV vasculopathy affecting cerebral and peripheral arteries.
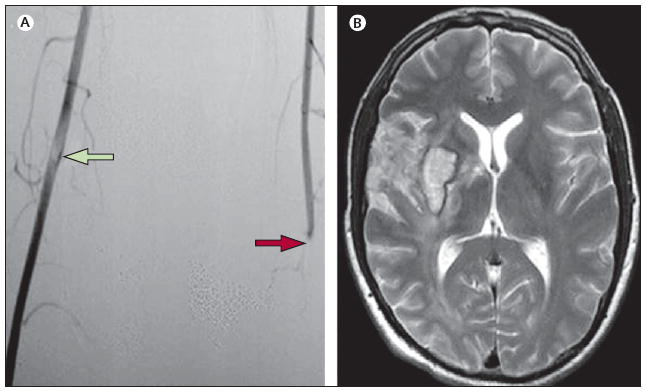
(A) Lower-limb angiography shows occlusion of the left femoral artery (red arrow) and the presence of luminal non-occlusive thrombi in the right femoral artery (green arrow). (B) Axial T2-weighted brain MRI of the same patient revealed an ischaemic lesion involving the right striatocapsular region, adjacent white matter, and frontotemporal cortex laterally and superiorly. Small haemorrhagic foci were seen inside the ischaemic lesion; magnetic resonance angiogram revealed proximal occlusion of the right middle cerebral artery (not shown). No other pathological findings were apparent (left side of picture, right side of patient). Reproduced from Massano and co-workers,32 with permission from John Wiley. VZV=varicella zoster virus.
VZV-associated cranial nerve disease
Cranial neuropathies after zoster are not rare, often occurring many days to weeks after the rash. Although the mechanism of disease is unknown, the late onset parallels the development of VZV vasculopathy in the brain weeks after zoster. Cranial neuropathies, particularly when multiple, might be secondary to vasculopathy of arteries supplying one or more cranial nerves. Three arterial systems have a role in the vascularisation of cranial nerves.33 First is the inferolateral trunk, which most often arises from the internal carotid artery and supplies cranial nerve III, as well as nerves IV, V1, and VI; the vascularisation of cranial nerve III provides an explanation for its involvement in diabetes mellitus. Second is the middle meningeal system, which derives from the external carotid artery and supplies cranial nerves V2, V3, and VII. The intrapetrous portion of nerve VII has two defined vascular territories: the first (stylomastoid artery) is limited to nerve VII and the second (middle meningeal) supplies nerves VII and V. Lastly, the ascending pharyngeal system, which also derives from the external carotid artery, supplies nerves IX, X, XI, and XII; cranial nerve XI has a dual vascularisation, which explains why this nerve can either be spared, as described in an angiographic accident, or involved, as in a case of herpes zoster. A vascular mechanism should be considered when cranial nerve lesions occur in polyneuritis cranialis syndromes caused by diabetes or zoster.
Giant-cell (temporal) arteritis
VZV mostly reactivates in the elderly (the same age-group in which giant-cell arteritis predominates), often affects arteries, and produces multinucleated giant cells in acutely infected tissue. Thus, clinicians have raised the question of whether VZV might cause giant-cell arteritis. However, an analysis of arteries from ten pathologically verified cases of giant-cell arteritis for the presence of VZV antigen and DNA was negative.34 A later study confirmed the absence of any association of VZV with giant-cell arteritis.35
VZV vasculopathy in children
As in adults, VZV vasculopathy in children also often occurs weeks to months after zoster or varicella. There are many case reports; the first, reported by Kamholz and Tremblay,36 was of a patient with varicella followed by hemiparesis with angiographic evidence of vasculopathy. Another case of loss of balance and increasing right-sided weakness occurred in a 17-month-old boy who had had a vesicular rash in the ophthalmic and mandibular division of the left trigeminal nerve 4 weeks earlier; his mother had chickenpox at 8 months of gestation.37 Intracranial haemorrhage after primary VZV infection was described in a 7-month-old child.38 In another case, recurrent hemiplegia, virologically verified to be caused by VZV, developed in an infant whose mother had zoster infection during the third trimester.39
Pathology and mechanisms of disease
Large-vessel and small-vessel vasculopathy
Large infarctions that follow trigeminal-distribution or cervical-distribution zoster are usually ischaemic40 and are only rarely haemorrhagic.30 Clinical and radiological evidence of multifocal large and small infarction should suggest the possibility of VZV infection. Large calibre vessels at the base of the brain or over the convexities can be damaged, and large-sized ischaemic or haemorrhagic infarctions can be produced. Ovoid, well-demarcated lesions (figure 3), mostly at the grey–white matter junctions, are seen. As with large-vessel disease, the virus is also present in small blood vessels, as indicated by inclusions in endothelial cells41 or by in-situ hybridisation.42 Small ischaemic foci develop, followed by variable extents of necrosis and demyelination, depending on the degree of additional viral infection of oligodendrocytes.21 Previous investigators have emphasised either the demyelinative43,44 or the necrotic45 features of VZV small-vessel lesions. Myelin and axon stains suggest a relatively greater myelin preservation in some lesions, and pathology ranges from demyelination to necrosis produced by VZV. A large series of patients with VZV myelitis also showed combinations of vasculitis, necrosis, and demyelination in the spinal cord.21 Unlike large-vessel vasculopathy, which produces infarctions with few viral inclusions at their perimeter, small-vessel vasculopathy often yields lesions with several intranuclear Cowdry A inclusions (a hallmark of herpesvirus infection) in glia if there is a substantial vacuolated demyelinated perimeter. Lesions caused by small-vessel vasculopathy show central cavitation and macrophage influx secondary to the initial ischaemic event or to the additional damage caused by VZV infection of astrocytes and neurons. Disruption of the internal elastic lamina in cerebral arteries infected with VZV can result in a weakened vessel wall that leads to dolichoectasia or aneurysm with subarchnoid or intracerebral haemorrhage, or that contributes to arterial dissection.
