

Abstract
Inspired by the seminal work of Anfinsen, investigations of the folding of small water-soluble proteins have culminated in detailed insights into how these molecules attain and stabilize their native folds. In contrast, despite their overwhelming importance in biology, progress in understanding the folding and stability of membrane proteins remains relatively limited. Here we use mutational analysis to describe the transition state involved in the reversible folding of the β-barrel membrane protein PhoPQ-activated gene P (PagP) from a highly disordered state in 10 M urea to a native protein embedded in a lipid bilayer. Analysis of the equilibrium stability and unfolding kinetics of 19 variants that span all eight β-strands of this 163-residue protein revealed that the transition-state structure is a highly polarized, partly formed β-barrel. The results provide unique and detailed insights into the transition-state structure for β-barrel membrane protein folding into a lipid bilayer and are consistent with a model for outer membrane protein folding via a tilted insertion mechanism.
Keywords: beta barrel, membrane protein, PagP, phi-value analysis, protein folding
Membrane proteins are encoded by up to 30% of genes in most organisms (1) and are crucial to a wide diversity of essential activities in biology (for example, ref. 2 and references therein). Identifying the forces that govern the folding and stability of membrane proteins is thus fundamental to understanding their functions and elucidating how sequence changes give rise to altered functional properties and, for some such proteins, grave disease (3). Membrane proteins divide into two major classes: proteins that are α-helical in their transmembrane domains and are ubiquitously distributed and proteins that span the membrane in the form of a β-barrel and are found within the outer membranes of Gram-negative bacteria, chloroplasts, and mitochondria. α-Helical membrane proteins are difficult to unfold to an unstructured state (4), hampering detailed investigations of their folding mechanisms into the lipid bilayer (5). Important progress in folding studies on this class of membrane proteins has been made recently, however, using detergent-solubilized proteins (6). In particular, partial denaturation of bacteriorhodopsin followed by refolding/unfolding into mixed detergent/lipid micelles by titration of the destabilizing detergent sodium dodecyl sulfate has provided insights into the packing of preformed helices around a critically maintained helical core in the denatured state of the protein (7–10). Such an approach cannot be employed, however, to investigate folding of membrane proteins into a lipid bilayer, the physicochemical properties of which are known to have profound effects on membrane protein stability and assembly (10–14). By contrast with the recalcitrance of α-helical membrane proteins to complete denaturation, β-barrel transmembrane proteins are readily unfolded using membrane-compatible chaotropes (14–16). For outer membrane protein A (OmpA), this has allowed energetic contributions of functionally important hydrogen bonds (17) and of residues in the anchoring aromatic girdles (18) on stability to be quantified. Despite their experimental tractability and ubiquity in nature, however, a detailed structural description of the folding transition state of any β-barrel membrane protein remained to be achieved.
Here we have exploited the experimental amenability of the Escherichia coli outer membrane protein PhoPQ-activated gene P (PagP) to provide a detailed analysis of the folding mechanism of a β-barrel transmembrane protein. PagP comprises a monomeric eight-stranded β-barrel with an N-terminal, amphiphathic α-helix (19, 20) (Fig. 1A). The protein functions as an enzyme as part of the bacterial stress response mechanism, reinforcing the outer membrane by transferring a palmitoyl chain from phospholipids to lipopolysaccharides (21). We have previously shown that PagP folds spontaneously into lipid bilayers, commencing from a highly denatured state that lacks residual secondary structure in 6 M guanidinium chloride (Gdn-HCl). Having established conditions under which this transition is fully reversible, we here present an integrated thermodynamic and kinetic analysis of the folding and unfolding transition of the wild-type protein. Combined with analysis of the stability and folding properties of 19 variants of the protein that contain single-point mutations spanning all eight β-strands of the native β-barrel, we describe the transition state for folding of a β-barrel membrane protein into a lipid bilayer in residue-specific detail. The results reveal a transition-state structure that is highly polarized, involving at least partial structure formation in all eight β-strands of the native β-barrel, and suggest a route by which a folding polypeptide chain can both enter and traverse a bilayer to adopt its native state.
Fig. 1.

