

Abstract
Peripheral inflammation induces persistent central sensitization characterized by mechanical allodynia and heat hyperalgesia that are mediated by distinct mechanisms. Compared to well-demonstrated mechanisms of heat hyperalgesia, mechanisms underlying the development of mechanical allodynia and contralateral pain are incompletely known. In this study, we investigated the distinct role of spinal JNK in heat hyperalgesia, mechanical allodynia, and contralateral pain in an inflammatory pain model. Intraplantar injection of complete Freund’s adjuvant (CFA) induced bilateral mechanical allodynia but unilateral heat hyperalgesia. CFA also induced a bilateral activation (phosphorylation) of JNK in the spinal cord, and the phospho JNK1 (pJNK1) levels were much higher than that of pJNK2. Notably, both pJNK and JNK1 were expressed in GFAP-positive astrocytes. Intrathecal infusion of a selective peptide inhibitor of JNK, D-JNKI-1, starting before inflammation via an osmotic pump, reduced CFA-induced mechanical allodynia in the maintenance phase but had no effect on CFA-induced heat hyperalgesia. A bolus intrathecal injection of D-JNKI-1 or SP600126, a small molecule inhibitor of JNK also reversed mechanical allodynia bilaterally. In contrast, peripheral (intraplantar) administration of D-JNKI-1 reduced the induction of CFA-induced heat hyperalgesia but did not change mechanical allodynia. Finally, CFA-induced bilateral mechanical allodynia was attenuated in mice lacking JNK1 but not JNK2. Taken together, our data suggest that spinal JNK, in particular JNK1 plays an important role in the maintenance of persistent inflammatory pain. Our findings also reveal a unique role of JNK1 and astrocyte network in regulating tactile allodynia and contralateral pain.
Keywords: c-Jun-N-terminal kinase, MAP kinase, Astrocytes, Spinal cord, Contralateral pain, Inflammation, Tactile allodynia
1. Introduction
Increasing evidence indicates that mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinases (ERKs), p38, and c-Jun N-terminal kinases (JNK), play important roles in persistent pain sensitization after tissue and nerve injuries [17,26,28,53]. These MAPKs also have multiple isoforms, such as ERK1, ERK2, ERK5, p38α, p38β, p38γ, p38δ, JNK1, JNK2, and JNK3 [33]. Currently available inhibitors do not distinguish among different isoforms. By using antisense oligodeoxynucleotides and RNA interference, the role of some isoforms, such as ERK2, ERK5, and p38β in inflammatory pain has been revealed [36,63,68]. However, the specific role of different JNK isoforms in inflammatory pain remains elusive.
Tissue damage-induced inflammatory pain is characterized by heat hyperalgesia (increased responsiveness to noxious stimuli) and mechanical allodynia (painful responses to normally innocuous stimuli). It is generally believed that peripheral mechanisms, such as activation of transient receptor potential (TRP) and sodium channels in primary sensory neurons, play an essential role in the induction of heat hyperalgesia [34,67]. But the mechanisms underlying the development of mechanical allodynia are not well defined. It appears that central sensitization and large Aβ fiber activation are important for the induction of mechanical allodynia [29,41,55,67]. Importantly, mechanical allodynia is more characteristic than thermal hypersensitivity in chronic pain conditions. Mechanical allodynia is not only produced in the injured tissue, but also spread to the adjacent non-injured regions and even contralateral side [38,47,67]. For example, mechanical allodynia develops in the contralateral paws after CFA inflammation [3,5,51,57]. Further, inflammation produces bilateral increase in the phosphorylation of the transcription factor cAMP response element binding protein (CREB) [30,45] and expression of cyclooxygenase-2 (COX2) [59] in the spinal cord.
Accumulating evidence indicates that spinal cord astrocytes become reactive after nerve injury and CFA inflammation [1,14,57,71]. Upon activation, astrocytes may promote pain by producing cytokines [e.g., interleukin-1β (IL-1β)] [20]) and chemokines [e.g., monocyte chemoattractant protein-1 (MCP-1)] [18]. Astrocytes are characterized by forming astroglial networks (gap junctions) between them [19]. Interestingly, gap junction blocker carbenoxolone suppresses “mirror pain” (mechanical allodynia) in the contralateral paw [62]. In a previous study we showed that intrathecal injection of L-alpha-aminoadipate, a cytotoxin for astrocytes, can reverse nerve injury-induced mechanical allodynia [71]. Further, nerve injury activates JNK in spinal astrocytes, and intrathecal infusion of JNK inhibitors effectively attenuates nerve injury-induced mechanical allodynia [71]. In the present study, we investigated whether JNK, in particular JNK1 in spinal astrocytes has a distinct role in regulating inflammation-induced heat hyperalgesia, mechanical allodynia, and contralateral pain (spread of pain).
2. Materials and Methods
2.1 Animals
Male adult Sprague Dawley rats (220–260 g), CD1 mice (20–30 g), and mice lacking Jnk1 (JNK1−/−) and Jnk2 (JNK2−/−), obtained from Jackson Laboratories with C57/Bl6 genetic background, and their littermate control mice, were used in this study. All animal studies were performed under Harvard Medical School Animal Care institutional guidelines. Peripheral inflammation was induced by intraplantar injection of CFA (Sigma, 100 μl for rats and 20 μl for mice) in the left hind paws under brief anesthesia with sevofluorane.
2.2 Drug administration
The peptide inhibitor of JNK, D-JNKI-1was provided by Dr. Christophe Bonny, University of Lausanne, Switzerland [6] and dissolved in saline. JNK inhibitor SP600125 was purchased from Calbiochem and dissolved in 10% DMSO. For single intrathecal injection, spinal cord puncture was made under brief sevofluorane anesthesia with a 30 gauge needle between the L5 and L6 level to deliver the reagents (20μl for rats) to the CSF [71]. The doses of JNK inhibitors we used for bolus injection (0.1–4 nmol of D-JNKI-1 and 20 nmol of SP600125) and pump infusion (100 μM of D-JNKI-1) were based on our previous study [71]. Immediately after the needle entry into subarachnoid space (change in resistance), a brisk tail flick could be observed. For sustained spinal drug infusion in rats, osmotic pumps (1μl/h for 7 days, Alzet, Cupertino, CA) were implanted under the back skin. Each pump was connected to a polyethylene (PE10) catheter that was inserted into subarachnoid space between L5-L6 vertebrate. The drug filled pumps were soaked in saline for 3 h before implantation [25].