The presence of multifocal lesions at the grey–white matter junction suggests that VZV can spread haematogenously. However, such lesions were also seen in a patient with AIDS who developed encephalitis, and in whom pathological and virological evidence of VZV was found in the eye, optic chiasm, lateral geniculate bodies, and occipital cortex, indicating anterograde spread of VZV throughout the visual system.46 The presence of lesions at the grey–white matter junction after primary involvement of visual pathways might be explained by transaxonal spread of the virus from the eye to the brain, followed by local infection of small blood vessels that ultimately leads to ischaemic infarction and necrosis.
Virological analysis of a patient who died after VZV vasculopathy showed both VZV DNA (figure 5A) and VZV-specific antigen (figure 5B) in the media of cerebral arteries.20 VZV DNA47 and VZV antigen48 were detected in the cerebral arteries (and CSF) of two other patients with VZV vasculopathy in the same year. The detection of VZV DNA and VZV antigen supported the earlier detection of multinucleated giant cells (figure 5C) and herpes virions (figure 5D) in smooth-muscle cells of the middle cerebral artery49 and basilar artery50 of patients with VZV vasculopathy, as well as the detection of VZV antigen in two patients with VZV vasculopathy.42,51
Figure 5. Pathological and virological findings in arteries of patients who died from VZV vasculopathy.
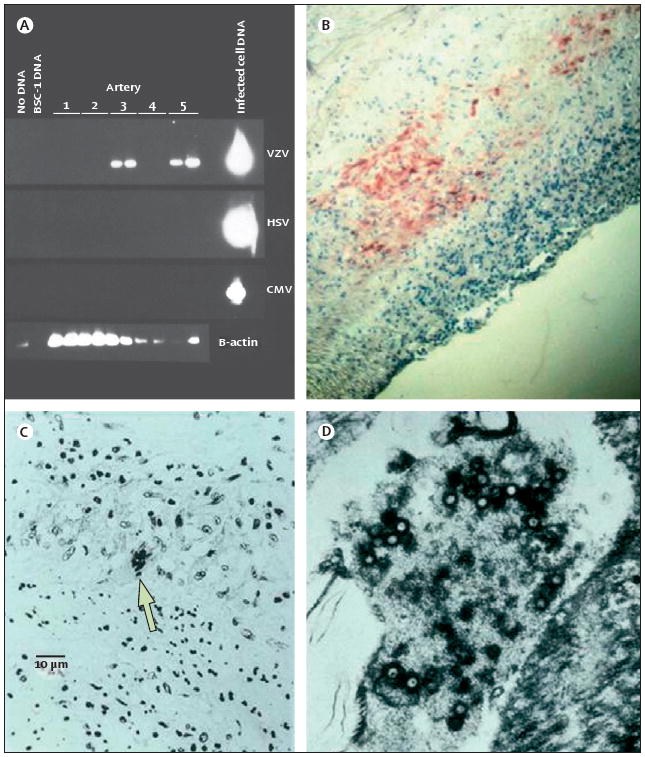
(A) VZV DNA in the posterior cerebral artery (lane 3) and basilar artery (lane 5). (B) VZV antigen (red) in the media of a cerebral artery. Panels A and B are reproduced from Gilden and co-workers,20 with permission from the American Academy of Neurology. (C) Cerebral artery with multinucleated giant cells (arrow). (D) Multiple herpes virions within a cerebral artery. CMV=cytomegalovirus. HSV=herpes zoster virus 1. VZV=varicella zoster virus.
