

Abstract
Histones are frequently decorated with covalent modifications. These histone modifications are thought to be involved in various chromatin-dependent processes including transcription. To elucidate the relationship between histone modifications and transcription, we derived quantitative models to predict the expression level of genes from histone modification levels. We found that histone modification levels and gene expression are very well correlated. Moreover, we show that only a small number of histone modifications are necessary to accurately predict gene expression. We show that different sets of histone modifications are necessary to predict gene expression driven by high CpG content promoters (HCPs) or low CpG content promoters (LCPs). Quantitative models involving H3K4me3 and H3K79me1 are the most predictive of the expression levels in LCPs, whereas HCPs require H3K27ac and H4K20me1. Finally, we show that the connections between histone modifications and gene expression seem to be general, as we were able to predict gene expression levels of one cell type using a model trained on another one.
Keywords: high CpG content promoter, low CpG content promoter, regression analysis, transcription
The DNA of eukaryotic organisms is packaged into chromatin, whose basic repeating unit is the nucleosome. A nucleosome is formed by wrapping 147 base pairs of DNA around an octamer of four core histones, H2A, H2B, H3, and H4 (1–5) which are subject to a number of posttranslational covalent modifications [(6); for review, see ref. 7]. These modifications can alter the chromatin structure and function by changing the charge of the nucleosome particle, and/or by recruiting protein complexes either individually or in combination (8). Hence, histone modifications are thought to constitute a “Histone Code,” which is read out by proteins to bring about specific downstream effects (9, 10).
Histone modifications have been linked to a number of chromatin-dependent processes, including replication, DNA-repair, and transcription. The link between histone modifications and transcription has been particularly intensively studied. It has been found that individual modifications can be associated with transcriptional activation or repression. Acetylation and phosphorylation generally accompany transcription; sumoylation, deimination, and proline isomerization are usually found in transcriptionally silent regions; methylation and ubiquitination are implicated in both activation and repression of transcription (8). Furthermore, the establishment of some modifications is dependent on the presence of other modifications, e.g., the catalysis of H3K4me3 requires the presence of H2BK120ub1 (the so-called trans-tail pathway) and the phosphorylation on serine 5 on the C-terminal domain of RNA polymerase II (pol II) (for review, see ref. 11, which also reviews other examples for the combinatorial action of histone modifications).
Transcription proceeds in a series of steps, also referred to as transcription cycle, starting with preinitiation complex formation, pol II recruitment, the transition to an initiating and thereafter elongating pol II, elongation, and finally termination (for review, see refs. 12, 13). The first four steps take place at the promoter and are tightly regulated to achieve a precise control of gene expression. The regulatory mechanisms depend on the action of transcription factors, which facilitate the recruitment of pol II and/or chromatin modifying complexes. Histone modifications can therefore be viewed as a read out of the activity of transcription factors. In line with this idea, there are established links between the distinct steps in the transcription cycle and some histone modifications, e.g., H3K4me3 is associated with transcription initiation [(14) and references therein], H3K27me3 with the repression of transcription elongation [(15) and references therein], and H3K36me3 with the removal of histone acetylations in the wake of an elongating pol II (for review, see ref. 11). However, in general, little is known about the relationship between histone modifications and the transcriptional process.
Here, we systematically study this relationship. We analyzed the recently published genome-wide localization data of 38 histone modifications and one histone variant measured in human CD4+ T-cells (16, 17). We address four major questions: (i) Is there a quantitative relationship between histone modifications levels and transcription? (ii) Are there histone modifications that are more important than others to predict transcript levels? (iii) Are there different requirements for different promoter types? (iv) Are the relationships general? To answer these questions, we derived models that quantitatively relate the measured expression level of genes (18) to the level of modifications at their promoters. We show that our models faithfully capture the measured expression levels of genes, suggesting that the levels of modifications are quantitatively related to gene expression and that there is a tight link between these histone modifications and the transcriptional process. Furthermore, combinations of only two to three modifications are sufficient to build models that give rise to at least 95% of the performance obtained by using all modifications. The separation of low CpG content promoters (LCPs) from high CpG content promoters (HCPs) revealed that different histone modifications are identified dependent on the CpG content of the promoters. Finally, gene expression levels of another cell type were accurately predicted using a model trained on the CD4+ T-cells, suggesting that the relationship between histone modifications and gene expression is a general one.