2.3 Western blots
As described previously [70], animals were rapidly killed after deep anesthesia with isoflurane, and the L4-5 spinal segments from both sides were quickly removed and homogenized in a SDS sample buffer containing a mixture of proteinase and phosphatase inhibitors (Sigma). Protein samples (25 μg) were separated on SDS-PAGE gel and transferred to nitrocellulose blots. The blots were blocked with 5% milk and incubated overnight at 4°C with antibody against phosphorylated JNK (pJNK, rabbit, 1:1000; Cell Signaling), phosphorylated c-Jun (rabbit, 1:300; Cell Signaling), JNK (rabbit, 1:1000; Cell Signaling), GAPDH (mouse, 1:20000; Millipore), and β-tubulin (mouse, 1:5000; Sigma). These blots were further incubated with HRP-conjugated secondary antibody, developed in ECL solution, and exposed onto Hyperfilm (Amersham Biosciences) for 1–10 min. Specific bands were evaluated by apparent molecular size. The intensity of the selected bands was captured and analyzed using Image J software (NIH).
2.4 Immunohistochemistry
Animals were terminally anesthetized with isoflurane and perfused through the ascending aorta with saline followed by 4% paraformaldehyde/1.5% picric acid. After the perfusion, the spinal cord segments (L4-5) were removed and postfixed in the same fixative overnight, then replaced with 15% sucrose overnight. Spinal sections (transverse, free-floating, 30 μm) were cut in a cryostat. For double immunostaining of pJNK and GFAP, the sections were blocked with 2% goat serum in 0.3% Triton for 1 h at RT and incubated over night at 4°C with a mixture of primary antibodies (anti-pJNK, 1:1000, rabbit, cell signaling; and anti-GFAP, 1:5000, mouse, Millipore). The sections were then incubated for 1 h at RT with a mixture of Cy3- and FITC-conjugated secondary antibodies (1:400, Jackson ImmunoResearch). For double staining of JNK1 and GFAP, the Tyramide Signaling Amplification (TSA) kit (PerkinElmer) was used to amplify immunofluorescence signal of JNK1. The spinal sections were blocked with TNB buffer and incubated with JNK1 antibody (1:1000; rabbit, Cell Signaling) overnight at 4°C, followed by a biotinylated secondary antibody (1:200) for 2 h at room temperature. The sections were then incubated with StreptaAvidin (1:100) for 30 min and FITC-conjugated tyramide (1:50) for 10 min at room temperature. After JNK1 immunostaining, the sections were further processed with GFAP immunostaining with a mouse-anti-GFAP antibody (1:5000; Millipore). The stained sections were examined with a Nikon fluorescence microscope, and images were captured with a CCD Spot camera.
2.5 Behavioral analysis
Animals were habituated to the testing environment daily for at least 2 d before baseline testing. The room temperature and humidity remained stable for all experiments. For testing mechanical sensitivity, animals were put under inverted plastic boxes (11×13×24 cm) on an elevated mesh floor and allowed 30 min for habituation, before the threshold testing. Mechanical allodynia was tested using von Frey hairs. The paw was pressed with one of a series of von Frey hairs with logarithmically incrementing stiffness (0.6, 1, 1.4, 2, 4, 6, 8, 10, 15, and 26 g for rats, and 0.02, 0.04, 0.08, 0.16, 0.32, 0.64, 1.28, and 2.56 g for mice) (Stoelting, Kiel, WI), presented perpendicular to the plantar surface. Each hair was applied for 5–6 s on rats and 1–2 s on mice [41]. The 50% withdrawal threshold was determined using Dixon’s up–down method [9]. For testing heat sensitivity, animals were put in a plastic box placed on a glass plate, and the plantar surface was exposed to a beam of radiant heat through a transparent Perspex surface [22]. The baseline latencies were adjusted to 12–15 sec with a maximum of 20 sec as cutoff to prevent potential injury. The latencies were averaged over three trials, separated by a 3 min interval.
2.6 Quantification and statistics
For the quantification of Western Blot, the density of specific bands for pJNK1 and JNK1 (46 kDa), pJNK2 and JNK2 (54 kDa), p-c-Jun, β-tubulin, and GAPDH was measured with imaging analysis software (Image J, NIH). The size of rectangle was fixed for each band and the background near that band was subtracted. pJNK1/2 or JNK1/2 levels were normalized to loading controls (β-tubulin or GAPDH). The Western data were analyzed with one way ANOVA followed by Newman-Keuls test for post hoc analysis. For behavioral studies, the data of paw withdrawal latencies and paw withdrawal thresholds (determined by up-down method) passed normality test, thus were suitable for parametric statistics. The data were analyzed with student’s t-test (two groups only) or one way ANOVA followed by Newman-Keuls test for post hoc analysis. All the data were presented as mean ± SEM, and P<0.05 was considered statistically significant in all cases.
3. Results
3.1 Time course of inflammation-induced bilateral mechanical allodynia and unilateral heat hyperalgesia
Unilateral injection of 100 μl of undiluted CFA into a hindpaw of rats produces marked inflammation and inflammatory pain [25,31]. CFA-induced mechanical allodynia on the ipsilateral paw was very rapid (< 1 hour) (Fig. 1A). The paw withdrawal threshold (PWT) dropped from 18.5 ± 2.5 g before CFA injection (baseline) to 0.7 ± 0.1 g at 1 hour after CFA injection. The mechanical allodynia remained at the peak level for more than 14 d. Mechanical allodynia was also found in the contralateral paws. But the contralateral allodynia was delayed, starting at 3 h, and the magnitude of allodynia in the contralateral paws was also smaller than that on the ipsilateral paws (Fig. 1A).