Pathway of VZV infection of cerebral arteries
Results from several studies have shown afferent fibres from trigeminal and other ganglia to both intracranial and extracranial blood vessels, thus providing an anatomical pathway for the transaxonal spread of virus. Retrograde axonal transport studies in felines52 showed that the middle cerebral artery receives its sensory innervation from the ipsilateral trigeminal ganglia; horseradish peroxidase labelling of the middle cerebral artery revealed labelled neurons exclusively in the most medial part of the trigeminal ganglia, corresponding to the ophthalmic division of the trigeminal nerve. Furthermore, addition of horseradish peroxidase to dural structures, such as the middle meningeal artery or the transverse or superior sagittal sinus, revealed labelled cells in trigeminal ganglionic neurons that were always ipsilateral to the site of application.53
Later studies measured concentrations of immunoreactive substance P in the pial arteries of cats after bilateral removal of C1–3 dorsal root ganglia or trigeminal ganglia.54 Removal of the C1–3 ganglia resulted in decreased concentrations of tachykinin substance P in the vertebral and basilar arteries and their branches by 72% and 50–66%, respectively. Bilateral removal of the trigeminal ganglia decreased concentrations of substance P in all forebrain vessels, including the rostral basilar artery. Overall, the vertebrobasilar arteries and their tributaries are innervated by substance P-containing fibres originating from the upper cervical dorsal root ganglia, and the anterior cerebral artery is innervated by both trigeminal ganglia.54 Assuming a similar anatomy in human beings, the data provide a pathway for spread of VZV from cervical dorsal root ganglia to posterior circulation arteries and from trigeminal afferents to anterior circulation arteries.
Treatment
VZV vasculopathy is caused by productive viral infection in arteries, as evidenced by the presence of multinucleated giant cells, Cowdry A inclusion bodies and herpes virus particles, and VZV antigen and VZV DNA in cerebral vessels.20 Thus, all patients are typically treated with intravenous aciclovir on the basis of category 3 evidence (ie, opinions of respected authorities based on clinical experience, descriptive studies, or reports of expert committees). The largest study so far consists of 30 patients. Of all the patients treated with aciclovir alone, 20 improved or stabilised, compared with 23 who improved or stabilised when treated with both aciclovir and steroids.9 Because the patients received different treatment regimens at different institutions in an uncontrolled setting, the determination of optimum dose, duration of antiviral treatment, and benefit of concurrent steroid therapy awaits prospective studies with larger case numbers.
The clinical diagnosis of VZV vasculopathy is strongly suspected when a patient with a recent history of varicella or zoster has a transient ischaemic attack or stroke corroborated by MRI abnormalities, particularly at the grey–white matter junction. The presence of a CSF mononuclear pleocytosis, including red blood cells in the CSF, along with angiography that reveals focal narrowing and beading in cerebral vessels, further support the clinical diagnosis. When the diagnosis of VZV vasculopathy is being considered, and we are awaiting CSF studies of anti-VZV IgG antibody or VZV DNA to confirm the diagnosis, we treat all patients immediately with intravenous aciclovir, 10–15 mg/kg three times daily for a minimum of 14 days. Because there is often an inflammatory response in cerebral arteries, we also give oral prednisone, 1 mg/kg daily for 5 days; no taper is needed. We never treat patients with VZV vasculopathy with steroids for more than a week because long-term treatment can potentiate virus infection. Finally, we have encountered several patients with VZV vasculopathy who continue to have neurological symptoms after intravenous aciclovir treatment. Most, but not all, of these patients were HIV positive or had AIDS; one patient had diabetes. In these situations, we have recommended oral valaciclovir, 1 g three times daily for an additional 1–2 months, which resulted in cessation of symptoms.
Questions and controversies
VZV vasculopathy versus encephalitis
Nearly all CNS diseases caused by VZV infection include stroke. Most diseases are not primary encephalitis, but are instead caused by unifocal or multifocal infarction that develops secondary to productive virus infection in large and small cerebral arteries. The question arises as to whether primary VZV encephalitis exists at all. Earlier reports of clinical cases of encephalitis associated with varicella55 and zoster56 do not provide a definitive answer. For example, MRI, which allows antemortem diagnosis of the focal nature of VZV vasculopathy, had not yet been developed at the time of these earlier studies, and none of the reports detailed histopathological findings. Nevertheless, there are patients with zoster who develop a transient encephalopathy and have a CSF pleocytosis. Normally, this presentation would suggest the possibility of mild pure VZV encephalitis. However, many patients with zoster have a CSF pleocytosis so it is impossible to be certain that the presence of the cells in the CSF is due to zoster alone or caused by encephalitis. Overall, although mild VZV encephalitis associated with zoster probably exists, it is far less common than VZV vasculopathy.