(A) Cartoon representation of PagP [Protein Data Bank (PDB) ID code 1THQ (19); W. L. DeLano, http://www.pymol.org (2002)]; β-strands are labeled A–H; periplasmic turns t1–3. (B) Equilibrium refolding (▪) and unfolding (□) of wild-type PagP. The unfolding transitions of PagP variants bearing mutations in residues in (B) the hydrophobic surface, (C) the aromatic girdles, and (D) the barrel interior are also shown. For these variants residues mutated that are located in the N-terminal half of the protein sequence are indicated by closed symbols, whereas residues in the C-terminal half of the protein sequence are shown as open symbols. Solid lines are global fits to a two-state mechanism yielding a common MUN = 6.86 ± 0.20 kJ/mol/M. All experiments were performed using 0.4 μM PagP in diC12∶0PC-liposomes at an LPR of 3200∶1 in 50 mM sodium phosphate buffer pH 8 at 25 °C.
Results
Thermodynamic and Kinetic Measurements of the Folding and Unfolding Transition of Wild-Type PagP.
In previous work we have shown that PagP folds spontaneously from a completely unfolded conformation in 6 M Gdn-HCl into di-lauroyl-phosphatidylcholine (diC12∶0PC)-liposomes at a lipid-to-protein ratio (LPR) of 800∶1 in the presence of 7 M urea (22). Here, using tryptophan (Trp)-fluorescence as a probe for correct folding, we established that 0.4 μM PagP denatured in 10 M urea folds efficiently to its native state in 100 nm diC12∶0PC-liposomes at an LPR of 3200∶1 in a transition that is completely reversible and is independent of both the protein and lipid concentration under the conditions employed (Fig. 1B) (SI Text and Fig. S1). The resulting equilibrium curve fitted well to a two-state transition from which the thermodynamic properties of the protein were determined, with a free energy for unfolding, 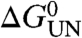 , of 60.17 ± 1.80 kJ/mol and an MUN of 6.86 ± 0.20 kJ·mol-1·M-1, the latter being a measure for the buried surface area in the native structure (Fig. 1B and Table S1). Refolding experiments using lipids within the gel phase at the temperature used (25 °C with diC16∶0PC-liposomes or 10 °C with diC14∶0PC-liposomes) indicated that the folding reaction commences from a highly unfolded state in 10 M urea that is lipid-associated and lacks β-sheet secondary structure (SI Text and Fig. S2). Full reversibility, independent of both the protein and lipid concentration, was not obtained at lower LPRs or in liposomes of other composition or size (see SI Text for further details).
, of 60.17 ± 1.80 kJ/mol and an MUN of 6.86 ± 0.20 kJ·mol-1·M-1, the latter being a measure for the buried surface area in the native structure (Fig. 1B and Table S1). Refolding experiments using lipids within the gel phase at the temperature used (25 °C with diC16∶0PC-liposomes or 10 °C with diC14∶0PC-liposomes) indicated that the folding reaction commences from a highly unfolded state in 10 M urea that is lipid-associated and lacks β-sheet secondary structure (SI Text and Fig. S2). Full reversibility, independent of both the protein and lipid concentration, was not obtained at lower LPRs or in liposomes of other composition or size (see SI Text for further details).
Using these conditions, the (un)folding kinetics of wild-type PagP were examined using Trp-fluorescence as a probe of folding/unfolding into/out of 100 nm diC12∶0PC-vesicles. In 7 M urea the kinetics of folding revealed a large burst phase followed by two exponential phases (Table S2). Closer to the transition midpoint (at [urea]≥7.8 M) the folding kinetics fitted well to a single exponential phase that described the total expected kinetic amplitude (Fig. 2A). The kinetics of unfolding fitted well to a single exponential at all urea concentrations examined (Fig. 2B). Combining both sets of data, a chevron plot describing the urea-concentration dependence of the (un)folding rate constants was constructed (Fig. 2C). This revealed a linear unfolding branch that intersected with the folding branch close to the expected midpoint (Fig. 2C). The slopes of the folding and unfolding branches in the chevron plot define the m values associated with the folding and unfolding transitions and are proportional to the changes in solvent accessibility of the protein between the unfolded state and the transition state, and the transition state and the native state, respectively. The sum of the kinetic m values associated with the folding and unfolding transitions (mf = 3.90 ± 0.17 kJ·mol-1·M-1 and mu = 2.81 ± 0.29 kJ·mol-1·M-1, respectively) agreed well with the MUN value obtained from equilibrium unfolding data (6.86 ± 0.20 kJ·mol-1·M-1), and the free energy of unfolding (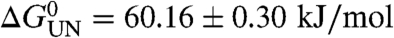 ) agreed well with -RT ln(kf/ku) (58.56 ± 3.12 kJ/mol), indicating that folding and unfolding occur by a reversible two-state reaction at the urea concentrations used (see Materials and Methods and ref. 23). An important property of the transition-state ensemble is how much surface area is buried in this species compared with the solvent accessibility of the native state. This can be calculated from the βT-value, which equals mf/MUN (see Materials and Methods and ref. 24). The βT value of the transition state of PagP of approximately 0.6 thus reveals a transition state for the folding of PagP that is placed midway on the reaction coordinate between the unfolded state in 10 M urea and lipid-embedded, native PagP.