Figure 1.

Time course of CFA-induced mechanical allodynia (A) and heat hypealgesia (B) in rats. CFA produces rapid (<1 hour) and persistent (>2 weeks) mechanical allodynia in both ipsilateral and contralateral paws (A). CFA only induces ipsilateral heat hyperalgesia, which largely recovers after 2 weeks (B). *, P<0.05; **, P<0.01, compared to pre-CFA baseline, n = 6.
CFA also induced rapid (< 1 h) heat hyperalgesia (Fig. 1B). The heat hyperalgesia peaked at 6 h. The paw withdrawal latency (PWL) decreased from 13.1 ± 0.6 s before inflammation to 3.3 ± 0.3 s at 6 h after CFA injection. Unlike mechanical allodynia, heat hyperalgesia largely recovered at 14 d, although PWL was still lower than that of the baseline. Strikingly, heat hyperalgesia was exclusively unilateral: there was no change in PWLs in the contralateral paws at all the time points examined (Fig. 1B). Consistently, we did not notice any obvious sign of inflammation in the contralateral paws, suggesting that the CFA model we produced is a monoarthritis model.
3.2 Bilateral activation of JNK in the spinal cord after CFA inflammation
Like other MAPKs JNK is activated by phosphorylation [17]. Thus, we examined JNK phosphorylation at different times after inflammation. Western blot analysis showed a low level of basal phosphorylation of JNK1/2. Notably, the basal pJNK1 (46 kDa) levels were much higher than basal pJNK2 (52 kDa) levels (Fig. 2A,B). According to the time course of CFA-induced pain behavior, we examined JNK activation in the bilateral lumbar spinal cord (L4-5) at 6 h, 1 d, 3 d, 7 d, and 14 d after CFA injection. CFA induced a rapid increase in pJNK1 and pJNK2 levels in both the ipsilateral (Fig. 2A) and contralateral (Fig. 2B) spinal cord. The increase in pJNK1 and pJNK2 started at 6 h and maintained at 14 d (Fig. 2A,B). In contrast, total levels (phosphorylated plus non-phosphorylated) of JNK1 and JNK2 did not change after CFA (Fig. 2C), indicating that increases in pJNK levels result from increases in JNK phosphorylation. Although both pJNK1 and pJNK2 increased after inflammation, pJNK1 levels were much higher than pJNK2 levels at all the time points examined (Fig. 2A,B), suggesting that pJNK1 is the predominant form of pJNK in the spinal cord.
Figure 2.

Western blot analysis showing time course of CFA-induced expression of pJNK1, pJNK2, JNK1, and JNK2 in the spinal cord of rats. JNK1 and JNK2 are rapidly activated and maintained for >14 days in both ipsilateral (A) and contralateral (B) lumbar spinal cord. Low panels in A and B show densities of pJNK1 and pJNK2 bands after being normalized to tubulin. Total levels of JNK1 and JNK2 do not change after inflammation (C). Low panels in C shows densities of JNK1 and JNK2 against GAPDH. *, P<0.05; compared to naive animals, n = 4–6.
3.3 Expression of pJNK and JNK1 in spinal cord astrocytes
Immunohistochemistry also reveal an increase in pJNK immunoreactivity in the superficial spinal cord after inflammation (Fig. 3A,B). Double staining further indicated that pJNK was exclusively colocalized with GFAP (Fig. 3C), a cellular marker for astrocytes. Thus, pJNK is also expressed in spinal cord astrocytes after inflammation, in support of previous studies in neuropathic conditions [42,71]. Furthermore, JNK1 was expressed GFAP-positive astrocytes in the spinal cord (Fig. 3D–F), again in agreement with previous studies [35,71].
Figure 3.

Immunohistochemistry showing pJNK and JNK1 expression in spinal cord astrocytes of rats after CFA injection. (A, B) CFA increases expression of pJNK in the spinal cord 3 days after inflammation. (C) Colocalization of pJNK and GFAP (marker for astroglia) 3 days after CFA injection. (D–F) Double staining shows a colocalization of JNK1 (D) and GFAP (E) in the superficial spinal cord (laminae I–III) 3 days after CFA injection. Scale bars, 50 μm.
3.4 Inhibition of CFA-induced mechanical allodynia in the maintenance phase by intrathecal pretreatment of a peptide inhibitor of JNK
To determine the role of JNK in inflammatory heat hyperalgesia and mechanical allodynia, we inhibited the action of JNK with a highly selective JNK inhibitor, D-JNKI-1, which does not affect the activity of >40 other kinases including ERK and p38 [6]. D-JNKI-1 is a peptide derived from JNK binding domain of JNK-interacting protein-1 (JIP-1), and selectively blocks the access of JNK to its substrates by a competitive mechanism (Fig. 4A). Further, a 10-amino acid HIV Tat (48–57) transporter sequence is linked to the JBD of JIP-1 so that the peptide is cell membrane permeable. Finally, a convert to D-form amino acids makes the peptide resistant to proteinases (Fig. 4A) [6].
Figure 4.
