

Abstract
With rare exceptions, spontaneous tumors originate from a single cell. Yet, at the time of clinical diagnosis, the majority of human tumors display startling heterogeneity in many morphological and physiological features, such as expression of cell surface receptors, proliferative and angiogenic potential. To a substantial extent, this heterogeneity might be attributed to morphological and epigenetic plasticity, but there is also strong evidence for the co-existence of genetically divergent tumor cell clones within tumors. In this perspective, we summarize the sources of intra-tumor phenotypic heterogeneity with emphasis on genetic heterogeneity. We review experimental evidence for the existence of both intra-tumor clonal heterogeneity as well as frequent evolutionary divergence between primary tumors and metastatic outgrowths. Furthermore, we discuss potential biological and clinical implications of intra-tumor clonal heterogeneity.
Keywords: diversity, selection, evolution
1. Introduction
Human cancers frequently display substantial intra-tumor heterogeneity in virtually all distinguishable phenotypic features, such as cellular morphology, gene expression (including the expression of cell surface markers and growth factor and hormonal receptors), metabolism, motility, and angiogenic, proliferative, immunogenic, and metastatic potential [1–4].
It is widely accepted that tumor development can be regarded as a process of Darwinian evolution. The evolutionary perspective is contingent on population thinking, where selection forces work on a population of individuals with different heritable traits. Indeed, large numbers of cell divisions, required for the emergence of full-blown malignancies and increased genetic instability, seen in most cancers, present plenty of opportunities for the emergence of multiple mutants. This genetic heterogeneity translates into phenotypic heterogeneity, and heritable phenotypes will in turn provide material for selection forces to work on. However, it is likely that a substantial fraction of phenotypic heterogeneity seen in tumors can arise from phenotypic plasticity and differentiation of cancer stem cells and is therefore non-heritable.
Although the existence of intra-tumor phenotypic heterogeneity has been recognized from the early days of experimental cancer research, the relative contributions of heritable and non-heritable mechanisms are still not clear, and yet the nature of tumor heterogeneity can have profound implications both for tumor development and therapeutic outcomes.
2. Non-heritable sources of diversity in tumor cell populations
2.1. Cancer stem cells
The concept of cancer stem cells was proposed to explain heterogeneity of cancer cells more than three decades ago (for an excellent historical perspective, see [2]). However, it has emerged as a mainstream idea only relatively recently, initially for hematopoietic malignancies, after the discovery that a phenotypically distinct subset of human acute myeloid leukemia cells (CD34+CD38−) can reconstitute the disease upon transplantation into immunodeficient mice in limiting dilution assays, whereas all other leukemic cells are devoid of this ability [5]. Subsequently, the cancer stem cell concept has been expanded to encompass solid malignancies: the CD44+CD24− subset of human breast cancer cells was shown to be highly enriched for the ability to initiate tumors upon transplantation into mammary fat pads of immunocompromised mice [6], and the existence of phenotypically defined subsets of cells enriched for tumor-initiating ability was later demonstrated for other solid cancers (reviewed in [7]).
According to the cancer stem cell concept, only a minor fraction of tumor cells, termed “tumor stem cells” is responsible for the maintenance and progression of tumors because these cells possess the unique ability to both self-renew and differentiate into the bulk of tumor “non-stem” cells. Thus, the majority of tumor cells are considered to be products of abnormal differentiation of cancer stem cells, and, although some might be capable of limited proliferation, they represent evolutionary “dead ends.” It is important to stress that the concepts of clonal evolution and cancer stem cells are complementary rather than mutually exclusive. It can hardly be doubted that tumor progression is contingent on acquiring specific mutations in oncogenes and tumor suppressor genes. From an evolutionary perspective, limitation of self-replicating capacity to a fraction of tumor cells means that the effective population size is restricted to this stem-like compartment, rather than encompassing a bulk of tumor cells. The implication is that phenotypic and genetic heterogeneity, associated with tumor stem cell differentiation, are irrelevant for tumor progression (as long as they do not affect the tumor stem cell subpopulation or lead to stem cell conversion), as selection can only work on the heritable phenotypes of cancer stem cells.
The existence of morphologically defined subsets of cancer cells which are enriched for the ability to form tumors in xenograft models has been demonstrated by multiple groups for many hematopoietic and solid malignancies. Still, the concept of cancer stem cells remains a subject of debate, especially in solid tumors. A major challenge to the cancer stem cell concept originates from the observation that its definition is based on experimental evidence (e.g., tumor initiation at limited dilution) that is highly subject to how the assay was performed. For example, in experimentally induced or spontaneous cancers, the majority of cancer cells are capable of initiating tumors in either syngenic or xenograft models [8, 9] and thus can be considered cancer stem cells. When most of the cells in a tumor are cancer stem cells, singling them out becomes meaningless. It has also been noted that numerical considerations frequently reveal inconsistencies with data interpretation in experiments transplanting sorted cell populations [10]. In addition, comparisons of the genetic composition of breast cancer CD44+CD24− cells, which are presumed to be “cancer stem cells,” versus CD44−CD24+ cells, which are considered to be “non-stem cells,” have revealed that in some cases these subpopulations are genetically divergent [11, 12], which is inconsistent with a model of simple differentiation.
2.2. Phenotypic plasticity
An alternative explanation for the differential ability of specific subsets of tumor cells to initiate tumors in xenograft models is the concept of tumor cell plasticity. According to this idea, the majority of tumor cells can be viewed as stem cells with varying degrees of “stemness,” where the “stemness” is influenced by microenvironmental cues and perhaps some stochastic cell-autonomous mechanisms [13]. In fact, even for normal tissue stem cells, the simplistic model of a differentiation hierarchy through rigid, unambiguously definable stages, seems to be poorly supported by experimental data. Observation of substantial phenotypic and functional heterogeneity within normal stem/progenitor cell pools has led to the proposal of alternative models of the organization of stem cell compartments that incorporate self-organization, flexibility, and plasticity of stem cell properties [14–16]. Importantly, mathematical models that involve plasticity of the stem cell phenotype provide a much better match to experimental data on stem cell dynamics than does the concept of rigid differentiation hierarchy [17].
Phenotypic plasticity is one of the basic features of organisms, as genotypes do not specify a strictly defined, invariable phenotype, but instead a range of phenotypic manifestations within a norm of reaction. The ability of tumor cells to alter their phenotype in response to microenvironmental cues can be illustrated by the dramatic observation that teratocarcinoma cells injected into normal blastocysts, can contribute morphologically normal cells into different cellular lineages and tissues [18]. At this point, it is clear that interactions of tumor cells with the microenvironment both shape malignant behavior and promote tumor progression [19]. The microenvironment within a tumor is not completely homogeneous: different regions of a tumor can have different densities of blood and lymphatic vasculature, different numbers and types of infiltrating normal cells, and different composition of extracellular matrix. Therefore, tumor cells within a given tumor are expected to experience a range of microenvironmental cues, which would in turn translate into a range of phenotypic manifestations.
In addition to phenotypic differences that arise in response to differences in environmental conditions, cells can show heterogeneity of features even within apparently homogeneous environments. Such heterogeneity, arising from noise in gene expression and existence of meta-stable configuration of intracellular networks, is a very basic and evolutionarily ancient feature of all living cells. For example, a minor fraction of genetically homogeneous bacterial populations displays marked resistance to antibiotic treatment, a phenomenon termed “persistence.” Notably, this resistance is maintained only over a couple of cell divisions, and populations formed by the progeny of persistent bacteria have the same frequency of persistent cells as the original population [20]. Observation of single-cell dynamics with the help of microfluidic devices has linked persistence with reduced proliferation rates [21]. Thus, explanation of heterogeneity in both growth rates and resistance to cytotoxic agents needs not to involve stem cells.