) agreed well with -RT ln(kf/ku) (58.56 ± 3.12 kJ/mol), indicating that folding and unfolding occur by a reversible two-state reaction at the urea concentrations used (see Materials and Methods and ref. 23). An important property of the transition-state ensemble is how much surface area is buried in this species compared with the solvent accessibility of the native state. This can be calculated from the βT-value, which equals mf/MUN (see Materials and Methods and ref. 24). The βT value of the transition state of PagP of approximately 0.6 thus reveals a transition state for the folding of PagP that is placed midway on the reaction coordinate between the unfolded state in 10 M urea and lipid-embedded, native PagP.
Fig. 2.

(A) Folding and (B) unfolding kinetics of 0.4 μM PagP in diC12∶0PC-liposomes (LPR 3200∶1) measured by Trp-fluorescence. The arrows indicate increasing urea concentrations (7.8 to 8.8 M and 9 to 10 M urea in steps of 0.2 M for folding and unfolding, respectively). (C) Urea-concentration dependence of the rate constants of folding (▪) and unfolding (□). Lines represent linear fits to each dataset. (D) Urea-concentration dependence of the unfolding rate constant of PagP from diC12∶0PC-liposomes measured using Trp-fluorescence [0.4 μM PagP; LPR 3200∶1 (□) or 800∶1 (Δ)] and using CD-spectroscopy (5 μM PagP; LPR 800∶1) at 232 nm (×) and 218 nm (+). Inset, CD-spectra of native (N) and unfolded, membrane-associated (U) PagP [CD-signal = mean residue[Θ] × 103 (deg cm2 dmol-1)].
Mutant Design.
Structural insights into the folding transition states of many small, water-soluble proteins have been obtained using ϕ-value analysis (25, 26), which combines measurements of the effects of mutation on the thermodynamic stability of the native state and the folding and/or unfolding kinetics for a series of site-directed mutants. To determine the nature of the (un)folding transition state of PagP using ϕ-value analysis, 19-point mutants were generated that spanned all eight β-strands that comprise the native β-barrel (Fig. 3A and B). Ten of the mutations targeted residues buried in the β-barrel interior, seven of which disrupted hydrogen bonds (S58A, E90A, T108A, S130A, T137A, Q139A, and Q160A), whereas D24N, R94A, and M72A were created to disrupt a salt bridge, a cation-π interaction, or to probe the catalytic binding pocket, respectively. Four residues were mutated in the aromatic girdles defining the PagP surface at the membrane interface. Two (W51A and Y153A) are in the extracellular girdle, and two (W17A and Y23A) are in the periplasmic girdle, of which W17A is on the N-terminal amphiphatic α-helix. The remaining five residues mutated were in the hydrophobic band on the β-barrel surface (F55A, A85G, Y87A, L105A, and M157A) (Fig. 3A and B, Table S1, and Fig. S3).
Fig. 3.

(A) Cartoon representation of a cross-section through PagP [PDB ID code 1THQ (19); W. L. DeLano, http://www.pymol.org (2002)]. A schematic is shown above to allow orientation. Mutated residues on the hydrophobic surface are highlighted in blue, those in the aromatic girdles are shown in green, and residues in the β-barrel interior are in red. (B) Topology map of PagP highlighting mutated residues colored as in (A) with their specific interactions (hydrogen or π-cation bonds) with other side chains (red dashed lines; see Fig. S3 for details). (C–E) Examples of unfolding transients between 8.8 and 10 M urea (0.4 μM PagP; LPR 3200∶1) of F55A, Y153A, and R94A, respectively. The arrows indicate increasing urea concentrations in 0.2 M steps. (F–H) Urea-concentration dependence of the unfolding rate constants of wild-type PagP and PagP variants bearing mutations in the hydrophobic exterior of the barrel, the aromatic girdles, and the barrel interior, respectively. Residues located in the N-terminal half of the β-barrel are indicated by closed symbols, residues in the C-terminal half by open symbols. The data were globally fitted yielding a common mu = 2.52 ± 0.11 kJ·mol-1·M-1.