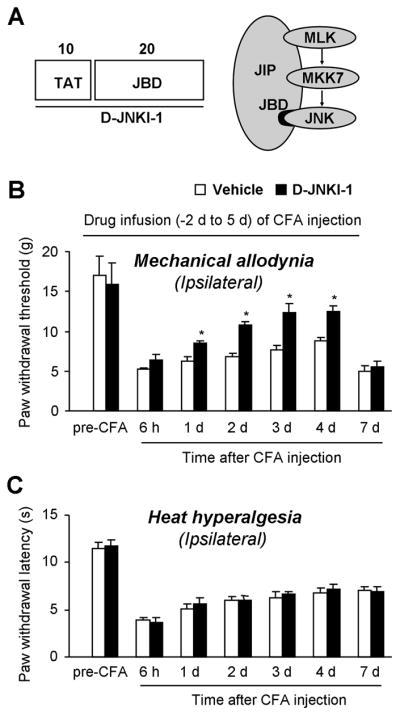
Intrathecal infusion of D-JNKI-1 via an osmotic pump reduces CFA-induced mechanical allodynia in the maintenance phase in rats. (A) Structural organization of the D-JNKI-1 peptide. The D-JNKI-1 (30 amino acids, aa) is a chimeric peptide starting with a 10-aa TAT transporter sequence, followed by a 20-aa minimal JBD (JNK binding domain) sequence from the JIP1 molecule. (B) Pretreatment of D-JNKI-1, starting 2 days before inflammation, inhibits CFA-induced mechanical allodynia in the maintenance phase (day 1 to day 4) but not in the induction phase (6 h). (C) Pretreatment of D-JNKI-1 fails to alter CFA-induced heat hyperalgesia. D-JNKI-1 (100 μM) or vehicle (PBS) infusion (1 μl/h, 1 week duration) started 2 days before CFA injection and ended 5 days after CFA injection. *, P<0.05, compared to vehicle control, n = 5.
To examine whether D-JNKI-1 can prevent inflammatory pain, we infused D-JNKI-1 at a low concentration (100 μM) via intrathecal osmotic pump (1 μl/h) for 1 week, starting 2 d before CFA injection. Notably, a previous study used a much higher concentration of SP600125 (8 mM) for pump infusion [54]. Intrathecal infusion of D-JNKI-1 to spinal fluid did not change basal paw withdrawal threshold (Fig. 4B) and paw withdrawal latency (Fig. 4C) tested before CFA injection (P>0.05), indicating that JNK does not regulate basal pain perception. Neither did D-JNKI-1 affect the induction of mechanical allodynia tested at 6 h after CFA injection (Fig. 4B). However, mechanical allodynia in the maintenance phase (day 1 to day 4) was attenuated by D-JNKI-1. Two days after the pump infusion stopped, mechanical allodynia developed again on day 7 (Fig. 4B). We can not exclude the possibility that the peptide inhibitor is degraded over time at the body temperature. In sharp contrast, CFA-induced heat hyperalgesia on the ipsilateral paws was not altered by the inhibitor at all the time points examined (Fig. 4C).
3.5 Reverse of bilateral mechanical allodynia by intrathecal inhibition of JNK
To further determine whether spinal JNK is required for the maintenance of inflammatory pain, we applied a bolus injection of D-JNKI-1 intrathecally (i.t.) on post-CFA day 3 and tested mechanical and heat sensitivity 3 h after the injection. Our previous study showed that D-JNKI-1 produced the best anti-allodynic effect at 3 h after bolus injection in the spinal nerve ligation model [71]. D-JNKI-1 produced a dose-dependent reversal of mechanical allodynia in both ipsilateral and contralateral paws (ipsilateral, P < 0.01; contralateral, P < 0.01). Even a low dose (0.4 nmol) of D-JNKI-1 had a significant anti-allodynic effect (P < 0.05). Strikingly, the anti-allodynic effect of D-JNKI-1 was more robust on the contralateral side, compared to the ipsilateral side (Fig. 5A,B). D-JNKI-1 only reversed CFA-induced ipsilateral heat hyperalgesia at a high dose (4 nmol) but not low doses (0.1 and 0.4 nmol) (Fig. 5C).
Figure 5.

Reversal of CFA-induced mechanical allodynia in rats by intrathecal D-JNKI-1 and SP600125. (A–C) A bolus injection of D-JNKI-1, given 3 days after CFA injection, dose-dependently reduces mechanical allodynia on the ipsilateral (A) and contralateral (B) paws. Notice that the anti-allodynic effects of D-JNKI-1 is more robust in the contralateral paws. D-JNKI-1 only reduces CFA-induced unilateral heat hyperalgesia at the highest dose (C). *, P<0.05; **, P<0.01; compared to corresponding saline control, n = 6–9. (D–F) A bolus injection of SP600125 (20 nmol), given 3 days after CFA injection, reduces ipsilateral (D) and contralateral (E) mechanical allodynia but not unilateral heat hyperalgesia (F). *, P<0.05, compared to corresponding vehicle (10% DMSO) control, n = 5–6. (G) Western blotting showing a reduction of spinal p-c-Jun levels, 3 h after SP600125 treatment. Right panel shows the density of p-c-Jun band after being normalized to GAPDH. *, P<0.05, n = 6.
To confirm a specific role of JNK in the maintenance of inflammatory pain, we i.t. injected another JNK inhibitor, SP600125 on post-CFA day 3. As D-JNKI-1, SP600125 (20 nmol) partially reversed CFA-induced mechanical allodynia in both ipsilateral and contralateral paws (Fig. 5D,E), with no effect on heat hyperalgesia (P > 0.05) (Fig. 5F). The data are also in agreement with the previous report showing that D-JNKI-1 is more potent than SP600126 in producing anti-allodynic action [71].
To determine whether the JNK pathway is inhibited by SP600125, we collected spinal cord tissues after behavioral testing (3 h after drug injection) and examined the phosphorylation of c-jun, a critical downstream target of JNK. Western blotting showed that SP600125 significantly reduced CFA-induced p-c-jun expression by 34% (P<0.05, Fig. 5G), suggesting that the spinal JNK pathway is indeed inhibited by SP600125.
3.6 Attenuation of CFA-induced heat hyperalgesia by intraplantar inhibition of JNK
To examine whether JNK has a peripheral role in inflammatory pain, we injected D-JNKI-1 (10 nmol) into the plantar surface of a hindpaw, 30 min before CFA injection. This pretreatment of D-JNKI-1 partially delayed the onset of CFA-induced heat hyperalgesia at 15 min, 3 h, and 6 h but not at 24 h (Fig. 6A). Pretreatment of intraplantar D-JNKI-1 had no effect on CFA-induced mechanical allodynia (Fig. 6B). Further, posttreatment of intraplantar D-JNKI-1 (10 nmol) on post-CFA day 3 failed to alter heat hyperalgesia and mechanical allodynia (Fig. 6C, D). These data suggest that peripheral JNK only contributes to the induction of inflammatory heat hyperalgesia.