As the nomenclature of clinical unifocal and multifocal VZV vasculopathy becomes recognised, the fact that syndromes of large-vessel and small-vessel vasculopathy are not always distinct should be noted. Both might be involved and produce waxing and waning neurological symptoms and signs.20 In the future, reference to patients, such as case 1 in the study by Häusler and co-workers,57 as having VZV multifocal vasculopathy rather than VZV encephalitis would be prudent. Results from a neuropathological study of 32 fatal cases of varicella, which excluded cases of Reye's syndrome (acute encephalopathy with fatty degeneration of the liver), showed that intranuclear inclusions were restricted to areas of previous necrosis, whereas direct viral invasion was noted to be contiguous to areas where cerebral blood vessels or the blood–brain barrier were impaired. The notion of a primary vasculopathy was not mentioned.58 In a clinical study of four children with varicella who developed serious neurological disease, CT and MRI revealed bilateral basal ganglionic infarction in all brains.59 Finally, in one of the first cases of VZV encephalomyelitis that developed in a boy with leukaemia in whom VZV was isolated from brain tissue, microscopic findings revealed multiple areas of acute necrotic infarction.60 Four later cases of fatal VZV multifocal leucoencephalitis (three in patients with leukaemia and one in a patient with ovarian cancer) revealed multifocal abnormalities both on brain imaging and at autopsy,61–63 characteristic of multifocal VZV vasculopathy. In some cases, the virus was also found in the cerebral parenchyma; however, in protracted cases, the fact that the virus spreads beyond cerebral vessels (the primary site of pathology) to the brain parenchyma is not surprising. Essentially, in earlier reports of VZV encephalitis, VZV leucoencephalitis, and post-varicella encephalitis, the primary nature of the vasculopathy (reviewed elsewhere64) was not recognised. VZV vasculopathy is distinct from the entity of post-infectious encephalomyelitis, a rare CNS complication of chickenpox.
Panel: Major features of the VZV vasculopathies.
Unifocal or multifocal
Deep-seated infarction more common than superficial infarction
White matter more commonly affected than grey matter
Grey–white matter junctions commonly affected
Involvement of large or small arteries, but more commonly both
Rash not required for diagnosis
CSF pleocytosis can include increased red blood cells
CSF pleocytosis absent in a third of cases
Detection of VZV antibody superior to that of VZV DNA for diagnosis
Can cause spinal-cord infarction
Can cause aneurysm
Might present with subarachnoid haemorrhage
Might present with cerebral haemorrhage
Might present with carotid dissection
Might cause peripheral arterial disease
All of the above can occur in children or adults after varicella infection
CSF=cerebrospinal fluid. VZV=varicella zoster virus.
Is VZV the only viral cause of vasculopathy?
VZV is the only virus in human beings that has been shown to replicate in arteries and produce vasculopathy. Speculations about cytomegalovirus and HIV as causes of vasculopathy in human beings are not supported by pathological changes or by indication of productive virus infection of cerebral arteries in patients with transient ischaemic attacks or stroke. Many cases of HIV-associated vasculopathy most probably indicate an undiagnosed VZV vasculopathy, as supported by clinical features, CSF findings, brain imaging, and angiographic features. Thus, patients presenting with neurological deficits that are possibly suggestive of a vasculopathy, even in the absence of rash or CSF pleocytosis, should undergo virological testing of the CSF for both VZV DNA and anti-VZV IgG antibody as part of the diagnostic assessment.
Search strategy and selection criteria.
References for this Review were obtained from personal reprint files, supplemented by searches of PubMed with the search terms “varicella zoster virus”, “varicella zoster virus vasculopathy”, “VZV vasculopathy”, “varicella encephalitis”, “varicella zoster encephalomyelitis”, “herpes zoster encephalitis”, “herpes zoster myelitis”, “herpes zoster ophthalmicus”, “granulomatous angiitis”, and “cerebral angiitis” from 1948 to May, 2009. The bibliographies of the most recent articles were also screened to find other previously unidentified articles. Only articles published in English were reviewed.
Conclusions
VZV vasculopathies can complicate zoster or varicella and are caused by productive viral infection in cerebral arteries. The important features of VZV vasculopathy are summarised in the panel. Unlike most cases of acute viral encephalitis, VZV vasculopathy is often chronic and protracted. Lesions at grey–white matter junctions are a clue to diagnosis, which is best confirmed by the presence of anti-VZV IgG antibody in the CSF with reduced serum to CSF ratio of anti-VZV IgG compared with albumin or total IgG. Given a projected increase in both the ageing population and in individuals who are immunocompromised (eg, organ-transplant recipients or patients with cancer or AIDS), the incidence of VZV vasculopathy is likely to increase.
Among the protean neurological manifestations of VZV vasculopathy are aneurysm, subarachnoid and cerebral haemorrhage, arterial ectasia, and carotid dissection. Thus, in such patients, the clinician should determine whether there is a history of recent zoster or varicella rash and, if affirmative, then virological analysis for VZV should be done. Furthermore, because not all patients with VZV vasculopathy have a history of zoster or varicella rash, the CSF of all patients with unifocal or multifocal vasculopathy, as well as CNS angiitis of unknown aetiology, should be analysed for VZV DNA and anti-VZV IgG antibody. Rapid and accurate diagnosis can lead to effective treatment of VZV vasculopathy.