Results
Histone Modification Levels Are Predictive of Gene Expression in CD4+ T-Cells.
The presence or absence of certain histone modifications has been shown to correlate with the expression status of genes (19). To get a better understanding of the relationships between histone modifications and gene expression, we analyzed the publicly available genome-wide localization data for 38 histone modifications and one histone variant in human CD4+ T-cells (ChIP-seq data) (16, 17). We used the numbers of tags for each histone modification or variant, found in a region of 4,001 base pairs surrounding the transcription start sites of 14,802 RefSeq genes, as an estimation of the level of histone modifications. Furthermore, we examined microarray data measuring the transcript abundance of each of these genes in CD4+ T-cells (18), where the logarithm of the intensity served as expression value. Because open chromatin regions preferentially have higher tag counts than closed chromatin regions (20, 21), we also obtained ChIP-seq data for unspecific control ChIPs using goat and rabbit IgG antibodies in CD4+ T-cells (22) and mapped these tags to the same regions as the histone modification data.
We derived a simple model that relates the expression values to the histone modification levels (for details, see Methods). Briefly, we transformed the number of tags Nij for each modification i and promoter j to a logarithmic scale. Because some Nij were zero, we had to add a pseudocount αi to the levels of each modification to assure the logarithm would always be defined ( ). We chose the αi that maximizes the correlation of Nij to the expression value (estimated using 4,934 randomly selected promoters). We then built for the remaining 9,868 promoters a linear regression model where the entire set of modifications and the control IgG data served as input (Fig. 1; referred to as “full model”). We used 10-fold cross-validation to ascertain that a possible quantitative relationship is of general nature and not limited to a subset of genes. We evaluated the performance of the model by determining the Pearson correlation coefficient r between modeled and measured expression.
). We chose the αi that maximizes the correlation of Nij to the expression value (estimated using 4,934 randomly selected promoters). We then built for the remaining 9,868 promoters a linear regression model where the entire set of modifications and the control IgG data served as input (Fig. 1; referred to as “full model”). We used 10-fold cross-validation to ascertain that a possible quantitative relationship is of general nature and not limited to a subset of genes. We evaluated the performance of the model by determining the Pearson correlation coefficient r between modeled and measured expression.
Fig. 1.

Modeling framework. Models are equations that linearly relate the levels of histone modifications to the measured expression value.  corresponds to a vector of length L (the number of promoters), where the components are the transformed levels of a histone modification i (
corresponds to a vector of length L (the number of promoters), where the components are the transformed levels of a histone modification i ( , with Ni representing the number of tags in each promoter), a is the y intercept , and the bi to the slope associated with
, with Ni representing the number of tags in each promoter), a is the y intercept , and the bi to the slope associated with 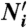 . y denotes a vector of length L whose components are the expression values. In the one-modification models, i can be any of the 39 modifications or two control IgG antibodies. In the two-modifications models, i and j are chosen to cover all combinations of two modifications without repetition. In the three-modifications models, i, j, and k are chosen to cover all combinations of three modifications without repetition. The full model incorporates all 41 variables.
. y denotes a vector of length L whose components are the expression values. In the one-modification models, i can be any of the 39 modifications or two control IgG antibodies. In the two-modifications models, i and j are chosen to cover all combinations of two modifications without repetition. In the three-modifications models, i, j, and k are chosen to cover all combinations of three modifications without repetition. The full model incorporates all 41 variables.
The full model is very well correlated to expression (r = 0.77, p-value of t-test < 2.2e-16; Fig. 2A), clearly demonstrating that the amounts of histone modifications at the promoter are well correlated to the expression level. Additional information about the slopes and the p-values of the slopes as well as the regression can be found in Tables S1 and S2. All correlation coefficients reported here have a p-value < 2.2e-16 (see Table S3 for a summary), so we omit p-values for the correlation coefficients in the remainder of the text. To check whether all modifications are required to model gene expression, we built models using combinations of one to three modifications (referred to as “one-modification models,” “two-modifications models,” etc.; Fig. 1).