Thermodynamic and Kinetic Analysis of the Equilibrium Stability and Unfolding Kinetics of PagP Variants.
For each variant of PagP created, the thermodynamic stability of the variant was determined using equilibrium titration with urea (Fig. 1B–D). Equilibrium unfolding curves for all variants were shown to be reversible with no hysteresis. In every case the resulting unfolding transition fitted well to a two-state transition. Due to the sparsity of the data in the pre- and posttransition baselines, the data were globally fitted yielding a value for MUN of 6.86 ± 0.20 kJ·mol-1·M-1. The stabilities of the variants were found to vary widely from the value of the wild-type protein from 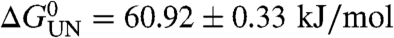 (e.g., for Q160A), to 53.75 ± 0.36 kJ/mol for the most destabilized mutant (R94A) (Table S1). With the sole exception of Q160A, all mutations were destabilizing (Fig. 1B–D, Table S1).
(e.g., for Q160A), to 53.75 ± 0.36 kJ/mol for the most destabilized mutant (R94A) (Table S1). With the sole exception of Q160A, all mutations were destabilizing (Fig. 1B–D, Table S1).
Given the complexity of the refolding kinetics below 7.8 M urea and the destabilized nature of most of the variants created, the unfolding kinetics, which were shown to be invariant over a broad range of PagP concentrations (Fig. 2D, Table S1), were determined using dilution of the native protein in 100 nm diC12∶0PC-liposomes into high concentrations of urea (see Materials and Methods) (Fig. 3C–H) and used for the calculation of the ϕ-values. For 18 of the 19 variants the unfolding kinetics of the lipid-embedded native protein were measured between 8.8 M urea (the unfolding midpoint for wild-type PagP) and 10 M urea, whereas for W17A unfolding was measured over a narrower urea-concentration range (Fig. 3G) because unfolding otherwise became too rapid to measure accurately by the manual mixing protocol used. Examples of unfolding transients are shown in Fig. 3C–E, and the corresponding urea-concentration dependences of the unfolding-rate constants are displayed in Fig. 3F–H. All curves were fitted globally yielding a common mu of 2.52 ± 0.11 kJ·mol-1·M-1, consistent with a conserved unfolding mechanism for all variants involving a transition-state ensemble with a βT of 0.6. Examination of the unfolding kinetics revealed two general observations. First, four of the five residues mutated that lie on the hydrophobic surface of the β-barrel increased the unfolding rate only marginally (< 2-fold), despite destabilizing the native state by circa 3–5.5 kJ/mol, suggesting that these residues are highly structured in the transition-state ensemble (Fig. 3F, Table S1). By contrast, the remaining variant in this region, Y87F, resulted in a 10-fold increase in the unfolding rate constant despite resulting in a similar 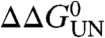 , providing important and unique evidence that the β-barrel is not complete within the transition-state structure (Table S1, Fig. S3B). Second, mutation of residues located in the N-terminal half of the PagP sequence (W17A, Y23A, D24N, W51A, F55A, S58A, Y87F, E90A, R94A) generally had a greater effect on the unfolding rate constants than mutation of residues located in the C-terminal half of the protein sequence (L105A, S130A, T137A, Q139A, Y153A, Q160A) (Fig. 3F–H), suggesting a polarized structure for the transition-state ensemble.
, providing important and unique evidence that the β-barrel is not complete within the transition-state structure (Table S1, Fig. S3B). Second, mutation of residues located in the N-terminal half of the PagP sequence (W17A, Y23A, D24N, W51A, F55A, S58A, Y87F, E90A, R94A) generally had a greater effect on the unfolding rate constants than mutation of residues located in the C-terminal half of the protein sequence (L105A, S130A, T137A, Q139A, Y153A, Q160A) (Fig. 3F–H), suggesting a polarized structure for the transition-state ensemble.
ϕ-Value Analysis.