Acknowledgments
This work was supported in part by grants NS32623 (DG, RJC, RM) and AG06127 (DG) from the National Institutes of Health. MAN is supported by training grant T32-NS07321 from the National Institutes of Health.
Footnotes
Contributors: DG planned the Review, undertook the literature search, and wrote the first draft. DG, RJC, RM, and MAN jointly edited and finalised the text.
Conflicts of interest: We have no conflicts of interest.
Contributor Information
Don Gilden, Departments of Neurology and Microbiology, University of Colorado Denver, School of Medicine, Aurora, CO, USA.
Randall J Cohrs, Department of Neurology, University of Colorado Denver, School of Medicine, Aurora, CO, USA.
Ravi Mahalingam, Department of Neurology, University of Colorado Denver, School of Medicine, Aurora, CO, USA.
Maria A Nagel, Department of Neurology, University of Colorado Denver, School of Medicine, Aurora, CO, USA.
References
- 1.Askalan R, Laughlin S, Mayank S, et al. Chicken pox and stroke in childhood. A study of frequency and causation. Stroke. 2001;32:1257–62. doi: 10.1161/01.str.32.6.1257. [DOI] [PubMed] [Google Scholar]
- 2.Braun KPJ, Bulder MMM, Chabrier S, et al. The course and outcome of unilateral intracranial arteriopathy in 79 children with ischaemic stroke. Brain. 2009;132:544–57. doi: 10.1093/brain/awn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cravioto H, Feigin I. Noninfectious granulomatous angiitis with a predilection for the nervous system. Neurology. 1959;9:599–608. doi: 10.1212/wnl.9.9.599. [DOI] [PubMed] [Google Scholar]
- 4.Rosenblum WI, Hadfield MG. Granulomatous angiitis of the nervous system in cases of herpes zoster and lymphosarcoma. Neurology. 1972;22:348–54. doi: 10.1212/wnl.22.4.348. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert GJ. Herpes zoster ophthalmicus and delayed contralateral hemiparesis. Relationships of the syndrome in central nervous system granulomatous angiitis. JAMA. 1974;229:302–04. [PubMed] [Google Scholar]
- 6.Hall S, Carlin L, Roach ES, McLean WT., Jr Herpes zoster and central retinal artery occlusion. Ann Neurol. 1983;13:217–18. doi: 10.1002/ana.410130226. [DOI] [PubMed] [Google Scholar]
- 7.Gilden DH, Lipton HL, Wolf JS, et al. Two patients with unusual forms of varicella-zoster virus vasculopathy. N Eng J Med. 2002;347:1500–03. doi: 10.1056/NEJMoa020841. [DOI] [PubMed] [Google Scholar]
- 8.Hilt DC, Buchholz D, Krumholz A, Weiss H, Wolinsky JS. Herpes zoster ophthalmicus and delayed contralateral hemiparesis caused by cerebral angiitis: diagnosis and management approaches. Ann Neurol. 1983;14:543–53. doi: 10.1002/ana.410140509. [DOI] [PubMed] [Google Scholar]
- 9.Nagel MA, Cohrs RJ, Mahalingam R, et al. The varicella zoster virus vasculopathies. Clinical, CSF, imaging, and virologic features. Neurology. 2008;70:853–60. doi: 10.1212/01.wnl.0000304747.38502.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ortiz GA, Koch S, Forteza A, Romano J. Ramsay Hunt syndrome followed by multifocal vasculopathy and posterior circulation strokes. Neurology. 2008;70:1049–51. doi: 10.1212/01.wnl.0000306634.51885.87. [DOI] [PubMed] [Google Scholar]
- 11.Kleinschmidt-DeMasters BK, Mahalingam R, Shimek C, et al. Profound cerebrospinal fluid pleocytosis and Froin's syndrome secondary to widespread necrotizing vasculitis in an HIV-positive patient with varicella zoster virus encephalomyelitis. J Neurol Sci. 1998;159:213–18. doi: 10.1016/s0022-510x(98)00171-3. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki Y, Riku Y, Goto Y, Mano K, Yoshida M, Hashizume Y. VZV vasculopathy associated with myelo-radiculoganglio-meningoencephalitis: an autopsy case of an immunocompetent 66-year-old male. J Neurol Sci. 2008;275:42–45. doi: 10.1016/j.jns.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 13.Gilden DH, Mahalingam R, Cohrs RJ, Kleinschmidt-DeMasters BK, Forghani B. The protean manifestations of varicella-zoster virus vasculopathy. J Neurovirol. 2002;8:75–79. doi: 10.1080/13550280290167902. [DOI] [PubMed] [Google Scholar]