Fig. 2.

Quantitative relationship between histone modification levels and expression. (A) Scatterplot with the predicted expression value in CD4+ T-cells using the full linear model on the x axis and the measured expression value in CD4+ T-cells on the y axis. The shades of blue indicate the density of points; the darker color, the more points. The red line indicates the linear fit between predicted and measured expression (y = 0.99x + 0.02), which are highly correlated (r = 0.77), indicating a quantitative relationship between levels of histone modifications at the promoter and gene expression levels. (B) Comparison of prediction accuracy between all possible one-modification, two-modifications, three-modifications models, and the full model for CD4+ T-cells. Models are sorted by ascending prediction accuracy along the x axis. The best models using only a small subset of modifications almost reach the prediction accuracy of the full linear model. (C) Bar plot showing the frequency of appearance of different histone modifications in best scoring three-modifications models (142 models) for CD4+ T-cells. Best scoring models are defined as reaching at least 95% of prediction accuracy of the full linear model. (D, E) Expression values of genes, which were at least 5-fold up or down regulated in CD36+ and CD133+ cells with respect to CD4+ T-cells, predicted using model parameters trained on data from CD4+ T-cells. The predicted and measured expression values are highly correlated for both CD36+ (D) (r = 0.75; 1,412 genes) and CD133+ (E) (r = 0.63; 1,243 genes) cells. The equations of the regression line for both CD36+ and CD133+ cells (y = 0.43x + 6.04 and y = 0.53x + 6.17, respectively) show a high value of the intercept and a slope different from one due to the fact that the levels of the histone modifications were not normalized across cell types.
We determined that the top one-modification (rmax = 0.72, H3K27ac), two-modifications (rmax = 0.74, H3K27ac + H4K20me1) and three-modifications models (rmax = 0.75, H3K27ac + H3K4me1 + H4K20me1) are very well correlated to expression (Fig. 2B). These results establish that not all modifications are equally important, possibly because of a high degree of redundancy. Moreover, the levels of a single modification (H3K27ac) can be used to faithfully model gene expression. However, the prediction accuracy increases as one goes from the best one-modification model increasing the number of modifications accounted for to the full model. This increase is not simply due to the higher model complexity (the more modifications considered, the more complex a model is), because the prediction accuracy is computed on test data. To confirm this, we used the Bayesian information criterion (BIC) (23), which also accounts for the model complexity. As long as the value of the BIC decreases, increasing the model complexity is beneficial. The BIC value keeps decreasing continuously, suggesting that it is not the model complexity which governs the increase in prediction accuracy. However, the BIC values decrease only slightly after using more than four modifications (Fig. S1A). Our results suggest that the levels of as few as three modifications at the promoter are enough to faithfully model expression of the associated gene.
To identify modifications, whose levels harbor most of the information about gene expression, we focused on the three-modifications models. We determined all three-modifications models whose Pearson correlation coefficient r reached at least 95% of the one obtained by the full model (rfull = 0.77). There were 142 models that satisfied this criterion, which is a sufficiently high number to justify an overrepresentation analysis by computing the probability of observing a particular modification in that many subsets due to chance alone. Our results show that four histone modifications, H4K20me1, H3K27ac, H3K79me1, and H2BK5ac (Fig. 2C), are significantly overrepresented in the set of models (p-values of the hypergeometric test 7.58e-50, 8.95e-46, 7.83e-30, and 2.88e-27, respectively), each of them appearing in roughly half the studied models (57.7%, 54.9.5%, 42.9%, and 40.8%, respectively). The remaining histone modifications appear in at most 7% of the models, a frequency expected from random sampling (p-value of the hypergeometric test 0.47). Goat and rabbit IgG were found in only a small number of the best models (2.11% and 3.52%, p-values of the hypergeometric test 0.99 and 0.95, respectively), which shows they do not contribute significantly to the prediction accuracy. This, along with the fact that the prediction accuracy of one-modification models trained on these variables is low (rgoat.IgG = 0.15; rrabbit.IgG = 0.09; Fig. S1B), shows that the high prediction accuracy of linear models using histone modifications as predictors is not merely a consequence of higher accessibility of open chromatin. The result of the overrepresentation analysis is robust to variations of the threshold (Fig. S2A and B), presuming that the set of best scoring models does not exceed 20% of the total number of models, which then naturally leads to random inclusion of other histone modifications. Thus, H4K20me1, H3K27ac, H3K79me1, and H2BK5ac appear to be the most important modifications associated with gene expression levels.