The existence of similar heterogeneity has also been demonstrated in mammalian cells. Normal and cancer cell lines display substantial heterogeneity in timing of apoptotic response to TRAIL ligand. This heterogeneity does not depend on genetic or epigenetic mechanisms but is instead caused by apparently noise-driven differences in levels of protein expression [22]. Plasticity of tumor cell phenotype is not limited to apoptotic response. For example, genetically homogeneous tumor cell lines display morphological heterogeneity, as mixtures of immotile, rounded cells and motile, fibroblast-like ones can be found both in vitro and in vivo. In this case, the phenotypic differences result from different, mutually exclusive, and inter-convertible activation of Rac and Rho GTPases [23].
Concepts of cancer stem cells and phenotypic plasticity need not to be mutually exclusive. Even if the majority of tumor cells in some (or many) cancers are incapable of sustained proliferation and, therefore, can be described as non-stem cells, the stem cell compartment can still be phenotypically diverse and plastic. The subjects of tumor stem cells and stem cell plasticity constitute a focus of very intensive research; therefore, it is likely that some of the current controversies will be resolved relatively soon. Regardless of the outcome of the cancer stem cell debate, it is likely that non-heritable mechanisms are responsible for a large fraction of intra-tumor heterogeneity of cellular phenotypes.
3. Heritable heterogeneity
3.1. Intra-tumor clonal diversity
Non-heritable mechanisms of cellular heterogeneity, however, cannot be the only contributors to the diversity of tumor cell phenotypes. Cancers are not static entities: they start from a genetically normal cell and conclude with billions of malignant cells that have accumulated large numbers of mutations in “driver” and “passenger” genes [24].
For the sake of simplicity, tumor evolution is often depicted as a clean succession of clonal expansion rounds, where every new round is driven by the acquisition of an additional mutational event, which leads to a selective sweep (Fig. 1A). This depiction aptly conveys the sequence of the key mutations that drive tumor progression, as viewed on the macroscopic scale from the endpoint of a malignant tumor. However, this depiction is unlikely to reflect the dynamics of tumor evolution at the more fine scale. The acquisition of mutations is a stochastic process, and there is no choreographer to determine which mutation should occur next so that the tumor can progress to the next stage. Instead, random mutations are constantly produced as a result of proliferation and increased genomic instability and then “tested” by Darwinian selection. Only minority of random mutations are selectively advantageous, while a large fraction of mutations will be discarded by selection. Furthermore, many neutral or even mildly disadvantageous mutations can be retained in the population or even undergo some expansion due to genetic drift. Moreover, the long-term evolutionary success of mutations providing a positive selective advantage is not granted. Darwinian selection is context-specific and oblivious to the future. As a consequence, some of the mutations that are selectively advantageous at certain stages of tumor progression and can trigger substantial clonal expansion may lead to evolutionary dead ends and, therefore, may not be present in a fully malignant tumor. The complexity of tumor evolution is further influenced by the ongoing alterations of tumor microenvironment associated with tumor progression [25], which are likely to alter the selective pressures experienced by tumor cells. Therefore, at the microscopic level, tumor evolution is likely to be non-linear, and substantial genetic heterogeneity is expected in tumor cell populations (Fig. 1B).
Fig. 1. Schematic view of monoclonal and multiclonal models of tumor progression.

Increasing color intensity correlates with tumor progression, whereas different colors reflect different clones. A: Traditional, linear model of clonal succession, where progressive mutations in oncogenes and tumor suppressor genes drive linear succession of rounds of clonal expansion, manifested as tumor progression. B: Multi-clonal model of tumor progression: although all cells in tumors originate from a single initiated cell, the evolution of the tumor is more “messy”, with genetically divergent tumor clones co-existing within tumors for substantial periods of time. The population sizes and characteristics of clones change as tumors evolve, with some clone populations expanding in size and others remaining unchanged or becoming extinct. In advanced stages of tumor evolution, tumors might become dominated by single clones.
It is useful to distinguish cellular genetic heterogeneity from clonal genetic heterogeneity [26]. The former refers to genetic differences at the level of single tumor cells, whereas the latter refers to genetic differences that have been amplified by clonal expansion. Focusing on clonal heterogeneity instead of cellular heterogeneity eliminates some of the “noise” of tumor evolution, as many of the variants detectable at the level of individual cells fail to clonally expand because of their occurrence in a cell that has lost stem cell properties, unfavorable effects on fitness, or simple stochastic reasons. However, “clonal heterogeneity” will not necessarily be completely “noise-free,” as clonal expansion does not necessarily prove the selective value of a mutation.
It should also be noted that, from a perspective of selection operating in the evolution of tumors, stable, heritable changes in gene expression due to epigenetic alterations are indistinguishable from similar changes caused by alterations in DNA sequences. Silencing of gene expression by hypermethylation of promoter regions is frequently observed in cancers [27]; therefore, heritable epigenetic changes should be included in considerations of clonal heterogeneity.
The existence of clonal heterogeneity has been documented for a variety of malignancies, including leukemias [28]; breast [11, 29–34], prostate [35–37], colon [38–40], brain [41, 42], esophagus [26], head and neck [43], bladder [44], and gynecological carcinomas [45]; liposarcoma [46]; and multiple myeloma [47]. Thus, the experimental evidence for clonal heterogeneity is overwhelming. However, due to multiple technical challenges, the available data are mostly fragmentary and likely represent only the “tip of the iceberg,” with the extent of clonal heterogeneity and the dependence of clonal heterogeneity on tumor type, subtype, and disease stage remaining mostly unexplored.
4. Methods of studying clonal heterogeneity
Apart from the lack of general interest, the scarcity of definitive publications on clonal heterogeneity can be attributed to the substantial technical challenges faced by researchers who want to study genetic heterogeneity in human cancers. In this section, we present an overview of the approaches that have been used to study genetic heterogeneity, pointing out their strengths and limitations. In addition, we discuss the issue of inadequate sampling and technical caveats arising from the frequent necessity to analyze limited amounts of starting material. It is useful to divide the approaches to study clonal heterogeneity into focused and genome-wide methods.
4.1. Focused approaches
In focused approaches, analysis is limited to a particular genetic locus or a set of loci. The advantage of focused approaches is that they allow for the analysis of specific genes that have been implicated as drivers of tumor progression. Current knowledge of cancer genetics permits focusing on genetic lesions specific for types and subtypes of a particular cancer. The obvious disadvantage of these methods is that limiting analysis to a few loci will miss other potentially important differences and will likely underestimate the extent and patterns of clonal heterogeneity.
Most of the published reports on intra-tumor clonal heterogeneity have relied on the detection of allelic imbalances. Most commonly, this analysis is performed on microdissected tumor samples to ensure lack of contamination by normal cells and to provide adequate sample sizes. The DNA is then isolated from the captured cells, and PCR is performed using primers against microsatellite markers specific to regions that have previously been characterized as being frequently altered in the particular type or subtype of cancer under study. Comparison to controls, germline samples from the same patients, allows for the detection of alleles that have been lost somatically. Although obtaining data is relatively simple, straightforward interpretation is obscured by multiple technical caveats, such as frequently poor correlation between chromosomal deletion/amplification and under/overrepresentation of relevant microsatellite markers (for in-depth discussion, see [48]). Therefore, data obtained by analysis of allelic imbalances often requires confirmation by independent techniques.
Studies of clonal heterogeneity may rely on the detection of amplification of specific chromosomal regions by fluorescent in situ hybridization (FISH) [11, 26, 42, 44, 47] or immuno-FISH (FISH coupled with immunofluorescence) analysis [12]. In FISH analysis, fluorescently labeled BAC (bacterial artificial chromosome) probes against regions amplified in the particular cancer type, as well as control probes for centromeric regions, are hybridized to cancer cells or tissue sections. Combination with immunofluorescence provides the advantage of focusing on specific subtypes of tumor cells. FISH analysis is less vulnerable to artifacts than are other approaches, such as analysis of allelic imbalances; thus, it can be the approach of choice for the validation of differences initially detected by other means. Disadvantages include its labor-intensiveness, as large numbers of cells need to be analyzed in order to generate statistically meaningful data. Also, although potentially suitable for the analysis of chromosomal deletions, detection of amplified regions is more practical.