The unfolding rate constants of wild-type PagP and each of its variants were compared at 8.8 M urea, and the differences in the activation energies for unfolding, 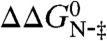 , were calculated. The ratio between
, were calculated. The ratio between 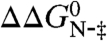 and
and 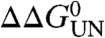 was then used to calculate the ϕ-value for folding, ϕF, where ϕF = 1 - ϕU (25). A value of 0 suggests side chains that are structurally unfolded in the transition-state ensemble, whereas a value of 1 suggests a highly native-like environment of the side chain. In the PagP transition state, eight of the calculated ϕF-values were > 0.5 and six were < 0.3 (Table S1). Of the variants with ϕF > 0.5, five were located in the C-terminal strands (E–H) and one was in the periplasmic turn between strands D and E, suggesting that the C-terminal half of the β-barrel is well structured in the transition-state ensemble (Fig. 4A and B). However, the low ϕF-value (≤ 0.30) of T108A and Q139A in strands E and G, respectively, indicate that the β-barrel is only partially formed. Indeed, whereas T108 is involved in a hydrogen bond to Y70 on strand C in the native structure of PagP, Q139 is involved in a wider hydrogen bonding network involving residues on strands D (E90), F (S130), and H (R158) in native PagP (Fig. S3C, G, and H). All other variants with ϕF < 0.3 (W17A, Y23A, D24N, S58A, Y87F; Table S1) are spatially close to the N-terminal α-helix in native PagP (Fig. 4A), consistent with the docking of this helix and the completion of the β-barrel occurring late in the folding process after the transition state has been traversed within the lipid bilayer (22, 27). Interestingly, W17A and T108A had negative ϕF-values, suggesting that these residues make nonnative stabilizing contacts in the transition-state ensemble, although the identity of these contacts remains to be elucidated (28). Overall, the data revealed a transition-state ensemble that is highly polarized and in which the β-barrel is largely formed but remains open and relatively unstructured toward the periplasmic side, where interactions with the N-terminal helix ultimately will occur as the native state develops.
was then used to calculate the ϕ-value for folding, ϕF, where ϕF = 1 - ϕU (25). A value of 0 suggests side chains that are structurally unfolded in the transition-state ensemble, whereas a value of 1 suggests a highly native-like environment of the side chain. In the PagP transition state, eight of the calculated ϕF-values were > 0.5 and six were < 0.3 (Table S1). Of the variants with ϕF > 0.5, five were located in the C-terminal strands (E–H) and one was in the periplasmic turn between strands D and E, suggesting that the C-terminal half of the β-barrel is well structured in the transition-state ensemble (Fig. 4A and B). However, the low ϕF-value (≤ 0.30) of T108A and Q139A in strands E and G, respectively, indicate that the β-barrel is only partially formed. Indeed, whereas T108 is involved in a hydrogen bond to Y70 on strand C in the native structure of PagP, Q139 is involved in a wider hydrogen bonding network involving residues on strands D (E90), F (S130), and H (R158) in native PagP (Fig. S3C, G, and H). All other variants with ϕF < 0.3 (W17A, Y23A, D24N, S58A, Y87F; Table S1) are spatially close to the N-terminal α-helix in native PagP (Fig. 4A), consistent with the docking of this helix and the completion of the β-barrel occurring late in the folding process after the transition state has been traversed within the lipid bilayer (22, 27). Interestingly, W17A and T108A had negative ϕF-values, suggesting that these residues make nonnative stabilizing contacts in the transition-state ensemble, although the identity of these contacts remains to be elucidated (28). Overall, the data revealed a transition-state ensemble that is highly polarized and in which the β-barrel is largely formed but remains open and relatively unstructured toward the periplasmic side, where interactions with the N-terminal helix ultimately will occur as the native state develops.
Fig. 4.

(A) Cartoon representation of PagP [PDB ID code 1THQ (19); W. L. DeLano, http://www.pymol.org (2002)] with ΦF-values mapped onto the native structure. (B) Topology map of PagP highlighting the ΦF-values of mutated residues. (C) Proposed folding mechanism of PagP into membranes: unfolded, but membrane-associated PagP (I); tilted insertion of the transition-state ensemble (II); and assembly of the helical clamp to yield native PagP (III). Note that the periplasmic halves of strands A, B, C, and D are depicted as loops to depict the low ΦF-values of side-chains in this region of the transition-state structure. In A–C, ΦF > 0.5 are in blue, 0.3 ≤ ΦF ≤ 0.5 are in purple, ΦF < 0.3 are in red, and ΦF < 0 are in orange. Residues 51, 72, 108, 137, and 160 gave 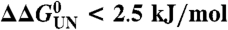 [the cut-off generally accepted to yield highly reliable ΦF-values (41)].
[the cut-off generally accepted to yield highly reliable ΦF-values (41)].