Interestingly, the prediction accuracies of the one-modification models, based on the overrepresented modifications only, greatly vary (rH3K27ac = 0.72, rH2BK5ac = 0.71, rH3K79me1 = 0.67, and rH4K20me1 = 0.55; Fig. S1B). Furthermore, the two-modifications with the highest individual information content, H3K27ac and H2BK5ac, appear only two times together in the set of best scoring models (Fig. S2C), suggesting that the information they provide is redundant, which is supported by the finding that their levels are highly correlated (r = 0.97). H4K20me1 and H3K79me1 occur together in only three of the 142 models, indicating that they are at least partially redundant. Moreover, we found that in almost all 142 models (92.9%), H3K27ac or H2BK5ac occur together with either H4K20me1 or H3K79me1.
Differential Requirement of Histone Modifications in High Vs. Low CpG Content Promoters.
Given the good agreement between modeled and measured expression values, we proceeded with further analysis of our models to infer the relationships between distinct histone modifications and different groups of promoters. More specifically, we separated the promoters into LCPs and HCPs. This was motivated by the fact that the nucleosomes in HCPs are almost always decorated with H3K4me3, whereas nucleosomes in LCPs carry this modification only when they are expressed (24). H3K4me3 is thought to be a mark of transcription initiation [(14) and references therein]. We reasoned that if these promoters are differently marked by histone modifications then the predictive power of histone modifications should also differ between these two groups of promoters.
We divided the promoters according to their CpG content, with 2,779 LCPs and 7,089 HCPs, and determined the regression parameters for the full model on both groups separately in a 10-fold cross-validation setting. As a first result, we observed that the prediction accuracy for LCPs (r = 0.72) is comparable to HCPs (r = 0.75) (Fig. S3).
We proceeded with building models with all combinations of one, two, and three modifications, for both sets of promoters separately. For HCPs, we found that the overall ranking of models remained very similar to the ranking of models determined for all promoters. This is hardly surprising because HCPs constitute 72% of all analyzed promoters, suggesting that the results for all promoters were dominated by HCPs. For LCPs, the ranking of the models changed compared to all promoters, although for the one-modification models, H3K27ac still remained the best correlated modification (r = 0.65). Strikingly, upon considering combinations of two modifications, we found that the model with the combination of H3K4me3 and H3K79me1 performed best (r = 0.69, compared to H3K27ac and H3K79me1 r = 0.67).
Next, we determined the overrepresented modifications in the best performing three-modifications models. H4K20me1 and H3K27ac (and possibly H2BK5ac) are significantly overrepresented among the best scoring models for HCPs (Fig. 3A; p-values of the hypergeometric test 9.97e-43, 2.58e-31, and 0.003, respectively), and H3K4me3 and H3K79me1 are significantly overrepresented in the LCPs (Fig. 3B; p-values of the hypergeometric test 9.71e-36 and 2.1e-34, respectively), demonstrating that different modifications are important for the prediction of expression of genes in these two groups.
Fig. 3.

Differences in transcriptional regulation between HCPs and LCPs. (A, B) Bar plots showing the frequency of appearance of different histone modifications in best scoring three-modifications models for HCPs (B) (50 models) and LCPs (C) (40 models) in CD4+ T-cells. Best scoring models are defined as reaching at least 95% of prediction accuracy of the full model trained on HCPs and LCPs, respectively. Only the top ten modifications are depicted. (C) Normalized cumulative tag counts in the region of -500 base pairs to 3,000 base pairs surrounding the transcription start site of RefSeq genes in CD4+ T-cells for the five important modifications identified by our analysis.