- 14.Russman AN, Lederman RJ, Calabrese LH, Embi PJ, Forghani B, Gilden DH. Multifocal varicella-zoster virus vasculopathy without rash. Arch Neurol. 2003;60:1607–09. doi: 10.1001/archneur.60.11.1607. [DOI] [PubMed] [Google Scholar]
- 15.Gilden DH. Infectious causes of multiple sclerosis. Lancet Neurol. 2005;4:195–202. doi: 10.1016/S1474-4422(05)01017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgoon MP, Hammack BN, Owens GP, Maybach AL, Eikelenboom MJ, Gilden DH. Oligoclonal immunoglobulins in cerebrospinal fluid during varicella zoster virus (VZV) vasculopathy are directed against VZV. Ann Neurol. 2003;54:459–63. doi: 10.1002/ana.10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagel MA, Forghani B, Mahalingam R, et al. The value of detecting anti-VZV antibody in CSF to diagnose VZV vasculopathy. Neurology. 2007;68:1069–73. doi: 10.1212/01.wnl.0000258549.13334.16. [DOI] [PubMed] [Google Scholar]
- 18.Aurelius E, Johansson B, Skoldenberg B, Staland A, Forsgren M. Rapid diagnosis of herpes simplex encephalitis by nested polymerase chain reaction assay of cerebrospinal fluid. Lancet. 1991;337:189–92. doi: 10.1016/0140-6736(91)92155-u. [DOI] [PubMed] [Google Scholar]
- 19.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 5-1995. A 73-year-old man with focal brain lesions and peripheral-nerve disease. N Engl J Med. 1995;332:452–59. doi: 10.1056/NEJM199502163320708. [DOI] [PubMed] [Google Scholar]
- 20.Gilden DH, Kleinschmidt-DeMasters BK, Wellish M, Hedley-Whyte ET, Rentier B, Mahalingam R. Varicella zoster virus, a cause of waxing and waning vasculitis: the New England Journal of Medicine case 5-1995 revisited. Neurology. 1996;47:1441–46. doi: 10.1212/wnl.47.6.1441. [DOI] [PubMed] [Google Scholar]
- 21.Devinsky O, Cho ES, Petito CK, Price RW. Herpes zoster myelitis. Brain. 1991;114:1181–96. doi: 10.1093/brain/114.3.1181. [DOI] [PubMed] [Google Scholar]
- 22.Kenyon LC, Dulaney E, Montone KT, Goldberg HI, Liu GT, Lavi E. Varicella-zoster ventriculoencephalitis and spinal cord infarction in a patient with AIDS. Acta Neuropathol. 1996;92:202–05. doi: 10.1007/s004010050509. [DOI] [PubMed] [Google Scholar]
- 23.Thurnher MM, Bammer R. Diffusion-weighted MR imaging (DWI) in spinal cord ischemia. Neuroradiology. 2006;48:795–801. doi: 10.1007/s00234-006-0130-z. [DOI] [PubMed] [Google Scholar]
- 24.Orme HT, Smith AG, Nagel MA, Bert RJ, Mickelson TS, Gilden DH. VZV spinal cord infarction identified by diffusion-weighted MRI (DWI) Neurology. 2007;69:398–400. doi: 10.1212/01.wnl.0000266390.27177.7b. [DOI] [PubMed] [Google Scholar]
- 25.Gürsoy G, Aktin E, Bahar S, Tolun R, Özden B. Post-herpetic aneurysm in the intrapetrosal portion of the internal carotid artery. Neuroradiology. 1980;19:279–82. doi: 10.1007/BF00347809. [DOI] [PubMed] [Google Scholar]
- 26.de Broucker T, Verollet D, Schoindre Y, et al. Cerebral vasculitis with aneurysms caused by varicella-zoster virus infection during AIDS: a new clinicoangiographical syndrome. Rev Neurol. 2008;164:61–71. doi: 10.1016/j.neurol.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Bhayani N, Ranade P, Clark NM, McGuinn M. Varicella-zoster virus and cerebral aneurysm: case report and review of the literature. Clin Infect Dis. 2008;47:e1–3. doi: 10.1086/588842. [DOI] [PubMed] [Google Scholar]
- 28.Constantinescu CS. Association of varicella-zoster virus with cervical artery dissection in 2 cases. Arch Neurol. 2000;57:427. doi: 10.1001/archneur.57.3.427. [DOI] [PubMed] [Google Scholar]
- 29.Dalton CM, Jäger HR, Losseff NA, Greenwood RJ. Varicella zoster virus and intracranial dolichoectasia in a late adult cancer survivor. J Neurol Neurosurg Psychiatry. 2008;79:573–74. doi: 10.1136/jnnp.2007.120725. [DOI] [PubMed] [Google Scholar]