Clonal heterogeneity can be assessed by sequencing specific genes known to be frequently mutated in a particular cancer [26, 38, 43]. This analysis usually involves microdissection and PCR amplification of the samples. The major advantage of sequencing is that it allows for focusing on specific “driver” genes; thus, any heterogeneity will most likely be of functional importance. Intensive studies in the field of cancer genetics, and recent sequencing of cancer genomes [49–51] allowed the identification of cancer-type-specific subsets of genes that “drive” cancer progression. Therefore, it should be feasible to analyze samples for sets of genes causally implicated in a given cancer. However, to the best of our knowledge, no such studies have been reported thus far. Also, instead of the detection of specific nucleotide changes, bisulfite sequencing of specific alleles can be used to detect heritable methylation changes either in promoters of “driver” genes or in a larger set of “passenger” sites, which can be used as markers to trace clonal expansion patterns [52].
Lymphoid malignancies sometimes present another unique opportunity for focused clonality analysis. Early steps of development of B and T cells involve step-wise rearrangements of immunoglobulin (Ig) and T-cell receptor (TCR) genes, respectively. Germline loci for Ig and TCR genes contain multiple alternative gene segments, encoding for gene domains, that are randomly chosen for rearrangements to generate thousands to millions (depending on the gene) potential mature rearranged products [53] distinguishable by PCR [54]. While, in most cases, a given leukemia or lymphoma arises from a cell with completed rearrangement, and, thus, all of the cells in the malignancy start with an identical rearrangement, the immunogene loci often undergo further rearrangements through recombination or somatic hypermutation, providing a pattern that can be used to analyze evolutionary relationships among distinct clones. Combining PCR and deep sequencing appears to be an especially promising way to analyze this, as it allows for the detection of rare subclones without a priori knowledge of the products of rearrangements [28]. One potential drawback is that the subclonal changes might have no effect on fitness, i.e., they are evolutionarily neutral. However, the approach is still useful given that it allows for establishing evolutionary relationships between primary disease and relapses [55–58] and permits tracing the evolutionary history of leukemias. Moreover, substantial evidence suggests that Ig proteins and B cell receptors are actively involved in the development of hematopoietic malignancies [59]; thus, at least in some cases, the subclonal changes might be of functional importance.
4.2. Genome-wide approaches
Instead of focusing on particular loci, whole genomes can also be analyzed in clonal heterogeneity studies. An obvious advantage of these approaches is that their success is not contingent on a priori knowledge; therefore, potentially important differences, missed by focused approaches can be detected. The major disadvantages of these methods are that the functional impact of detected differences is often unclear and that the clonal differences might be evolutionarily neutral. In addition, many genome-wide approaches suffer from limited resolution.
Karyotypic analysis based on chromosomal banding was frequently used in early reports on clonal heterogeneity [33, 41, 60, 61]. In this technique, tumor cells are subjected to short-term culture followed by fixation and staining, which reveals a pattern of chromosomal banding in metaphase chromosomes. This method allows for the detection of large chromosomal abnormalities, but it has several pitfalls. Karyotypic analysis requires culturing the cells, which may lead to preferential outgrowth of selected tumor cell subpopulations, changing the representation of the original tumor. It is also labor-intensive and has low resolution.
More recent publications usually rely on comparative genomic hybridization (CGH) to detect chromosomal aberrations. DNA from tumor and normal cells is differentially labeled with fluorescent probes and, in the classical form of the assay, hybridized to normal metaphase chromosomes. Fluorescent signal is then analyzed using specialized software, revealing regions of chromosomal losses and gains. CGH is one of the most widely used techniques in studies of intra-tumor clonal heterogeneity. It provides unbiased coverage of the whole genome. However, hybridization to metaphase chromosomes gives limited resolution, as small regions of deletion and amplification can be missed. Instead of metaphase chromosomes, differentially labeled DNA can be hybridized to genomic arrays, allowing for the detection of smaller-scale allelic imbalances up to the single-nucleotide level using single nuclear polymorphism (SNP) arrays [11]. Notably, analysis of chromosomal imbalances can be performed on the level of specific chromosomes (using chromosomal arrays) rather than on a whole-genome scale [29]. Finally, clonal heterogeneity can potentially be detected by the analysis of DNA content, where tumor cells are stained with DNA-labeling dye and subjected to flow cytometric analysis (FACS). This approach reveals ploidy status and the distribution of cells in different phases of the cell cycle, and, in cases of large differences, allows for the detection of populations with distinct aneuploidy based on their shift from normal ploidy peaks [26, 40, 41, 62]. The advantages of this method are that sampling issues are not much of a problem (discussed below) and that it is capable of resolving even highly intermixed subpopulations. However, its resolution is limited to the detection of clones that differ substantially in DNA content. Therefore, the method provides limited molecular insights unless distinct subpopulations are sorted and subsequently analyzed with more sensitive methods.
4.3. Sampling issues
One of the obvious challenges of studying clonal heterogeneity in human malignancies is the issue of sampling. One aspect of this problem is the availability of representative biopsies. Core biopsies, obtained for diagnostic purposes, sample only small regions of tumors, and, therefore, are not likely to be informative about the clonal composition of each tumor as a whole. Thus, adequate analysis of intra-tumor clonal heterogeneity often relies on access to post-surgical samples from different regions of tumors.
Sample size is another important issue. When a tumor specimen is too large, most methods will only provide an average picture, usually reflecting the dominant clone. Thus, potential heterogeneity would be missed: while large numbers of cells pooled will quench the “noise,” signal from minor subpopulations will be lost. The exception is deep sequencing of whole genomes, which allows for the detection of mutant alleles present in a small fraction of tumor cells [32]. On the other end of the spectrum, many of the analyses suitable for the characterization of clonal heterogeneity can be performed with starting material of just single cells [63]. Focusing on single cells, however, brings about the problem of dealing with cell-to-cell variability (the cell-to-cell genetic heterogeneity discussed above), where some of the distinguishable variants have no chances of clonal expansion and thus constitute evolutionary “dead ends.” This problem of biological noise can potentially be dealt with by analyzing larger numbers of individual cells, but scaling up the analysis will inevitably drive up the cost and labor required, making these studies impractical. Moreover, single cell analysis suffers from increased vulnerability to technical caveats related to the necessity to amplify single genomes prior to the analysis (discussed below).
Therefore, with respect to sample size, there is a need to balance signal-to-noise ratios. Many studies achieve this balance by focusing on relatively small regions of tumors, ideally representing morphologically distinct units, that are isolated by microdissection. The validity of this approach is supported by the observation of the clustering of populations that differ in gene expression [64] and genetic composition [38]. Moreover, in xenograft experiments, when tumors are produced from a mixture of distinct clones, these clones can segregate into distinct zones, and the segregation appears to increase with progression [65]. However, sampling small regions of tumors can still miss relatively small patches of genetically distinct cells, unless large numbers of samples are taken from the individual tumors. In addition, if genetically distinct clones are intermixed within small anatomically distinct units, such heterogeneity will be missed due to pooling of cells.
Another approach to sampling is based on the analysis of phenotypically distinct subpopulations after singling them out by FACS or by immunobead separation. For example, analysis of the genomic composition of CD44+CD24− and CD44−CD24+ breast carcinoma cells has revealed the existence of unexpected clonal divergence between the two populations in some tumor samples [11]. Obviously, this approach can be valid only for the cases when phenotypic differences can be related to differences in genetic composition, and genetic heterogeneity within the populations will be missed.