To gain further insight into the possible functions of the histone modifications that were highly correlated with gene expression for HCPs and LCPs, we examined the average normalized tag densities for these five modifications in the region surrounding the transcription start site (TSS) (Fig. 3C), referred to as localization analysis. We found that H3K4me3, H3K27ac, and H2BK5ac have the highest levels at the promoter, with the highest peaks around 100 base pairs downstream of the TSS. H3K79me1 is enriched along the gene body, and H4K20me1 shows two distinct patterns: a peak close to the promoter at a similar position to H3K4me3 and H3K27ac, and a further enrichment across the gene body region.
Histone Modification Levels Are Predictive of Gene Expression Across Different Cell Types.
We showed that models incorporating only the information of four histone modifications can accurately predict gene expression levels within a given cell type. Next, we wanted to check whether models trained on the data of one cell type can be used to predict gene expression in another cell type. We obtained genome-wide localization data for histone modifications as well as microarray gene expression values in CD36+ and CD133+ cells (25). Here, our analysis was restricted to the nine histone modifications (H3K4me1/3, H3K27me1/3, H2A.Z, H4K20me1, H3K9me1/3, and H3K36me3) measured in all three cell types. We trained a linear model on the CD4+ data. Using the trained model parameters, we predicted gene expression levels from histone modification data measured in CD36+ and CD133+ cells.
Because the gene expression profiles of CD36+ and CD133+ cells are highly correlated to CD4+ T-cells (r = 0.79 and r = 0.82, respectively) (Fig. S4A and B), we restricted the prediction to genes with a fold change higher than five. The correlation of predicted and measured expression values is high for both CD36+ (r = 0.75; Fig. 2D) and CD133+ (r = 0.63; Fig. 2E) cells, and does not vary significantly when genes with fold changes higher or lower than five are selected (Fig. S4C–H). This result strongly suggests that the relationship between histone modifications and gene expression is general and not dependent on the cellular context (see Discussion).
In summary, we found that the levels of histone modifications are well correlated to gene expression and that this relationship can be generalized across different cell types. Moreover, our analysis revealed that the number of important modifications can be reduced from 39 to four, indicating that these four modifications may play a crucial role in the transcriptional process, both reinforcing each other or in a combinatorial manner. We also found that, upon separating promoters into LCPs and HCPs, different sets of modifications were found to be important, which indicates that these promoters are regulated differently (see Discussion).
Discussion
We have shown that the levels of histone modifications at a promoter proximal region are well correlated to the expression of genes. Other studies classified the promoters for each modification into groups (17, 26), e.g., modification X is present or absent. Discretization ought to have two beneficial effects, namely the reduction of noise and parameters. Although discretization is necessary in some modeling approaches to reduce the number of parameters, e.g., learning a Bayesian network (26), in our approach, it increases the number of parameters, because one has to choose at least one threshold for each modification in addition to the slopes in the linear regression model. If discretization is indeed beneficial for modeling gene expression, we expect that the results of a discrete model should be better than a corresponding continuous model. Thus, we compared full models incorporating either the levels directly (continuous model) or a binary classification of them (discrete model). Although the correlation is not significantly different (Fig. 2A and Fig. S5A), the mean squared error (MSE) increased from 1.54 for the continuous model to 1.71 for the discrete model. The same is true for the best three-modification continuous and discrete models. Here, the discrete model is only able to reproduce the general trend in expression values and thus has a higher MSE (MSE = 1.84; Fig. S5B) than the continuous model (MSE = 1.68, which is even lower than the MSE for the full discrete model; Fig. S5C). We conclude that discretization has no beneficial effect on the prediction accuracy and argue that in our modeling framework discretization is not necessary and is even reducing the predictive power at the cost of increasing the number of parameters.