- 30.Elble RJ. Intracerebral hemorrhage with herpes zoster ophthalmicus. Ann Neurol. 1983;14:591–92. doi: 10.1002/ana.410140518. [DOI] [PubMed] [Google Scholar]
- 31.Jain R, Deveikis J, Hickenbottom S, Mukherji SK. Varicella-zoster vasculitis presenting with intracranial hemorrhage. Am J Neuroradiol. 2003;24:971–74. [PMC free article] [PubMed] [Google Scholar]
- 32.Massano J, Ferreira D, Toledo T, Mansilha A, Azevedo E, Carvalho M. Stroke and multiple peripheral thrombotic events in an adult with varicella. Eur J Neurol. 2008;15:e90–91. doi: 10.1111/j.1468-1331.2008.02267.x. [DOI] [PubMed] [Google Scholar]
- 33.Lapresle J, Lasjaunias P. Cranial nerve ischaemic arterial syndromes: a review. Brain. 1986;109:207–15. doi: 10.1093/brain/109.1.207. [DOI] [PubMed] [Google Scholar]
- 34.Nordborg C, Nordborg E, Petursdottir V, et al. Search for varicella zoster virus in giant cell arteritis. Ann Neurol. 1998;44:413–14. doi: 10.1002/ana.410440323. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy PGE, Grinfeld E, Esiri MM. Absence of detection of varicella-zoster virus DNA in temporal artery biopsies obtained from patients with giant cell arteritis. J Neurol Sci. 2003;215:27–29. doi: 10.1016/s0022-510x(03)00167-9. [DOI] [PubMed] [Google Scholar]
- 36.Kamholz J, Tremblay G. Chickenpox with delayed contralateral hemiparesis caused by cerebral angiitis. Ann Neurol. 1985;18:358–60. doi: 10.1002/ana.410180317. [DOI] [PubMed] [Google Scholar]
- 37.Leis AA, Butler IJ. Infantile herpes zoster ophthalmicus and acute hemiparesis following intrauterine chickenpox. Neurology. 1987;37:1537–38. doi: 10.1212/wnl.37.9.1537. [DOI] [PubMed] [Google Scholar]
- 38.Danchaivijitr N, Miravet E, Saunders DE, Cox T, Ganesan V. Post-varicella intracranial haemorrhage in a child. Dev Med Child Neurol. 2006;48:139–42. doi: 10.1017/S0012162206000302. [DOI] [PubMed] [Google Scholar]
- 39.West SL, Newton RW, Baildam EM, Turner AJ, Arkwright PD. Recurrent hemiplegia associated with cerebral vasculopathy following third trimester maternal herpes zoster infection. Dev Med Child Neurol. 2006;48:991–93. doi: 10.1017/S0012162206002179. [DOI] [PubMed] [Google Scholar]
- 40.Kuroiwa Y, Furukawa T. Hemispheric infarction after herpes zoster ophthalmicus: computed tomography and angiography. Neurology. 1981;31:1030–32. doi: 10.1212/wnl.31.8.1030. [DOI] [PubMed] [Google Scholar]
- 41.Jemsek J, Greenberg SB, Taber L, Harvey D, Gershon A, Couch RB. Herpes zoster-associated encephalitis: clinicopathologic report of 12 cases and review of the literature. Medicine. 1983;62:81–97. [PubMed] [Google Scholar]
- 42.Schmidbauer M, Budka H, Pilz P, Kurata T, Hondo R. Presence, distribution and spread of productive varicella zoster virus infection in nervous tissues. Brain. 1992;115:383–98. doi: 10.1093/brain/115.2.383. [DOI] [PubMed] [Google Scholar]
- 43.Chrétien F, Gray F, Lescs MC, et al. Acute varicella-zoster virus ventriculitis and meningo-myelo-radiculitis in acquired immunodeficiency syndrome. Acta Neuropathol. 1993;86:659–65. doi: 10.1007/BF00294307. [DOI] [PubMed] [Google Scholar]
- 44.Morgello S, Block GA, Price RW, Petito CK. Varicella-zoster virus leukoencephalitis and cerebral vasculopathy. Arch Pathol Lab Med. 1988;112:173–77. [PubMed] [Google Scholar]
- 45.Gray F, Mohr M, Rozenberg F, et al. Varicella-zoster virus encephalitis in acquired immunodeficiency syndrome: report of four cases. Neuropathol Appl Neurobiol. 1992;18:502–14. doi: 10.1111/j.1365-2990.1992.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 46.Gray F, Bélec L, Lescs MC, et al. Varicella-zoster virus infection of the central nervous system in the acquired immune deficiency syndrome. Brain. 1994;117:987–99. doi: 10.1093/brain/117.5.987. [DOI] [PubMed] [Google Scholar]