Figure 6.

Peripheral (intraplantar) inhibition of JNK in rats reduces CFA-induced heat hyperalgesia in the induction phase. (A, B) Pretreatment of D-JNKI-1 (10 nmol), 30 minutes before CFA injection, attenuates CFA-induced heat hyperalgesia (A) but not mechanical allodynia (B). (C, D) Posttreatment of intraplantar D-JNKI-1, 3 days after CFA, fails to reverse established heat hyperalgesia (C) or mechanical allodynia (D). *, P<0.05, compared to saline control, n = 5.
3.7 CFA-induced bilateral mechanical allodynia is attenuated in mice lacking JNK1 but not JNK2
We also examined CFA-induced JNK activation in the spinal cord of mice. Like rats, mice exhibited persistent increase of pJNK1 and pJNK2 in both the ipsilateral and contralteral spinal cord. The increase started on day 1 and maintained on day 28. Although both pJNK1 and pJNK2 showed increases after inflammation, the pJNK1 levels were much higher than pJNK2 levels (Fig. 7A–C), confirming that pJNK1 is the predominant form of pJNK in the mouse spinal cord.
Figure 7.

Western blotting showing time course of CFA-induced pJNK1 and pJNK2 expression in the spinal cord of mice. (A) CFA induced persistent increase (> 4 weeks) of JNK1 and JNK2 in both ipsilateral and contralateral spinal cord. (B, C) Densities of pJNK1 (B) and pJNK2 (C) in the spinal cord after being normalized to GAPDH. *, P<0.05, compared to naïve animals, n = 4–6.
To define the distinct role of JNK1 and JNK2 in inflammatory pain, we examined CFA-induced heat hyperalgesia and mechanical allodynia in Jnk1 or Jnk2 knockout mice. Like rats, wild-type C57Bl6 mice exhibited a persistent (> 28 d) and bilateral mechanical allodynia (Fig. 8A) and a transient (< 21 d) and unilateral heat hyperalgesia (Fig. 8B) following CFA inflammation. In mice lacking JNK1, CFA-induced mechanical allodynia was partially but significantly attenuated in both ipsilateral (Fig. 8A) and contralateral paws (Fig. 8B), especially in the maintenance phase. In sharp contrast, CFA-induced heat hyperalgesia was comparable in Jnk1 knockout mice and wild-type mice (Fig. 8C, D). In Jnk2 knockout mice, both CFA-induced mechanical allodynia and heat hyperalgesia were comparable to that of wild-type mice (Fig. 9A–D).
Figure 8.

CFA-induced mechanical allodynia is reduced in mice lacking JNK1. CFA produced rapid and persistent bilateral mechanical allodynia (A, B) and ipsilateral heat hyperalgesia (C, D) in wild-type mice. But in JNK1−/− mice, mechanical allodynia is attenuated bilaterally, especially in the maintenance phase (A, B). CFA-induced heat hyperalgeisa is comparable in wild-type and JNK1−/− mice (C, D). *, P<0.05; **, P<0.01; compared to wild type mice at the corresponding time points. Notice that the baseline pain sensitivity is normal in the knockout mice. n = 7–8.
Figure 9.

CFA-induced inflammatory pain is normal in mice lacking JNK2. CFA-induced mechanical allodynia (A, B) and heat hyperalgesia (C, D) are comparable between JNK2−/− and wild-type mice in all the time points examined. n = 5–6.
It is worthy to notice that the baseline thermal and mechanical thresholds in mice lacking JNK1 or JNK2 were not different from that of wild-type mice (Fig. 8 and 9), suggesting that JNK1 and JNK2 have no role in regulating pain sensitivity in normal conditions.
Discussion
We have made several interesting findings in this study. First, unilateral CFA injection in a hindpaw (monoarthritis) induced bilateral mechanical allodynia, bilateral JNK activation in the spinal cord, but ipsilateral heat hyperalgesia. Second, intrathecal infusion of the JNK inhibitors D-JNKI-1 and SP600125 at low doses, reduced CFA-induced mechanical allodynia bilaterally in the maintenance phase, but had no effect on CFA-induced heat hyperalgesia. Third, peripheral injection of D-JNKI-1 reduced CFA-induced heat hyperalgesia in the induction phase without any effect on mechanical allodynia. Fourth, JNK1 was expressed in spinal cord astrocytes and represented the predominant isoform of JNK in the spinal cord. Finally, mice lacking JNK1 but not JNK2 exhibited reduced mechanical allodynia bilaterally in the maintenance phase. These data indicate a unique role of spinal JNK1 in the maintenance of mechanical allodynia.
4.1 Distinct roles of central and peripheral JNK in regulating mechanical allodynia and heat hyperalgesia
Obata et al. showed that pre- and post-treatment of SP600125 reduced spinal nerve ligation (SNL)-induced mechanical allodynia but failed to alter SNL-induced heat hyperalgesia [54]. Intrathecal infusion of a peptide inhibitor of JNK, D-JNKI-1 also attenuated SNL-induced mechanical allodynia [71]. Compared to SP600125, D-JNKI-1 is more selective [6] and potent in reversing nerve injury-induced mechanical allodynia [71]. The present study showed that pretreatment of D-JNKI-1 via continuous i.t. infusion reduced CFA-induced mechanical allodynia in the maintenance phase but failed to change CFA-induced heat hyperalgesia. Posttreatment of D-JNKI-1 (0.4 and 4 nmol, i.t.) or SP600125 (20 nmol, i.t.) further reversed mechanical allodynia, whereas heat hyperalgesia was only reduced by a high dose of D-JNKI-1 (4 nmol, i.t.). Our results further suggest spinal JNK is critical for maintaining mechanical allodynia.