4.4 Issues of quantity and quality
iscussed above, many methods of the analysis of tumor heterogeneity rely on microdissection of small, anatomically distinct sections of tumors from frozen or archived tissues. Microdissection allows for focusing the analysis on small, morphologically homogeneous populations and avoids contamination with non-malignant cells. However, the technique leads to increased DNA damage and thus reduced quality. The issue of DNA quality is especially pronounced in the analysis of archived formalin-fixed paraffin embedded tissues.
Due to the low amount of starting material, many tumor heterogeneity analysis methods, especially those involving whole-genome analysis, rely on prior amplification of whole genomes. Several efficient methods for whole-genome amplification, relying either on PCR-based or isothermal DNA amplification, have been developed over the last decades [66]. However, whole-genome amplification produces only a representation, not a replica, of the genome, so, inevitably, representation bias is introduced. This bias might be more pronounced in PCR-based techniques due to different efficiencies of amplification of fragments of different sizes and nucleotide composition. However, even isothermal, linear amplification techniques have been shown to suffer from substantial representation bias [67]. The issue of representation bias is most pronounced when the starting material is small (<10 cells) [66], culminating at the single-cell level. To some extent, bias issues can be reduced by using identically manipulated control samples. However, acquiring robust data often requires confirmation by independent methods, such as FISH analyses.
5. Sources of clonal heterogeneity
Tumor initiation and progression are dependent on rare, stochastic mutational events, and the chances of any given individual getting diagnosed with a particular cancer within a limited timeframe are relatively low, suggesting that cancers have to be clonal in origin, i.e., they are likely to arise from a single cell gone rogue. This consideration, supported by substantial experimental evidence, has led to a wide acceptance of the clonal origin of cancers [68, 69], although there might be some rare exceptions in cases of hereditary cancers [70]. Even though most cancers start from a single mutated cell, mechanisms that constrain clonal evolution in populations of normal progenitor cells are no longer fully functional in tumors, allowing for an ongoing evolutionary process in populations of tumor cells [71, 72]. Multiple rounds of proliferation, often counter-balanced by cell death, are required to produce macroscopic tumors, and genomic instability, observed in most cancers, is expected to constantly produce new mutations, which serve as raw material on which tumor evolution can work.
Given that any particular type of cancer progresses through a relatively narrow evolutionary trajectory [73], “jackpot” mutations, which endow mutant clones with the ability to achieve selective sweeps, are likely to be very rare. Most of the spontaneous mutations that occur are neutral or deleterious, and, while grossly disadvantageous mutations will be discarded, neutral or near-neutral mutations can undergo expansions due to genetic drift [74] and can sometimes reach fixation in small tumors [75, 76]. Therefore, for the rare mutations that confer competitive advantage and thus can be selected for by Darwinian forces, achieving clonal dominance can take a long time. Moreover, spatial constraints that exist within a tumor can slow down the achievement of clonal dominance by substantially inhibiting competition among different tumor cell clones. The importance of spatial constraints for the emergence and maintenance of clonal heterogeneity is supported by experimental observations in colon cancer as well as in silico modeling [38].
In addition to providing barriers to competition, spatial intra-tumor heterogeneity can support the co-existence of genetically distinct clones by affecting selective pressures. Most tumors represent at least partially structured habitats where individual cells within a tumor are experiencing differences in interactions with extracellular matrix, physical contact with other cells (both tumor and non-tumor), and gradients of oxygen, nutritional and growth factors, and metabolites. Therefore, tumor cells within different parts of tumors could be expected to experience different selective pressures, leading to the selection of different sets of mutations.
Mechanisms of competitive co-existence of different species have been a focus of intense theoretical research in ecology [77, 78], and some of the predictions of this research were validated in experimental studies on bacterial cell populations. It has been demonstrated that a structured habitat in experimental microbial ecosystems, as opposed to a homogeneous habitat, allows for the emergence of stable genetic diversity and that selection forces are important for the maintenance of this diversity [79, 80].
Mathematical modeling suggests that, even in the absence of substantial spatial heterogeneity, evolving pre-malignant tumors are likely to be characterized by quasi-steady states with the coexistence of multiple populations with different “strategies.” However, a clone that evolves toward invasive cancer will be capable of invading and destroying pre-malignant populations [81]. Another way to consider different “strategies” is through the prism of ecological interactions, which we will discuss below.
6. Heterogeneity of distant metastases
Clonal heterogeneity within primary tumors is only one of the aspects of clonal heterogeneity of cancers. Cancer is a systemic disease: malignant cancers shed large numbers of cells into the blood stream and lymph vessels; some of these cells settle down in distant sites and develop into metastases. Therefore, an understanding of clonal heterogeneity of cancers is incomplete without an analysis of metastatic tumors and disseminated tumor cells. Moreover, distant metastasis is the most fearsome aspect of cancer, since it is responsible for the majority of cancer-related deaths, and, therefore, understanding the underlying biological mechanisms of it is of primary importance.
When considering the clonal heterogeneity of metastatic tumors, two key questions need to be considered:
1) What is the clonal relationship between primary and metastatic tumors: are tumor cells of metastatic lesions direct descendants of clones present in advanced primary tumors, or have they diverged at earlier stages of tumor evolution?
2) What is the degree of genetic heterogeneity of metastatic tumors: are they more or less clonally heterogeneous compared to primary tumors?
What is known about the clonal relationship between tumor cells in primary tumors and metastatic sites? According to a traditional model of tumor development, tissue constraints constitute a major evolutionary bottle-neck in cancer evolution [82]; thus, the acquisition of metastatic ability is considered to be the final step in tumor development, contingent on the acquisition of all of the other hallmarks of cancer [83]. This model implies that metastatic tumors should be genetically similar to the bulk of primary tumor cells.
Many studies that compared the genetic composition of primary tumors and secondary metastatic sites have found very close clonal relationships between the two in the majority of cases [34, 84–86]. Similarly, analysis of gene expression profiles revealed very similar patterns between primary tumors and metastatic sites, a scenario highly unlikely for genetically divergent clones [87–89]. Another prediction from the linear model of tumor progression is that different metastases should display close clonal relationships among each other. Indeed, this prediction is supported by a recent study that compared the genetic composition of anatomically distinct metastatic lesions in 29 prostate cancer patients using SNP arrays and CGH [90]. In all cases, different metastatic lesions within the same patients demonstrated close clonal relationships, signifying monoclonal origin [90]. This demonstration of monoclonality of metastatic cancers is especially interesting given that primary prostate cancers are frequently multi-focal [91, 92] and show substantial intra-tumor genetic heterogeneity [35, 36].
While the evidence of the close genetic relationship between primary and metastatic tumors is compelling, some cases display dramatic divergence, challenging the model where acquisition of metastasis is considered to be the last step of tumor progression. Radically different patterns of allelic losses, indicative of a high degree of genetic divergence, have been reported between primary tumors and lymph node metastases in prostate cancers [85] and between primary tumors and asynchronous metastases in breast cancers [86]. Highly divergent clonal evolution was also evident in a subset of cases in CGH studies of primary tumors versus lymph node metastases in breast cancers [34] and of primary tumors versus metastatic tumors in renal cell carcinomas [84]. A recent report, comparing sequences of primary tumors and metastases in lobular breast cancers, revealed multiple mutations present only in metastases and several other mutations with increased frequency in metastatic sites [32].