We demonstrated that only a few histone modifications are necessary to faithfully model gene expression. This finding can be understood if one assumes that the histone modifications belong to different groups, whose members are either involved in transcription or not. The modifications within the transcription-related groups provide almost the same information and our approach selects one representative modification. Alternatively, the selected histone modifications are involved in distinct steps during the transcription cycle. For example, they could recruit activities that are required to enable RNA pol II to progress from an initiating to an elongating state. In the light of the “Histone Code Hypothesis,” the latter idea is very attractive, but we would have much more confidence in supporting this idea if we were able to reproduce our results using an equally rich dataset in a preferentially independent cell type, which to our knowledge is currently not available.
We used three sets of promoters, namely all, LCPs, and HCPs to identify “important” modifications. Upon analyzing all promoters, we found that H2BK5ac, H3K27ac, H3K79me1, and H4K20me1 are overrepresented in models giving rise to the highest prediction accuracy in CD4+ T-cells. A recent study identified a common set of 17 modifications (mainly acetylations), referred to as the backbone. These modifications colocalize and their levels are well correlated (17). Genes with all of these backbone modifications present tend to be expressed, suggesting that either all or a subset of them are involved in transcription. Our analysis revealed only two of these modifications, H3K27ac and H2BK5ac, are important for modeling gene expression. This indicates that the remaining backbone modifications carry either redundant information or are less important for gene expression. Furthermore, the other two important modifications, H3K79me1 and H4K20me1, have been shown to be enriched in highly expressed genes, along with the modification backbone (17). This observation is in line with the idea that H3K79me1 and H4K20me1 are also involved in transcription. Thus, we conclude that our approach identified histone modifications which are likely to be key players in the transcriptional process.
We identified different sets of modifications important for modeling gene expression driven by LCPs or HCPs. In LCPs, we found that H3K4me3 and H3K79me1, while in HCPs H3K27ac and H4K20me1, were identified. These assignments can be reproduced using RNA-seq (27) instead of the microarray data, suggesting that a possible measurement bias due to the microarray technology is not a major factor. The prediction accuracy for modeling RNA-seq derived expression values is even higher (r = 0.81; Fig. S6A) than the one using microarray expression data (r = 0.77). The results of the overrepresentation analysis for all, HCPs, and LCPs are comparable between the RNA-seq and microarray-derived expression values. The only difference was that only H4K20me1, H3K27ac, and H2BK5ac, but not H3K79me1, are identified as being overrepresented in best scoring linear models for all promoters. However, when analyzing best scoring models for LCPs, H3K79me1 clearly comes up as overrepresented (Fig. S6B–D).
The reason for the difference in the important histone modifications in LCPs and HCPs is unclear, but indicates that different regulatory mechanisms act on these two promoter types. A possible clue for the function of the selected modifications is provided by the localization analysis (Fig. 3C). H3K4me3, H3K27ac, and H2BK5ac have the highest levels at the promoter, with the highest peak around 100 base pairs downstream of the TSS. H3K79me1 is enriched along the gene body, and H4K20me1 shows two distinct patterns: a peak close to the promoter at a similar position to H3K4me3 and H3K27ac, and an enrichment across the gene body region. The localization of these histone modifications suggests that H3K27ac, H2BK5ac, H3K4me3, and H4K20me1 function during transcription initiation and/or promoter clearance, whereas H3K79me1 and H4K20me1 are involved in transcription elongation.
Although for H3K4me3 a function during transcription initiation has been proposed (e.g., ref. 14 and references therein), a similar function has not been established for H3K27ac. A possible action of H3K27ac might be to prevent the repressive trimethylation of the same residue, because H3K27ac and H3K27me3 are mutually exclusive. Alternatively, H3K27ac itself could be recognized by a protein complex required for transcription. H3K79me1 is almost absent at the TSS and its levels increase in the gene body, indicating that it is involved in transcription elongation, in line with previous observations (28, 29). The functions of H2BK5ac and H4K20me1 in general, and in particular during transcription, are not well understood.