In contrast, peripheral administration of D-JNKI-1 in the hindpaw only reduced CFA-induced heat hyperalgesia in the induction phase (Fig. 6). Intraplantar injection of SP600125 also reduces inflammatory heat hyperalgesia induced by melittin, a major toxic peptide from bee venom [21]. However, CFA-induced mechanical allodynia was not affected by either pre- or post-treatment of intraplantar D-JNKI-1. Thus, peripheral JNK and central (spinal) JNK play different role in regulating inflammatory pain symptoms, and peripheral JNK contributes only to the induction of heat hyperalgesia.
4.2 JNK1 is the predominant isoform for mediating inflammatory pain
The JNK subfamily of MAPKs has three genes Jnk1, Jnk2 and Jnk3 encoding JNK1, JNK2, and JNK3, respectively. While JNK1 and JNK2 are expressed in all tissues, JNK3 is predominantly found in the CNS [44]. Mice deficient in a single isoform by targeted disruption of Jnk1, Jnk2, or Jnk3 gene are viable, but mice lacking both Jnk1 and Jnk2 are embryonic lethal [39]. Mice lacking Jnk3 display decreased susceptibility to kainic acid-induced seizures and cell death in the hippocampus [69]. Axotomy-induced death of neonatal DRG neurons is also reduced in Jnk3 deficient mice [37].
Our data showed that JNK1 is the major isoform activated in the spinal cord. Our previous western analysis primarily detected pJNK1 band in the spinal cord [71]. The present study also detected pJNK2 bands in the spinal cord, as a result of enhanced sensitivity of Western analysis. Further, pJNK2 showed a marked increase after inflammation. Thus the role of JNK2 in inflammatory pain should not be completely excluded. However, the levels of pJNK2 were much lower than that of pJNK1 in all the spinal cord samples we examined. Since both JNK1 and JNK2 mutant mice show normal basal pain thresholds, the development of nociceptive pathway does not require these JNK isoforms. However, CFA-induced mechanical allodynia is reduced in JNK1 but not JNK2 mutant mice, supporting a predominant role of JNK1 in mediating inflammatory pain. Notably, JNK1 knockout mice only show a moderate reduction in mechanical allodynia whereas JNK inhibitor largely reduces mechanical allodynia. This discrepancy may result from a developmental compensation in JNK1-deficient mice. In addition to JNK, other signaling molecules such ERK may also maintain mechanical allodynia [27].
4.3. A correlation between CFA-induced bilateral JNK activation and bilateral mechanical allodynia
Although CFA may induce polyarthritis several weeks later (>6 weeks) [7], we only observed monoarthritis in the first several weeks we examined.. Our time course study showed that unilateral CFA induced bilateral pain both in rats and mice, in support of previous studies in the CFA model [3,5,51,57], but see [12]. Strikingly, only mechanical allodynia is bilateral, whereas heat hyperalgesia remains to be ipsilateral. Moreover, heat hyperalgeisa is more transient than mechanical allodynia (Fig. 7). In numerous clinical pain syndromes and diverse animal models, pain hypersensitivity not only occurs in ipsilateral side, but also arises from contralateral side [2,13,24,43,47,60,61,64,]. In particular, contralateral mechanical allodynia is more common than heat hyperalgesia, although substance P, NMDA and non-NMDA receptors, and dynorphin have been implicated in contralateral heat hyperalgesia [10,13,47]. In a sciatic inflammatory neuropathy (SIN) model, Milligan et al show that weak immune activation produces unilateral allodynia whereas strong immune activation produces bilateral allodynia [47]. Strong circumstantial evidence also points to a central mechanism of mechanical allodynia [38].
Although many changes in mRNA/protein expression, such as spinal increase in c-Fos, prodynorphin, and NK-1 after inflammation are largely ipsilateral [15,16,25,56], some spinal changes are also bilateral. For example, formalin and CFA induce bilateral phosphorylation of CREB [30,45]. CFA also induces bilateral up-regulation of Cox-2 mRNA that depends on IL-1β [59]. Consistent with distinct role of peripheral and central JNK, COX-2 induction in the spinal cord is important for the development of mechanical allodynia, whereas peripheral COX-2 is certainly important for the development of heat hyperalgesia [65]. Our data showed that CFA-induced JNK phosphorylation is also bilateral. Thus, bilateral induction of COX-2 and pJNK in the spinal cord may share similar upstream mechanisms, such as cytokine dependence. Further, there is also a temporal correlation between bilateral JNK phosphorylation and bilateral mechanical allodynia. Importantly, spinal inhibition of JNK can attenuate CFA-induced bilateral mechanical allodynia. Indeed, the anti-allodynic effect of D-JNKI-1 was more robust on the contralateral side (Fig. 5A,B). Therefore, it is reasonable to postulate that bilateral JNK activation contributes to bilateral mechanical allodynia, especially in the maintenance phase and contralateral side.
4.4. JNK and astrocyte network for mechanical allodynia and bilateral pain
It is generally believed that activation of large A-fibers (Aβ) are necessary for inducing mechanical allodynia [29,41,55,67]. Low-threshold Aβ input can activate nociceptive pathways after intense noxious stimulation and tissue injury [4,8,32,58]. Innocuous input also activates PKCγ interneurons in the dorsal horn via myelinated afferent fibers [52]. Disinhibition of PKCγ neurons that receive innocuous input via Aβ fibers contributes to the activation of nociceptive specific neurons in the superficial dorsal horn [49,50]. However, it would be difficult to explain contralateral pain by these mechanisms.
Mounting evidence indicates that astrocytes play an important role in enhancing and maintaining chronic pain [11,27,48,66]. Spinal astrocytes are persistently activated after nerve injury, spinal cord injury, and tumor growth [27]. Astrocytes are the most abundant cell type in the CNS and have strong structural inter-relationship with neurons by enwrapping synaptic terminals, enabling signaling between glia and neurons. In particular, astrocytes form widespread networks via gap junctions and propagate calcium waves to a long distance [19,23]. Gap junction blockade inhibits “mirror pain” (mechanical allodynia) in the contralateral paw [62]. Thus, gap junction-mediated astrocyte networks may mediate mechanical allodynia and contralateral pain, i.e. spread of pain beyond the initial injury site.