Important clues about the clonal evolution of tumors and the relationship between primary tumors and metastatic sites can be gained by the analysis of disseminated tumor cells. Because peripheral blood, bone marrow, and lymph nodes normally do not contain cells of epithelial origin, cells that have disseminated from primary tumors can be detected by the use of epithelial-specific markers, cytokeratins and epithelial cell adhesion molecule (EpCAM) [93, 94]. Unexpected findings were reported by Klein and co-authors, who employed a PCR-based whole-genome amplification technique for the analysis of single disseminated tumor cells [63]. The authors demonstrated that disseminated tumor cells could be detected both in the bone marrow of patients and in mouse models of cancer at very early, pre-invasive stages of breast cancer development [95]. More support for the early dissemination of cancer cells came from CGH analysis of disseminated tumor cells from patients without overt metastases. These cells contained surprisingly few chromosomal aberrations [96], while higher-resolution LOH analysis did confirm the neoplastic origin of these cells and pointed to early events in tumor progression [97]. Interesting results were obtained for esophageal cancers, one of the most aggressive human carcinomas, where removal of primary tumors does not improve the chances of patient survival [98], suggesting the importance of relatively early metastatic spread. Lymph node and bone marrow metastases showed substantial divergence from primary tumors and among each other, suggesting parallel evolution shaped by microenvironment-specific selection forces. Interestingly, HER2 amplification in disseminated tumor cells was found to signify poor prognosis, whereas HER2 amplification in primary tumors had no prognostic value, further underlying the significance of divergent evolution between primary tumors and metastatic sites [99]. It should be noted, however, that since this analysis, based on the amplification of single-cell genomes, is associated with many technical caveats, critical interpretation of the data is highly warranted [100].
As discussed above, the obvious caveat of the studies of disseminated tumor cells is that the potential of any given disseminated single tumor cell to initiate a metastatic tumor is unknown. The mere fact that tumor cells survive at distant sites tells little about their ability to initiate a secondary tumor. Indeed, even normal epithelial cells were capable of extended survival in heterotopic sites in an animal model [101], and fetal cells are known to survive decades in women following pregnancy [102]. Also, most carcinomas display pronounced chromosomal instability, and, therefore, many disseminated cells might be a manifestation of “cell-to-cell heterogeneity,” i.e., they are products of genomic instability without any substantial chance of clonal expansion. Testing the relevance of these disseminated cells to secondary metastasis formation is not trivial. The most compelling evidence comes from a mouse model of breast cancer where the growth of lung metastases paralleled that of the primary site from early points of tumor progression, and excision of mammary glands when primary tumors were still at the in situ stages did not prevent or delay the development of metastases [95]. Tumor cells that disseminated from early-stage tumors into the bone marrow were capable of lethal proliferation upon transplantation into lethally irradiated mouse recipients [95]. While these findings are compelling, their relevance to human cancer is not clear, especially considering that the genetic heterogeneity of disseminated cancer cells in mouse tumors appears to be less pronounced than that in human tumors.
Another group demonstrated that bone marrow aspirates from carcinoma patients contain disseminated tumor cells that are capable of proliferation in cell culture and that the extent of this proliferation is inversely correlated with patient survival [103]. CGH analysis of disseminated tumor cells from breast cancer patients confirmed their high divergence from primary tumors [104]. This study demonstrated substantially higher numbers of genomic abnormalities in disseminated tumor cells compared to the studies of Klein et al., despite similar staging of the tumors analyzed, suggesting that the ability of disseminated cells to form metastases is contingent on the acquisition of a large number of genetic alterations. Additional evidence for parallel clonal evolution comes from the detection of metastatic tumors with unknown primary origin, which constitute a significant fraction of clinical cases [105]. In addition, analysis of the growth kinetics of primary and metastatic tumors is inconsistent with metastatic spread being a late event [106].
In summary, large amounts of evidence support the close clonal relationship of primary tumors and metastatic outgrowths. However, there is also strong evidence that suggests that metastatic spread can be an early event during tumor progression; thus, primary and metastatic tumors might become quite distinct genetically as they evolve. Some of the observed discrepancies might stem from technical issues, but it is plausible that the development of spontaneous human tumors is not confined to a single scenario. It is also possible that while many tumor cells are capable of early dissemination, only tumor cells that have acquired large numbers of oncogenic mutations are capable of initiating secondary tumors at metastatic sites.
Clearly, more studies are required to clarify the clonal relationship between primary and metastatic tumor cell populations, as this relationship could not only illuminate the evolutionary history of cancer development but also create a more solid ground for therapeutic decision making. Currently, the majority of therapeutic decisions are being made based on the analysis of primary tumor specimens [107]. Yet, this approach could only be justified in cases where the genetic compositions of primary and metastatic tumors are similar, especially when making decisions about emerging therapeutic approaches that target specific genetic changes. In addition, elucidating the issue of early versus late origin of metastasis-initiating cells is important for determining the usefulness of therapeutic approaches that target tumor cell invasion.
The question of clonal heterogeneity within metastases remains unexplored, and, notably, clonal heterogeneity within primary tumors has limited direct therapeutic relevance, as, in most cases primary tumors can be successfully removed by surgery, but this is most often not the case for metastatic outgrowths. Therefore, elimination of metastatic clones has to rely on adjuvant therapies, and it is likely that the success of these therapies will depend on the extent of the heterogeneity of these tumor cell populations, as we discuss below.
7. The biological impact of clonal heterogeneity
Despite strong evidence that many tumors are composed of mixtures of distinct subclones rather than being monoclonal, the knowledge about the extent of this heterogeneity at different stages of tumor development of different types and subtypes of cancer and before and after treatment is still lacking. One notable exception is Barrett's esophagus, where higher clonal diversity was shown to strongly correlate with risk of malignant progression [26], but, besides this example, the functional importance of clonal heterogeneity in human tumors remains largely unexplored. Clonal heterogeneity is likely to have a strong impact on tumor evolution via two different mechanisms. First, clonal diversity can provide a more diverse input material on which selection can work, thereby feeding tumor evolution during progression and therapeutic escape. Second, the co-existence of genetically distinct clones within a tumor can result in a network of biological interactions among the distinct clones. These biological interactions are likely to modulate progression and therapeutic responses; thus, the behavior of a tumor composed of distinct clones might be different from that of a monoclonal tumor or the behavior of the sum of the individual clones.
7.1. Clonal heterogeneity as a fuel of tumor evolution
Tumorigenesis is an evolutionary process, fueled by new mutations and driven by Darwinian selection [108, 109]. Therefore, higher genetic complexity of tumors is expected to provide more material on which selection can work, resulting in a faster pace of evolution. Although even genetically homogeneous tumors can be expected to constantly spur new clones, the complexity resulting from the branching of a monoclonal tumor will be less than the complexity generated by branching from a tumor composed of multiple distinct clones. Thus, clonally heterogeneous tumors can generate a larger variety of genetic variants to be tested by selection. In other words, genetically diverse tumors can explore larger areas of adaptive landscape, increasing the probability of clones reaching fitness peaks (Fig. 2A).
Fig. 2. The effect of clonal heterogeneity on the ability to explore the fitness landscape.
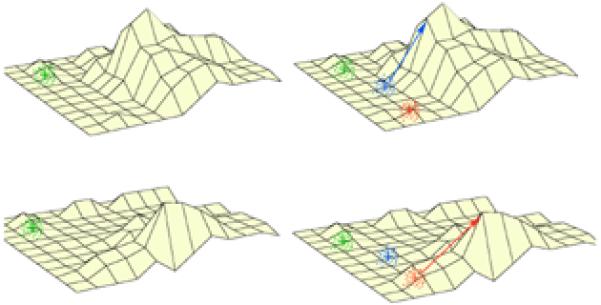
Fitness landscape (see glossary) is represented as a 3-D topographical map. Higher elevation represents higher fitness. Crosses of different colors represent genotypes of tumor cell clones. New mutations, branching from parental genotypes (represented by dots), can only explore genotypic space in the vicinity of their parental genotypes. If one of the progeny genotypes lands on a hill, it will be subjected to positive selection, which will enable further evolutionary progression toward the fitness peak. A: Clonal heterogeneity increases the probability of tumor progression. Left: the genotypic vicinity of the green clone includes regions of lower fitness and one small fitness peak. The probability that the mutant progeny of this clone reaches the hill of a larger fitness peak is very low. Right: when a tumor is composed of several genetically distinct populations (red, blue, and green), new mutants spurring from these clones can “sample” a larger region of fitness landscape, increasing their probability of reaching the hill of a larger fitness peak. In this case, some of the mutations branching from the blue clone are subjected to positive selection, allowing them to ultimately reach the fitness peak (indicated by blue arrow). B: Cancer therapy might lead to dramatic alterations of the adaptive landscape (compare fitness landscape in parts B and A). As a result, fitness of a given clone can become either higher or lower. A genetically heterogeneous tumor has higher probability that at least one of its clones will end up in a region with higher fitness value in the altered landscape (red clone in this case), enabling its evolution toward the fitness peak.