Discussion
The data presented above reveal unique insights into the transition state for folding of a β-barrel membrane protein. Importantly, the folding reaction is initiated from a membrane-adsorbed, yet highly unfolded conformation that lacks secondary structure (Fig. 4C, stage I) and results in a native protein embedded within a lipid bilayer. We showed, using mutational analysis, that the folding/unfolding transition-state ensemble involves formation of the extracellular half of the β-barrel, in which the C-terminal β-strands are highly structured, whereas the N-terminal β-strands and the α-helix remain poorly organized (Fig. 4). The data are consistent with tilted insertion into the membrane, suggested by the distinct pattern of ϕF-values in which residues unstructured in the transition-state ensemble are flanked by regions with high ϕF-values, some residues possessing ϕF-values close to 1.0 (Fig. 4C stage II). Subsequent assembly of the helical clamp and completion of the β-barrel then locks the native state of PagP into the membrane (22, 27) (Fig. 4C, stage III). Importantly, a similar tilted insertion mechanism was predicted by simulation of the assembly of OmpA into lipid bilayers (29). Such a mechanism is also consistent with experimental studies of OmpA insertion into lipid bilayers, in which the Trp-residues in the extracellular aromatic girdle were shown to migrate through the membrane in a concerted fashion (30). Together with the data presented here on PagP, we suggest that tilted insertion may be a generic route for folding and insertion of β-barrel membrane proteins into a lipid bilayer. Interestingly, tilted migration through the membrane has also been proposed for the α-helices in the voltage-sensor domain upon the activation of voltage-gated ion channels (31) and in the assembly of α-helical pore toxins (32), suggesting that tilted insertion may be also utilized for insertion of membrane proteins of other structural classes.
The extent to which the spontaneous in vitro folding mechanism of PagP resembles that of other β-barrel membrane proteins that have been investigated (15, 16) remains to be established. A second key question concerns the relationship of the mechanism of folding studied here in vitro with the biological folding mechanism, in which molecular chaperones have been purported to deliver partially folded outer membrane proteins to the membrane for their insertion (33). In one suggested mechanism, unfolded outer membrane proteins are targeted via a species-specific C-terminal motif to a polypeptide-transport-associated domain of the outer membrane YaeT-complex that then promotes the formation of β-strands via β-augmentation and spontaneous membrane insertion of the β-barrel (34, 35). Interestingly, in the present study the C-terminal transmembrane β-strands of PagP were found to be highly structured in the transition-state ensemble. Early folding of this region of the barrel could provide a mechanism for YaeT-facilitated membrane insertion in vivo. The challenges for the future will be to use the approaches developed here to investigate whether tilted insertion is a generic mechanism of β-barrel membrane protein folding and to elucidate precisely how accessory chaperones contribute to the folding of these proteins both in vitro and in vivo.
Methods
Expression and Purification of PagP-Mutants.
Site-directed mutations were introduced into the PagP gene sequence (22, 36) using primers listed in Table S3. Wild-type PagP and PagP variants were produced and purified from inclusion bodies and refolded into liposomes (22). Details are described in SI Text.
Equilibrium Unfolding and Refolding.
Equilibrium unfolding/refolding was investigated between 7 and 10 M urea using an LPR between 200∶1 and 4000∶1 in liposomes of different compositions at PagP concentrations between 0.1 and 0.4 μM. PagP was initially inserted into liposomes by diluting the protein in the presence of liposomes in 50 mM sodium phosphate buffer (pH 8) containing 7 M urea at 25 °C. For unfolding equilibrium curves refolded PagP was diluted threefold with appropriate amounts of urea. Refolding equilibrium curves were obtained by mixing unfolded PagP in the presence of liposomes in 10 M urea, but previously refolded in 7 M urea, with appropriate amounts of urea to yield identical reaction conditions to those for the unfolding curve. Samples were incubated overnight at 25 °C. Trp-fluorescence emission spectra were measured between 310 and 370 nm upon excitation of the samples at 280 nm with excitation and emission slit widths of 3 nm using a Photon Technology International fluorimeter (Ford, West-Sussex, United Kingdom). The average emission wavelength was calculated (37), and the resulting curves were fitted globally according to a two-state model (38). Details are given in SI Materials and Methods.
Folding and Unfolding Kinetics.