Previous studies reported that JNK is activated in spinal cord astrocytes after spinal nerve ligation [71], partial sciatic nerve ligation [42], and amyotrophic lateral sclerosis [46]. Our immunostaining results showed that pJNK and JNK1 are also expressed in spinal cord astrocytes after inflammation, in agreement with previous studies [35,71]. Consistently, JNK1 is the predominant JNK isoform in cultured astrocytes [27]. Although we should not completely exclude the possibility that JNK is activated in other spinal cell types after inflammation, our data suggest that JNK1 activation in spinal astrocyte networks plays an important role in mediating mechanical allodynia by producing proinflammatory and pronocicpetive mediators [17] or modulating glutamate uptake [11]. For example, JNK-induced MCP-1 production in spinal astrocytes can induce central sensitization and promote neuropathic pain [18].
4.5. Concluding remarks
Most drugs do not distinguish mechanical allodynia and heat hyperalgesia in animal models of persistent pain. However, mechanical allodynia is more persistent than heat hyperalgesia and spreads beyond the initial injury and even to the contralateral side. We have shown that unilateral CFA induces a persistent and bilateral mechanical allodynia and this allodynia is caused by bilateral activation of JNK, especially JNK1, in spinal cord astrocytes. Our data also suggest that mechanical allodynia and spread of pain (contralateral pain) share similar mechanisms, both involving JNK activation in astrocytes. In sharp contrast to spinal JNK that is critical for the maintenance of mechanical allodynia, peripheral JNK contributes to the induction of heat hyperalgesia. Therefore, targeting the JNK pathway may open a new avenue for chronic pain management. In particular, JNK inhibitors, such as D-JNKI-1, at very low doses, may be useful to alleviate some cardinal symptoms of chronic pain.
Acknowledgments
We thank Dr. Christophe Bonny of University of Lausanne, Switzerland for providing the JNK peptide inhibitor D-JNKI-1. This work was supported by NIH grants DE17794, NS54932, NS67686, and TW7180 to RRJ. This study was also partly supported by an IASP collaboration grant to RRJ and ID, National Natural Science Foundation of China (NSFC) 30500153 to YJG, and National Science Council of Taiwan (NSC97-2314-B-341-002-MY3) to YRW. YCL was supported by a fellowship from Cheng Kung University Medical College, Taiwan. All the authors do not have conflict of interest.
References
- 1.Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 2.Aloisi AM, Porro CA, Cavazzuti M, Baraldi P, Carli G. ‘Mirror pain’ in the formalin test: behavioral and 2-deoxyglucose studies. Pain. 1993;55:267–73. doi: 10.1016/0304-3959(93)90156-J. [DOI] [PubMed] [Google Scholar]
- 3.Ambalavanar R, Dessem D, Moutanni A, Yallampalli C, Yallampalli U, Gangula P, Bai G. Muscle inflammation induces a rapid increase in calcitonin gene-related peptide (CGRP) mRNA that temporally relates to CGRP immunoreactivity and nociceptive behavior. Neurosci. 2006;143:875–84. doi: 10.1016/j.neuroscience.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Baba H, Doubell TP, Woolf CJ. Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J Neurosci. 1999;19:859–67. doi: 10.1523/JNEUROSCI.19-02-00859.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertorelli R, Corradini L, Rafiq K, Tupper J, Calo G, Ongini E. Nociceptin and the ORL-1 ligand [Phe1psi (CH2-NH)Gly2]nociceptin(1-13)NH2 exert anti-opioid effects in the Freund’s adjuvant-induced arthritic rat model of chronic pain. Br J Pharmacol. 1999;128:1252–8. doi: 10.1038/sj.bjp.0702884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–6. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 7.Butler SH, Godefroy F, Besson JM, Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- 8.Cervero F, Laird JM. Mechanisms of allodynia: interactions between sensitive mechanoreceptors and nociceptors. Neuroreport. 1996;7:526–8. [PubMed] [Google Scholar]
- 9.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 10.Chen HS, Chen J, Sun YY. Contralateral heat hyperalgesia induced by unilaterally intraplantar bee venom injection is produced by central changes: a behavioral study in the conscious rat. Neurosci Lett. 2000;284:45–8. doi: 10.1016/s0304-3940(00)00955-1. [DOI] [PubMed] [Google Scholar]
- 11.Chiang CY, Wang J, Xie YF, Zhang S, Hu JW, Dostrovsky JO, Sessle BJ. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J Neurosci. 2007;22(27):9068–76. doi: 10.1523/JNEUROSCI.2260-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chillingworth NL, Donaldson LF. Characterisation of a Freund’s complete adjuvant-induced model of chronic arthritis in mice. J Neurosci Methods. 2003;128:45–52. doi: 10.1016/s0165-0270(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 13.Coderre TJ, Melzack R. Central neural mediators of secondary hyperalgesia following heat injury in rats: neuropeptides and excitatory amino acids. Neurosci Lett. 1991;131:71–4. doi: 10.1016/0304-3940(91)90339-u. [DOI] [PubMed] [Google Scholar]
- 14.DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 15.Draisci G, Iadarola MJ. Temporal analysis of increases in c-fos, preprodynorphin and preproenkephalin mRNAs in rat spinal cord. Brain Res. 1989;6:31–7. doi: 10.1016/0169-328x(89)90025-9. [DOI] [PubMed] [Google Scholar]
- 16.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 17.Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett. 2008;437:180–3. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;1(29):4096–108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giaume C, McCarthy KD. Control of gap-junctional communication in astrocytic networks. Trends Neurosci. 1996;19:319–25. doi: 10.1016/0166-2236(96)10046-1. [DOI] [PubMed] [Google Scholar]
- 20.Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–18. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao J, Liu MG, Yu YQ, Cao FL, Li Z, Lu ZM, Chen J. Roles of peripheral mitogen-activated protein kinases in melittin-induced nociception and hyperalgesia. Neuroscience. 2008;152:1067–75. doi: 10.1016/j.neuroscience.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 23.Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–93. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 24.Hunt JL, Winkelstein BA, Rutkowski MD, Weinstein JN, DeLeo JA. Repeated injury to the lumbar nerve roots produces enhanced mechanical allodynia and persistent spinal neuroinflammation. Spine. 2001;26:2073–9. doi: 10.1097/00007632-200110010-00005. [DOI] [PubMed] [Google Scholar]