Because we showed that histone modification levels are predictive of the gene expression levels in CD4+ T-cells, we further investigated whether this is a universal property which holds true for other cell types. We were able to successfully predict expression of genes in CD36+ and CD133+ cells, using histone modification data measured in these cells and model parameters trained on CD4+ data. Significantly, the prediction accuracy does not depend strongly on the level of change in expression in different cell types. Thus, our results establish the idea that the relationships between histone modification and gene expression are general. Furthermore, they underscore that the histone modifications and the transcriptional process are tightly connected to each other. We want to emphasize that our analysis as well as the data do not allow for deciding whether the histone modifications are cause or consequence of transcription, because the uncovered relationships are correlative in nature and therefore inherently undirected. However, our results imply that the histone modifications are very close to RNA pol II in the regulatory network controlling its activity. Whether they are upstream and/or downstream has to be elucidated in further experimental studies.
In summary, we have shown that the relationships between histone modification and transcription are well reproducible across different cell types. Furthermore, we singled out a small number of modifications, which together can account for a large portion of the expression variance. Whether these modifications play a crucial role during transcription, or whether they are representatives for groups of equally important modifications has to be clarified by further experimental studies. Regardless of which scenario turns out to be true, we can pinpoint a small number of modifications whose levels at the promoter can be used to infer gene expression and hence provide some information about the transcriptional process, which reduces the experimental effort to study the relationship between histone modifications and transcription.
Methods
Data.
The RefSeq Genes annotation track for the human genome sequence (hg18, March 2006) was downloaded from the University of California, Santa Cruz Genome Bioinformatics web site (http://genome.ucsc.edu/). The coordinates of uniquely mapped ChIP-Seq tags were taken from genome-wide studies of the distribution of 19 lysine or arginine histone methylations, one H2A.Z histone variant (16), and 19 histone acetylations in CD4+ T-cells (17). These coordinates were transformed by adding or subtracting 73 base pairs (for tags mapping to the + or - strand, respectively), thus centering the tags on the nucleosome, because only DNA corresponding to the ends of the nucleosome is sequenced. Tags were then mapped to a 4,001 base pair region surrounding the TSS of RefSeq genes. The tags in this region were summed and each gene was represented by 39 values (one per modification). ChIP-Seq data for goat and rabbit IgG was obtained (22) and mapped to the same regions as histone modifications data, but was not transformed by adding or substracting base pairs from the coordinates of the tags.
Expression microarray data for resting T-cells performed on Affymetrix Human Genome U133 Plus 2.0 GeneChip was taken from ref. 18. Raw expression values were averaged over all replicates. Only the RefSeq genes that could be uniquely mapped to an Affymetrix probe identifier were used in further analysis.
Selecting Promoters for Analysis.
To exclude the possibility that some of the RefSeq genes used in our analysis correspond to alternative transcripts of the same gene, all RefSeq genes were mapped to corresponding Unigene clusters (30), and only one RefSeq gene per cluster was kept, leaving 14,802 RefSeq genes for further analysis (see SI Methods for details)
Predicting Expression Using Linear Regression.
The whole dataset was divided into two random sets D1 (4,934 promoters) and D2 (9,868 promoters). Each modification i and promoter j was represented by the sum of tag counts Nij in the 4,001 base pair region surrounding the TSS and transformed to a logarithmic scale. Optimized pseudocounts αi were estimated on D1 (see SI Methods for details) and added to each Nij, to avoid undefined values of the logarithm when Nij equals zero. The 41 transformed values  in the D2 set were then used as predictor variables for training a linear regression model (full model) to predict the logarithm of expression. The Pearson correlation coefficient r between predicted and measured values was calculated in a 10-fold cross-validation setting (see SI Methods for details) and used as a measure of prediction accuracy.
in the D2 set were then used as predictor variables for training a linear regression model (full model) to predict the logarithm of expression. The Pearson correlation coefficient r between predicted and measured values was calculated in a 10-fold cross-validation setting (see SI Methods for details) and used as a measure of prediction accuracy.
All possible one-modification (41 models), two-modifications (820 models), and three-modifications models (10,660 models) were produced and their performance assessed as described above. The analysis was also repeated using RNA-Seq tag counts in CD4+ T-cells as a measure of expression (see SI Methods for details).
To test against overfitting, we produced linear models using all possible combinations of 1–5 and 37–41 modifications (there are too many 6–36 modification models, so we excluded them). In each group of models (corresponding to the number of modifications used in combination), the model with highest prediction accuracy was identified, and the tradeoff between model complexity and prediction accuracy assessed using the BIC (23).