The clonal composition of tumors can be especially important in determining responses to dramatic changes in the environment, such as changes induced by anti-cancer therapy. In this case, the pre-existence of resistant clones within a tumor can make the difference between tumor extinction (treatment success) and tumor evolutionary adaptation (treatment failure) (Fig. 2B).
A vivid illustration of the importance of clonal heterogeneity in therapeutic resistance can be found in chronic myelogenous leukemia (CML). CML is a leukemia caused by the chromosomal translocation that creates Bcr-Abl, a chimeric protein with abnormal kinase activity. CML patients, at least in the chronic phase, show remarkable response to an inhibitor of Bcr-Abl, imatinib mesylate. However, a significant fraction of these patients suffer from relapse of their disease, and the relapsed leukemias often contain mutations that make Bcr-Abl insensitive to inhibition by imatinib [110]. Analysis of patient samples collected before treatment often revealeds the presence of resistant subpopulations of leukemic cells; therefore, the treatment is thought to provide selective pressure that drives the expansion of these therapy-insensitive cells, causing the eventual relapse of the disease [111–113].
Mutational mechanisms of therapy resistance have been demonstrated for many other cancers. For example, gastrointestinal stromal tumors are predominantly driven by different activating mutations in KIT, many of which can be targeted by imatinib therapy. Despite strong clinical response, in many cases, tumors relapse with secondary resistant outgrowths resulting from additional resistance-conferring mutations in KIT. In some cases, different resistant mutants could be found in different nodules of the tumor, or even within the same nodules [114] (and references therein). A different type of resistance has been documented for tumors associated with inactivating mutations in BRCA1 and BRCA2. These tumors are deficient in homologous recombination-mediated DNA repair (HRR), which increases their genomic instability but makes them highly sensitive to platinum compounds and PARP (poly ADP ribose polymerase) inhibitors. Despite strong initial response to these agents, many patients suffer from relapse of drug-resistant tumors. Studies from the Ashworth and Tamaguchi groups have demonstrated that the mutations that confer resistance are usually deletions that restore BRCA protein activity and thus improve HRR [115, 116]: instead of preventing the primary action of the drugs, they restore the DNA repair ability of the tumor cells. These examples do not exhaust the documented cases for mutational mechanisms of resistance; however, a more detailed overview lies beyond the scope of this discussion.
Except for imatinib-sensitive Bcr-Abl mutants, pre-existence of resistant cell types prior to treatment has yet to be demonstrated. Moreover, the generality of the mutational mechanisms of acquiring therapeutic resistance remains an open question. For example, array-based CGH analysis of breast cancers pre- and post-therapy failed to show selection of non-modal cell lineages [117]. Nonetheless, intra-tumor heterogeneity is likely to represent a strong challenge to therapeutic success, as larger genetic diversity within a tumor would be expected to increase the probability of the pre-existence of resistant cell types that could be selected for by treatment and ultimately result in relapse of resistant tumors. Notably, in addition to the scenario of cancer therapy, selection for mutant variants rather than mutagenesis was proposed to be the key mechanism responsible for the carcinogenic action of a wide range of growth-limiting carcinogens [118–122].
7.2. Biological interactions among distinct tumor clones
Cancers can be viewed from an ecological perspective that focuses on interactions of organisms with their environment and among each other [72, 75, 123–125]. When applying an ecological perspective to human cancers, subclonal populations of tumor cells that differ in heritable traits are considered distinct “species,” whereas infiltrating normal cells, extracellular matrix, vessels, etc. are considered the environment. This perspective can be extended to include not only primary tumors, but also disseminated tumor cells and metastatic lesions, as they share the same systemic environment. Clearly, there are limits to the applicability of ecological concepts in understanding tumor biology. Ecological theory has been primarily developed for understanding the population dynamics of multicellular, sexual organisms, and many concepts and definitions used in general ecology need to be adapted in order to be applicable to tumor cells. In addition, the differentiation hierarchy within tumors complicates the applicability of the ecological perspective even further. Yet, there are strong parallels between populations of tumor cells and populations of asexual organisms, such as microbes, and considerations of these parallels can potentially improve our understanding of tumor biology and suggest novel therapeutic approaches. The importance of the ecological perspective to human tumors may be highlighted by blooming studies on the tumor microenvironment, as it is increasingly clear that understanding alterations within tumor cells is only part of the picture, and we need to understand interactions between tumors and their microenvironment in order to account for multiple aspects of tumor progression and therapeutic resilience [25, 126].
A major implication of the ecological perspective is that co-existence of phenotypically distinct clonal populations of tumor cells should inevitably lead to the formation of a network of biological interactions, which could be either direct or mediated by the tumor microenvironment. Some of the key interactions that are likely to exist between distinct tumor clones are summarized below.
Competition
is likely to be the strongest and most important biological interaction between tumor cells. Long-lived multicellular organisms have developed powerful mechanisms to limit the competitive outgrowth of mutant cells. These mechanisms include intrinsic tumor-suppressive mechanisms, wherein activation of strong oncogenes triggers activation of tumor-suppressive networks, resulting in senescence or death of mutated cells [127]. In addition, oncogenic mutations that can trigger stronger proliferation without engaging intracellular tumor suppressors often result in loss of stemness [128, 129], indicating additional, extrinsic tumor suppressor mechanisms. Therefore, it has been proposed that wild-type phenotype confers the highest fitness under normal conditions in populations of healthy, young progenitor/stem cells [130]. Moreover, the spatial organization of normal tissues can limit the role of competition between genetically distinct cells even further, confining stem cells to small pools [109]. However, carcinogenic and growth-limiting conditions can substantially modify the fitness landscape, allowing for the competitive outgrowth of oncogenically mutated cells, thereby initiating malignant evolution [122, 131–133]. Tumor progression is associated with further loss of tumor-suppressive mechanisms and disintegration of normal tissue morphology; thus, tumors start to resemble ecological systems rather than integrated tissues, and competition becomes the strongest biological interaction. The role of competition is further strengthened by the limited nature of resources: while, under tissue culture conditions, tumor cells are capable of limitless exponential growth, clonal expansion within tumors is severely constrained by the limited availability of oxygen, nutrients, growth factors, and space (habitable niches). Limited resources intensify competitive interactions both within and between interacting species (cancer subclones) [134]. In large, spatially homogeneous populations, competitive interaction results in the fixation of a clone with the highest fitness value [75, 109]. However, as discussed above, fixation can be inhibited by spatial organization, which limits competition to within clones. In addition, the existence of regions with distinct fitness values can mediate the co-existence of clonally distinct populations. Finally, a stable co-existence of multiple clones is also possible when fitness is density-dependent [135].