- 47.Melanson M, Chalk C, Georgevich L, et al. Varicella-zoster virus DNA in CSF and arteries in delayed contralateral hemiplegia: evidence for viral invasion of cerebral arteries. Neurology. 1996;47:569–70. doi: 10.1212/wnl.47.2.569. [DOI] [PubMed] [Google Scholar]
- 48.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 36-1996. A 37-year-old man with AIDS, neurologic deterioration, and multiple hemorrhagic cerebral lesions. N Engl J Med. 1996;335:1587–95. doi: 10.1056/NEJM199611213352109. [DOI] [PubMed] [Google Scholar]
- 49.Doyle PW, Gibson G, Dolman CL. Herpes zoster ophthalmicus with contralateral hemiplegia: identification of cause. Ann Neurol. 1983;14:84–85. doi: 10.1002/ana.410140115. [DOI] [PubMed] [Google Scholar]
- 50.Fukumoto S, Kinjo M, Hokamura K, Tanaka K. Subarachnoid hemorrhage and granulomatous angiitis of the basilar artery: demonstration of the varicella-zoster-virus in the basilar artery lesions. Stroke. 1986;17:1024–28. doi: 10.1161/01.str.17.5.1024. [DOI] [PubMed] [Google Scholar]
- 51.Eidelberg D, Sotrel A, Horoupian S, Neumann PE, Pumarola-Sune T, Price RW. Thrombotic cerebral vasculopathy associated with herpes zoster. Ann Neurol. 1986;19:7–14. doi: 10.1002/ana.410190103. [DOI] [PubMed] [Google Scholar]
- 52.Mayberg M, Langer RS, Zervas NT, Moskowitz MA. Perivascular meningeal projections from cat trigeminal ganglia: possible pathway for vascular headaches in man. Science. 1981;213:228–30. doi: 10.1126/science.6166046. [DOI] [PubMed] [Google Scholar]
- 53.Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol. 1984;16:157–68. doi: 10.1002/ana.410160202. [DOI] [PubMed] [Google Scholar]
- 54.Saito K, Moskowitz MA. Contributions from the upper cervical dorsal roots and trigeminal ganglia to the feline circle of Willis. Stroke. 1989;20:524–26. doi: 10.1161/01.str.20.4.524. [DOI] [PubMed] [Google Scholar]
- 55.Appelbaum E, Rachelson MH, Dolgopol VB. Varicella encephalitis. Am J Med. 1953;15:223–30. doi: 10.1016/0002-9343(53)90074-6. [DOI] [PubMed] [Google Scholar]
- 56.Appelbaum E, Kreps SI, Sunshine A. Herpes zoster encephalitis. Am J Med. 1962;32:25–31. doi: 10.1016/0002-9343(62)90179-1. [DOI] [PubMed] [Google Scholar]
- 57.Häusler M, Schaade L, Kemény S, Schweizer K, Schoenmackers C, Ramaekers VT. Encephalitis related to primary varicella-zoster virus infection in immunocompetent children. J Neurol Sci. 2002;195:111–16. doi: 10.1016/s0022-510x(02)00017-5. [DOI] [PubMed] [Google Scholar]
- 58.Takashima S, Becker LE. Neuropathology of fatal varicella. Arch Pathol Lab Med. 1979;103:209–13. [PubMed] [Google Scholar]
- 59.Darling CF, Larsen MB, Byrd SE, Radkowski MA, Palka PS, Allen ED. MR and CT imaging patterns in post-varicella encephalitis. Pediatr Radiol. 1995;25:241–44. doi: 10.1007/BF02011086. [DOI] [PubMed] [Google Scholar]
- 60.McCormick WF, Rodnitzky RL, Schochet SS, McKee AP. Varicella-zoster encephalomyelitis. Arch Neurol. 1969;21:559–70. doi: 10.1001/archneur.1969.00480180015001. [DOI] [PubMed] [Google Scholar]
- 61.Horten B, Price RW, Jimenez D. Multifocal varicella-zoster virus leukoencephalitis temporally remote from herpes zoster. Ann Neurol. 1981;9:251–66. doi: 10.1002/ana.410090308. [DOI] [PubMed] [Google Scholar]
- 62.Camack MA, Twiss J, Enzmann D, Amylon MD, Arvin AM. Multifocal leukoencephalitis caused by varicella-zoster virus in a child with leukemia: successful treatment with acyclovir. Pediatr Infect Dis J. 1993;12:402–06. doi: 10.1097/00006454-199305000-00011. [DOI] [PubMed] [Google Scholar]
- 63.Herrold JM, Hahn JS. Disseminated multifocal herpes zoster leukoencephalitis and subcortical hemorrhage in an immunosuppressed child. J Child Neurol. 1994;9:56–58. doi: 10.1177/088307389400900114. [DOI] [PubMed] [Google Scholar]
- 64.Gilden DH. Varicella zoster virus vasculopathy and disseminated encephalomyelitis. J Neurol Sci. 2002;195:99–101. doi: 10.1016/s0022-510x(02)00021-7. [DOI] [PubMed] [Google Scholar]