Analysis of the Importance and Combinatorial Influence of Modifications.
All 10,660 possible three-modifications models were produced and their prediction accuracy assessed. Best scoring models were defined as those for which the prediction accuracy reaches at least 95% of the prediction accuracy of the full model. The number of times each modification appears amongst this set of models was divided by the number of best scoring models to determine the fraction of appearance of each histone modification.
Prediction of Expression Across Different Cell Types.
Coordinates of ChIP-Seq tags for nine histone modifications and expression microarray data measured in CD36+ and CD133+ cells were taken from ref. 25. Expression values from both cell lines were normalized with respect to expression values in CD4+ T-cells, by first fitting a regression line between the two respective expression values and then transforming the expression values in either CD36+ or CD133+ cells in such a way that the equation of the regression line is equal to y = x. Optimal pseudocounts were determined and histone modifications data transformed as described above. A linear model was trained on data from CD4+ T-cells using nine histone modifications common to all three datasets. The linear model was then used to predict expression values in CD36+ and CD133+ cells using histone modifications data measured in each cell type as predictor variables.
Classification of Promoters According to CpG Content.
Normalized CpG content in the region of 3,000 base pairs surrounding the TSS was calculated as defined previously (31), with the promoters having a normalized CpG content > 0.4 being classified as HCP and the others being classified as LCP.
Supplementary Material
Acknowledgments.
We thank all members of the Computational Molecular Biology Department and especially Paz Polak for helpful comments and discussion. R.K. and K.V. acknowledge the European Molecular Biology Organization Young Investigator Program (Installation Grant 1431/2006 to K.V.), International Center for Genetic Engineering and Biotechnology (Italy) collaborative research program Grant CRP/CRO07-03, and Croatian Ministry of Science, Education and Sports Grant 119-0982913-1211. R.K. was funded by a fellowship from the International Max Planck Research School for Computational Biology and Scientific Computing.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909344107/DCSupplemental.
References
- 1.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319(5):1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD. Chromatin structure: A repeating unit of histones and DNA. Science. 1974;184(4139):868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 3.Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science. 1974;184(4139):865–868. doi: 10.1126/science.184.4139.865. [DOI] [PubMed] [Google Scholar]
- 4.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 5.Thomas JO, Kornberg RD. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci USA. 1975;72(7):2626–2630. doi: 10.1073/pnas.72.7.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 8.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 10.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs SM, Laribee RN, Strahl BD. Protein modifications in transcription elongation. Biochim Biophys Acta. 2009;1789(1):26–36. doi: 10.1016/j.bbagrm.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24(6):280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Svejstrup JQ. The RNA polymerase II transcription cycle: Cycling through chromatin. Biochim Biophys Acta. 2004;1677(1–3):64–73. doi: 10.1016/j.bbaexp.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130(1):77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29(1):69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellor J, Dudek P, Clynes D. A glimpse into the epigenetic landscape of gene regulation. Curr Opin Genet Dev. 2008;18(2):116–122. doi: 10.1016/j.gde.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Auerbach RK, et al. Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci USA. 2009;106(35):14926–14931. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teytelman L, et al. Impact of chromatin structures on DNA processing for genomic analyses. PLoS One. 2009;4(8):e6700. doi: 10.1371/journal.pone.0006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hastie T, Tibshirani R, Friedman T. The Elements of Statistical Learning: Data Mining, Inference and Prediction. New York: Springer–Verlag; 2001. pp. 206–208. [Google Scholar]
- 24.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui K, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4(1):80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu H, Zhu S, Zhou B, Xue H, Han JD. Inferring causal relationships among different histone modifications and gene expression. Genome Res. 2008;18(8):1314–1324. doi: 10.1101/gr.073080.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chepelev I, Wei G, Tang Q, Zhao K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic Acids Res. 2009;37(16):e106. doi: 10.1093/nar/gkp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seila AC, et al. Divergent transcription from active promoters. Science. 2008;322(5909):1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steger DJ, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28(8):2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wheeler DL, et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31(1):28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103(5):1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.