To measure the folding and/or unfolding rates of PagP, samples were prepared as above. Folding kinetics were measured in the presence of 7 M urea or between 7.8 and 8.8 M urea. Unfolding kinetics were measured between 8.8 and 10 M urea for wild-type PagP and all variants created except for the highly destabilized W17A variant, for which the rate of unfolding was measured between 8 and 8.8 M urea. Changes in Trp-fluorescence emission were measured at 335 nm upon excitation at 280 nm with excitation and emission slit widths of 3 nm using a Photon Technology International fluorimeter (Ford, West-Sussex, United Kingdom) equipped with a thermally controlled 4 cell changer. The temperature was regulated to 25 °C using a waterbath. The path length was 10 mm. Transients were followed for up to 4 h. Samples were mixed manually, resulting in a dead time of approximately 15 s. Traces were adjusted to account for photobleaching and fitted to a single or a double exponential function using Origin Pro version 7.5.
Analysis of Two-State Folding.
Empirically, the total free energy change upon unfolding, 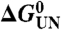 , shows a linear dependence on the denaturant concentration (24):
, shows a linear dependence on the denaturant concentration (24):
| [1] |
In a two-state folding process where only the native (N) and the unfolded (U) state are populated,
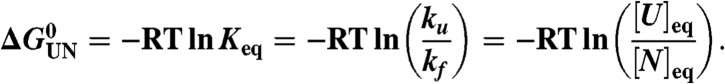 |
[2] |
Thus, the folding and unfolding rate constants also depend linearly on the denaturant concentration by the equation
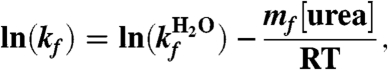 |
[3] |
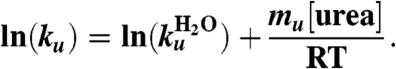 |
[4] |
The denaturant dependences of the (un)folding rate constants characterized by the m value for folding (mf) or unfolding (mu) relate to the MUN value measured in equilibrium experiments by
| [5] |
Folding and unfolding rate constants in the urea-concentration range measured were fitted using Eqs. 3 and 4, respectively, and the results were interpreted to verify Eqs. 2 and 5.
Calculation of ϕ-Values.
ϕ-value analysis assesses the relative changes in free energy of the folded state and the transition state relative to the unfolded reference state upon the mutation of one or more residues, i.e., the ratio 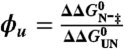 , in which ‡ denotes the transition state. The difference in equilibrium free energy upon mutation,
, in which ‡ denotes the transition state. The difference in equilibrium free energy upon mutation, 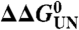 (
(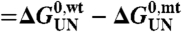 , in which wt is wild-type protein and mt is mutant protein), was determined from equilibrium unfolding experiments using the equality
, in which wt is wild-type protein and mt is mutant protein), was determined from equilibrium unfolding experiments using the equality
| [6] |
in which 〈MUN〉 is the average MUN (39). Changes in activation free energy for unfolding, 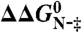 , were determined from the kinetic unfolding rate constants as follows (40):
, were determined from the kinetic unfolding rate constants as follows (40):
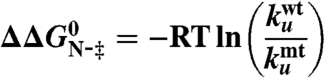 |
[7] |
and were calculated using the unfolding rate constants measured at 8.8 M urea to avoid the introduction of large errors by extrapolation. The ϕ-value for folding, ϕF, was calculated using ϕF = 1 - ϕU (25).
Supplementary Material
Acknowledgments.
We thank Russell Bishop for providing the plasmid pETCrcAHΔS and many helpful discussions. This work was funded by the Wellcome Trust (Grant 075514/Z/04/Z to G.H.M.H.). D.J.B. is an Engineering and Physical Sciences Research Council-funded White Rose Doctoral Training Centre lecturer.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 3947.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911904107/DCSupplemental.