- 25.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–85. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–69. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007;177:359–389. doi: 10.1007/978-3-540-33823-9_13. [DOI] [PubMed] [Google Scholar]
- 29.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–85. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;26(36):57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 32.Johanek LM, Simone DA. Cannabinoid agonist, CP 55,940, prevents capsaicin-induced sensitization of spinal cord dorsal horn neurons. J Neurophysiol. 2005;93:989–97. doi: 10.1152/jn.00673.2004. [DOI] [PubMed] [Google Scholar]
- 33.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 34.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 35.Katsura H, Obata K, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Transforming growth factor-activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia. 2008;56:723–33. doi: 10.1002/glia.20648. [DOI] [PubMed] [Google Scholar]
- 36.Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of extracellular signal-regulated protein kinases 5 in primary afferent neurons contributes to heat and cold hyperalgesia after inflammation. J Neurochem. 2007;102:1614–24. doi: 10.1111/j.1471-4159.2007.04698.x. [DOI] [PubMed] [Google Scholar]
- 37.Keramaris E, Vanderluit JL, Bahadori M, Mousavi K, Davis RJ, Flavell R, Slack RS, Park DS. c-Jun N-terminal kinase 3 deficiency protects neurons from axotomy-induced death in vivo through mechanisms independent of c-Jun phosphorylation. J Biol Chem. 2005;280:1132–41. doi: 10.1074/jbc.M410127200. [DOI] [PubMed] [Google Scholar]
- 38.Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–7. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- 39.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–76. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 40.Laird JM, Roza C, De Felipe C, Hunt SP, Cervero F. Role of central and peripheral tachykinin NK1 receptors in capsaicin-induced pain and hyperalgesia in mice. Pain. 2001;90:97–103. doi: 10.1016/s0304-3959(00)00394-8. [DOI] [PubMed] [Google Scholar]
- 41.LaMotte RH, Lundberg LE, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol. 1992;448:749–64. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–84. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- 43.Maleki J, LeBel AA, Bennett GJ, Schwartzman RJ. Patterns of spread in complex regional pain syndrome, type I (reflex sympathetic dystrophy) Pain. 2000;88:259–66. doi: 10.1016/S0304-3959(00)00332-8. [DOI] [PubMed] [Google Scholar]
- 44.Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res. 1996;35:47–7. doi: 10.1016/0169-328x(95)00181-q. [DOI] [PubMed] [Google Scholar]
- 45.Messersmith DJ, Kim DJ, Iadarola MJ. Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Brain Res. 1998;53:260–9. doi: 10.1016/s0169-328x(97)00308-2. [DOI] [PubMed] [Google Scholar]
- 46.Migheli A, Piva R, Atzori C, Troost D, Schiffer D. c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1997;56:1314–22. doi: 10.1097/00005072-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 47.Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–40. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PLoS ONE. 2007;2:e1116. doi: 10.1371/journal.pone.0001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miraucourt LS, Moisset X, Dallel R, Voisin DL. Glycine inhibitory dysfunction induces a selectively dynamic, morphine-resistant, and neurokinin 1 receptor- independent mechanical allodynia. J Neurosci. 2009;29:2519–27. doi: 10.1523/JNEUROSCI.3923-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagakura Y, Okada M, Kohara A, Kiso T, Toya T, Iwai A, Wanibuchi F, Yamaguchi T. Allodynia and hyperalgesia in adjuvant-induced arthritic rats: time course of progression and efficacy of analgesics. J Pharmacol Exp Ther. 2003;306:490–7. doi: 10.1124/jpet.103.050781. [DOI] [PubMed] [Google Scholar]
- 52.Neumann S, Braz JM, Skinner K, Llewellyn-Smith IJ, Basbaum AI. Innocuous, not noxious, input activates PKCgamma interneurons of the spinal dorsal horn via myelinated afferent fibers. J Neurosci. 2008;28:7936–44. doi: 10.1523/JNEUROSCI.1259-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–53. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–22. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–33. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- 56.Presley RW, Menétrey D, Levine JD, Basbaum AI. Systemic morphine suppresses noxious stimulus-evoked Fos protein-like immunoreactivity in the rat spinal cord. J Neurosci. 1990;10:323–35. doi: 10.1523/JNEUROSCI.10-01-00323.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–73. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 58.Ren K, Williams GM, Ruda MA, Dubner R. Inflammation and hyperalgesia in rats neonatally treated with capsaicin: effects on two classes of nociceptive neurons in the superficial dorsal horn. Pain. 1994;59:287–300. doi: 10.1016/0304-3959(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 59.Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 60.Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–18. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 61.Sinnott CJ, Garfield JM, Strichartz GR. Differential efficacy of intravenous lidocaine in alleviating ipsilateral versus contralateral neuropathic pain in the rat. Pain. 1999;80:521–31. doi: 10.1016/S0304-3959(98)00245-0. [DOI] [PubMed] [Google Scholar]
- 62.Spataro LE, Sloane EM, Milligan ED, Wieseler-Frank J, Schoeniger D, Jekich BM, Barrientos RM, Maier SF, Watkins LR. Spinal gap junctions: potential involvement in pain facilitation. J Pain. 2004;5:392–405. doi: 10.1016/j.jpain.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 63.Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem. 2005;92:1508–20. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- 64.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–82. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 65.Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119:287–94. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–5. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 67.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–9. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 68.Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund’s adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–96. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 70.Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–59. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 71.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–60. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]