Amensalism
is an interaction in which one interacting party is inhibited by the other without being affected itself. Under the competitive context, this interaction is also referred to as “interference competition.” As tumors grow under conditions of limited resources, amensalistic interactions can provide a competitive advantage to a clone that can inhibit other clones while being (at least relatively) resistant. A classical example of amensalism is an interaction between antibiotic-producing fungi and sensitive bacteria. An example of this type of interaction between genetically distinct human cells can be found in Bcr-Abl-driven leukemias: up-regulation of 24p3, an iron transporter, by Bcr-Abl leads to the depletion of iron and subsequent apoptosis in wild-type cells; Bcr-Abl expressing cells are resistant to this effect due to their downregulation of the ligand for 24p3 [136]. The existence of this type of interaction has also been documented among distinct tumor clonal populations, both in cell culture and in vivo, although the exact mechanisms are unclear [137, 138]. The concept of amensalism can be extended to interactions between primary and metastatic tumors, as many human and experimental tumors can suppress metastatic outgrowth by inhibiting angiogenesis [139, 140] or inducing dormancy of single disseminated tumor cells through uncharacterized mechanisms [141].
Thus, amensalism can work as a weapon in competitive “warfare” between distinct clonal populations. Although seemingly counterintuitive, this “warfare” can stabilize the co-existence of multiple genetically distinct variants. A vivid example for this stabilizing effect can be found in the natural community of soil microbes, which often display staggering species diversity despite inter-species competition for common resources. Mathematical modeling predicted the emergence and spatial organization of species that differ in production of antibiotic substances and antibiotic sensitivity [142]. Importantly, it was proposed that similar mechanisms can lead to the stable coexistence of distinct clones in tumors [143].
Antagonism
including predation and parasitism, is an interaction in which one interacting party can capture biomass from the other one. This interaction is widespread in natural ecosystems, and there are clear parallels between organism-tumor and host-parasite interactions. However, antagonism is unlikely to be relevant for the interactions between distinct tumor clones.
Commensalism
is a positive interaction in which one interacting party benefits the other without itself being affected. An example of this type of interaction can be found in normal tissues: mammary epithelial cells from estrogen receptor null mice (ERα −/−) fail to grow and do not develop branching structures upon transplantation into cleared fat pads. However, when mixed with wild-type epithelial progenitors, ERα −/− cells do proliferate and contribute to different lineages and different parts of the mammary gland [144]. A recent publication from the Weinberg lab demonstrated the relevance of this interaction in a human xenograft model, where an “instigator” tumor cell line augmented the proliferation and metastasis of genetically distinct “indolent” tumors that were incapable of forming macroscopic outgrowths on their own [145]. In this case, the “instigation” was mediated by systemic effects, which could at least partially be attributed to the secretion of SPP1 (osteopontin) by the “instigator” cells [145].
Although not experimentally validated, this interaction can account for the co-existence of clonal variants that differ in their angiogenic potential. If the fitness benefit of angiogenic factor production is higher than the associated fitness cost, an angiogenic clone can undergo competitive expansion and reach stable equilibrium with non-angiogenic “free rider” clones [146]. Interestingly, mathematical modeling shows that, under certain physiologic conditions, “free riders” capable of faster proliferation can out-compete the angiogenic clone, leading to the collapse of the tumor [135]. This prediction can potentially explain spontaneous regression of neuroblastomas accompanied by massive necrosis [147].
Mutualism
(cooperation) is a positive interaction in which both interacting parties can benefit from each other. Positive interactions between species have been widely documented in natural ecosystems. Mathematical modeling suggests that mutualistic interactions can lead to the coexistence of distinct species even under competitive contexts [148, 149]. It has been suggested that mutualistic interactions between distinct tumor clones can play an important role in tumor evolution by maintaining the survival and proliferation of tumor cells until one of the clones achieves a “full deck” of malignant mutations required for clonal dominance and full-blown malignancy [150, 151]. To the best of our knowledge, however, such interactions between distinct clonal tumor populations have not been experimentally documented.
Of note, most of the biological interactions are not mutually exclusive, and the net outcome for the interacting species will depend on the net sum of the different interactions. This net result can lead to both augmentation and retardation of overall tumor growth and progression. Elucidation of biological interactions between populations of tumor cells might be a very formidable task; however, it can deepen our understanding of tumor biology and uncover new therapeutic targets.
8. Clinical implications and future directions
The question of whether tumors are genetically homogeneous or rather consist of a number of genetically distinct clones is not a mere intellectual curiosity. Co-existence of distinct clones within a tumor can have profound clinical implications for disease progression, diagnosis, and therapeutic responses. Here, we will review the major clinical implications of tumor clonal heterogeneity and provide recommendations for future studies.
8.1. Sampling issues
Biopsies used for diagnostic purposes sample only small regions of tumors. Given that tumors are thought to evolve from a single initiated cell and that the identity of this initiated cell strongly influences the biology of cancer, small biopsies might be sufficient to accurately identify stages and subtypes of tumors. However, as the field of cancer treatment is moving toward the development of targeted therapeutic approaches, the question of whether the expression of a therapeutic target in a biopsy is representative of the whole corresponding tumor becomes of primary importance. For example, clonal heterogeneity involving different mutational statuses was documented for KRAS [37, 40] and TP53 [26, 37] genes, for which targeted therapy is currently being developed. Heterogeneous expression of the therapeutic target does not necessarily mean that the tumor will not be responsive to the treatment, especially if the targets are responsible for the “common good.” For example, targeting an angiogenic clone can also eliminate non-angiogenic “free rider” clones, which are dependent on the secretion of angiogenic factors. Still, clonal heterogeneity needs to be accounted for in the development of targeted therapy approaches.
8.2. Biomarkers
The index of clonal diversity is a strong predictor for malignant progression in Barrett's esophagus [26]; thus, it can be used for guiding treatment decisions. Although experimental evidence is still lacking, it is likely that Barrett's esophagus is not a biological oddity and that clonality measurements will turn out to be of predictive value for malignant progression in other cancers as well. However, sampling multiple regions from the same tumor is impractical for most cancer types, and it is not yet clear if obtaining accurate clonality measurements can be achieved using current routine diagnostic methods. Yet, it is possible that measurements of clonal heterogeneity can be useful irrespective of their utility in routine diagnostics. As we have discussed above, substantial clonal heterogeneity between primary and metastatic tumors can pose a barrier to targeted therapy. Since biopsies of metastases are frequently unavailable, it would be worth investigating if the clonal heterogeneity of primary tumors is indicative of clonal divergence in metastatic outgrowths. Also, though multiple biopsies from a single tumor might be impractical, the degree of cellular and molecular heterogeneity within a relatively small sample might also turn out to be of predictive value.
8.3. Therapeutic response
The ecological perspective suggests that biological interactions between tumor clones could have a dramatic impact on tumor evolution, both in natural progression and in evolution in response to selection forces created by therapeutic interventions. This suggestion is supported by experimental studies in mice that have demonstrated that the growth of tumors resulting from transplanting mixtures of clones cannot be predicted from the behavior of tumors developed from individual clones [65]. Similarly, experimental tumors composed of multiple clones display different sensitivity to cytotoxic drugs compared to monoclonal tumors, as clonal interactions can either potentiate or inhibit therapeutic efficacy. Therefore, clonal heterogeneity adds an additional level of complexity to our understanding of the biology of tumor development and poses challenges for the development of successful therapies. Moreover, as discussed above, high genetic heterogeneity of tumors means high probability of pre-existent clones that are resistant to therapeutic intervention and can be selected for by therapy, resulting in therapy failure.
On the other hand, clonal heterogeneity can potentially be exploited for therapeutic benefit. Maley et al. proposed two such strategies of anti-cancer therapy. First, boosting the selective advantage of benign clones can be used to drive malignant clones to extinction. Second, the selective advantage of clones sensitive to a particular therapy can be boosted, allowing them to achieve clonal dominance, and then treatment can be applied. In silico modeling confirmed that these strategies can be much more efficient compared to standard cytotoxic therapies [152]. Unfortunately, these strategies are not yet feasible, as we lack the knowledge of how to single out or boost benign or chemosensitive clones.