References
- 1.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hediger MA, et al. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch. 2004;447:465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 3.Sanders C, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Bioph Biom. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 4.Chen GQ, Gouaux E. Probing the folding and unfolding of wild-type and mutant forms of bacteriorhodopsin in micellar solutions: Evaluation of reversible unfolding conditions. Biochemistry. 1999;38:15380–15387. doi: 10.1021/bi9909039. [DOI] [PubMed] [Google Scholar]
- 5.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 6.Mackenzie KR. Folding and stability of alpha-helical integral membrane proteins. Chem Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 7.Joh NH, et al. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 9.Curnow P, Booth PJ. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci USA. 2007;104:18970–18975. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curnow P, Booth PJ. The transition state for integral membrane protein folding. Proc Natl Acad Sci USA. 2009;106:773–778. doi: 10.1073/pnas.0806953106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth PJ, Curnow P. Folding scene investigation: Membrane proteins. Curr Opin Struc Biol. 2009;19:8–13. doi: 10.1016/j.sbi.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta. 2004;1666:275–288. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Barrera FN, et al. Protein self-assembly and lipid binding in the folding of the potassium channel KcsA. Biochemistry. 2008;47:2123–2133. doi: 10.1021/bi700778c. [DOI] [PubMed] [Google Scholar]
- 14.Hong H, Tamm LK. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgess NK, Dao TP, Stanley AM, Fleming KG. Beta-barrel proteins that reside in the E. coli outer membrane in vivo demonstrate varied folding behavior in vitro. J Biol Chem. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanley AM, Fleming KG. The process of folding proteins into membranes: Challenges and progress. Arch Biochem Biophys. 2008;469:46–66. doi: 10.1016/j.abb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 17.Hong H, Szabo G, Tamm LK. Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nat Chem Biol. 2006;2:627–635. doi: 10.1038/nchembio827. [DOI] [PubMed] [Google Scholar]
- 18.Hong H, Park S, Jimenez RH, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 19.Ahn VE, et al. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J. 2004;23:2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang PM, et al. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Natl Acad Sci USA. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop RE. Structural biology of membrane-intrinsic beta-barrel enzymes: Sentinels of the bacterial outer membrane. Biochim Biophys Acta. 2008;1778:1881–1896. doi: 10.1016/j.bbamem.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huysmans GH, Radford SE, Brockwell DJ, Baldwin SA. The N-terminal helix is a post-assembly clamp in the bacterial outer membrane protein PagP. J Mol Biol. 2007;373:529–540. doi: 10.1016/j.jmb.2007.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 24.Tanford C. Protein denaturation. Adv Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
- 25.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 26.Raleigh DP, Plaxco KW. The protein folding transition state: What are Phi-values really telling us? Protein Peptide Lett. 2005;12:117–122. doi: 10.2174/0929866053005809. [DOI] [PubMed] [Google Scholar]
- 27.Cox K, Sansom MS. One membrane protein, two structures, and six environments: A comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Mol Membr Biol. 2009;26:205–214. doi: 10.1080/09687680902788967. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Mirny LA, Shakhnovich EI. Kinetics, thermodynamics and evolution of non-native interactions in a protein folding nucleus. Nat Struct Biol. 2000;7:336–342. doi: 10.1038/74111. [DOI] [PubMed] [Google Scholar]
- 29.Bond PJ, Sansom MS. Insertion and assembly of membrane proteins via simulation. J Am Chem Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinschmidt JH, den Blaauwen T, Driessen AJ, Tamm LK. Outer membrane protein A of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry. 1999;38:5006–5016. doi: 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- 31.Elinder F, Nilsson J, Arhem P. On the opening of voltage-gated ion channels. Physiol Behav. 2007;92:1–7. doi: 10.1016/j.physbeh.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 32.Bayley H. Membrane-protein structure: Piercing insights. Nature. 2009;459:651–652. doi: 10.1038/459651a. [DOI] [PubMed] [Google Scholar]
- 33.Knowles TJ, Scott-Tucker A, Overduin M, Henderson IR. Membrane protein architects: The role of the BAM complex in outer membrane protein assembly. Nat Rev Microbiol. 2009;7:206–214. doi: 10.1038/nrmicro2069. [DOI] [PubMed] [Google Scholar]
- 34.Knowles TJ, et al. Fold and function of polypeptide transport-associated domains responsible for delivering unfolded proteins to membranes. Mol Microbiol. 2008;68:1216–1227. doi: 10.1111/j.1365-2958.2008.06225.x. [DOI] [PubMed] [Google Scholar]
- 35.Robert V, et al. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Biol. 2006;4:e377. doi: 10.1371/journal.pbio.0040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bishop RE, et al. Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J. 2000;19:5071–80. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Royer CA, Mann CJ, Matthews CR. Resolution of the fluorescence equilibrium unfolding profile of Trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 39.Kellis JT, Jr, Nyberg K, Fersht AR. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry. 1989;28:4914–4922. doi: 10.1021/bi00437a058. [DOI] [PubMed] [Google Scholar]
- 40.Matouschek A, Kellis JT, Jr, Serrano L, Fersht AR. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 41.Sato S, Religa TL, Daggett V, Fersht AR. Testing protein-folding simulations by experiment: B domain of protein A. Proc Natl Acad Sci USA. 2004;101:6952–6956. doi: 10.1073/pnas.0401396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.