Gatenby and co-authors have recently proposed another strategy, termed adaptive therapy, which both accounts for the heterogeneity of tumor cells and takes advantage of it [153]. The conventional approach of adjuvant therapy is to hit tumors with the highest tolerated dose of cytotoxic drugs in a hope to kill the largest possible numbers of tumor cells, thereby decreasing the probability of the emergence of resistant mutants. However, this strategy would be expected to fail if resistant clones already exist before the treatment starts. Indeed, despite initial therapeutic responsiveness, treatment almost inevitably ends with the outgrowth of resistant tumors. Instead of completely eradicating tumors (which is not practically achievable in most cases), adaptive therapy aims to keep tumor size at bay by adjusting drug dose and timing of drug administration based on tumor response. This strategy prevents resistant variants from reaching fixation by maintaining the dominance of therapy-sensitive variants. Importantly, the feasibility of adaptive therapy is not contingent on knowledge of the exact mechanisms of the resistance, though the applicability of this idea has yet to be confirmed.
8.4. Future directions
Despite the substantial clinical significance of the issue of clonal heterogeneity, the subject remains very poorly explored. Therefore, we believe that a more systemic approach is needed to characterize the extent of clonal heterogeneity of different types and subtypes of cancers through different stages of tumor development, including disseminated tumor cells and metastases. Whether clonal heterogeneity changes in response to chemotherapy needs to be determined as well. Comprehensive characterization of tumor clonality is contingent on the development of approaches that can combine a focus on “driver” lesions with large-scale analysis. Advances in our understanding of the genetic composition of cancers make these analyses potentially feasible; however, methodology allowing for robust and cost-efficient analyses of small tumor samples is still lacking.
Accounting for the biological interactions among distinct clonal populations within tumors might pose the biggest challenge. The co-existence of multiple clones within a tumor leads to a complex network of interactions both among the clones and between the clones and the tumor microenvironment. Thus, tumors are likely to behave as complex systems and should be treated as such, especially given that more simplistic views of tumors as homogeneous entities appear to provide satisfactory results only in model systems where the complexity of experimental tumors is low. While deciphering the full extent of the interactions occurring within tumors may be an impossible task, we might be able to identify critical links and use this knowledge to control or eliminate tumors. Rich knowledge accumulated in the fields of evolutionary biology and ecology can provide useful insights for improving the clinical management of cancer patients.
ACKNOWLEDGEMENTS
We would like to thank Franzisca Michor and members of the Polyak lab for their critical reading of this manuscript. This work was supported by grants from the Breast Cancer Research Foundation, National Cancer Institute (P01CA080111), and US Department of Defense (W81XWH-09-1-0131 and BC087579). We apologize to all authors whose publication we may have omitted.
GLOSSARY
- Fitness
the ability of a cell to contribute to future generations (function of proliferation and survival). In the context of this discussion, fitness is only applicable to cells that have substantial self-renewal capacity and is relative to other cells in a population.
- Population
in our context, population refers to tumor cells, limited by tumor margins (localized tumors) or by a whole organism (disseminated tumors and metastatic outgrowths).
- Selection
change in the frequency of an allele in a population resulting from differences in the fitness values of the alleles.
- Genetic drift
change in the frequency of an allele in a population resulting from stochastic causes.
- Selective sweep
rapid increase in allele frequency driven by positive selection; results in fixation or near fixation of a mutation.
- Fixation
an allele reaching 100% frequency in a population.
- Norm of reaction
range of phenotypic manifestation of a given genotype in response to differences in the environment.
- Fitness (adaptive) landscape
mapping of relations between fitness values and genotypes. Each possible genotype and its corresponding fitness value are represented on the fitness landscape by a single spot. Fitness landscape is multi-dimensional, but visualization is limited to 2-D or 3-D. Higher fitness values are represented by peaks, lower ones by valleys. Similar genotypes are located on the adjacent coordinates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science. 1982;217:998–1003. doi: 10.1126/science.7112116. [DOI] [PubMed] [Google Scholar]
- [2].Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- [3].Nicolson GL. Generation of phenotypic diversity and progression in metastatic tumor cells. Cancer Metastasis Rev. 1984;3:25–42. doi: 10.1007/BF00047691. [DOI] [PubMed] [Google Scholar]
- [4].Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259–2265. [PubMed] [Google Scholar]
- [5].Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- [6].Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- [8].Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- [9].Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kern SE, Shibata D. The fuzzy math of solid tumor stem cells: a perspective. Cancer Res. 2007;67:8985–8988. doi: 10.1158/0008-5472.CAN-07-1971. [DOI] [PubMed] [Google Scholar]
- [11].Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- [12].Park SY, Gönen M, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ to invasive breast carcinoma. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 2006;66:1891–1895. doi: 10.1158/0008-5472.CAN-05-3450. discussion 1890. [DOI] [PubMed] [Google Scholar]
- [14].Loeffler M, Roeder I. Conceptual models to understand tissue stem cell organization. Curr Opin Hematol. 2004;11:81–87. doi: 10.1097/01.moh.0000133648.83991.af. [DOI] [PubMed] [Google Scholar]
- [15].Theise ND. Perspective: stem cells react! Cell lineages as complex adaptive systems. Exp Hematol. 2004;32:25–27. doi: 10.1016/j.exphem.2003.10.012. [DOI] [PubMed] [Google Scholar]
- [16].Colvin GA, Lambert JF, Abedi M, Dooner MS, Demers D, Moore BE, Greer D, Aliotta JM, Pimentel J, Cerny J, Lum LG, Quesenberry PJ. Differentiation hotspots: the deterioration of hierarchy and stochasm. Blood Cells Mol Dis. 2004;32:34–41. doi: 10.1016/j.bcmd.2003.09.013. [DOI] [PubMed] [Google Scholar]
- [17].Roeder I, Loeffler M. A novel dynamic model of hematopoietic stem cell organization based on the concept of within-tissue plasticity. Exp Hematol. 2002;30:853–861. doi: 10.1016/s0301-472x(02)00832-9. [DOI] [PubMed] [Google Scholar]
- [18].Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72:3585–3589. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000;6:324–329. doi: 10.1016/s1357-4310(00)01756-1. [DOI] [PubMed] [Google Scholar]
- [20].Gefen O, Balaban NQ. The importance of being persistent: heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol Rev. 2009;33:704–717. doi: 10.1111/j.1574-6976.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- [21].Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- [22].Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- [24].Parmigiani G, Boca S, Lin J, Kinzler KW, Velculescu V, Vogelstein B. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics. 2009;93:17–21. doi: 10.1016/j.ygeno.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- [26].Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, Reid BJ. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- [27].Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, Follows GA, Green AR, Futreal PA, Stratton MR. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benetkiewicz M, Piotrowski A, Diaz De Stahl T, Jankowski M, Bala D, Hoffman J, Srutek E, Laskowski R, Zegarski W, Dumanski JP. Chromosome 22 array-CGH profiling of breast cancer delimited minimal common regions of genomic imbalances and revealed frequent intra-tumoral genetic heterogeneity. Int J Oncol. 2006;29:935–945. [PubMed] [Google Scholar]
- [30].Fujii H, Marsh C, Cairns P, Sidransky D, Gabrielson E. Genetic divergence in the clonal evolution of breast cancer. Cancer Res. 1996;56:1493–1497. [PubMed] [Google Scholar]
- [31].Glockner S, Buurman H, Kleeberger W, Lehmann U, Kreipe H. Marked intratumoral heterogeneity of c-myc and cyclinD1 but not of c-erbB2 amplification in breast cancer. Lab Invest. 2002;82:1419–1426. doi: 10.1097/01.lab.0000032371.16521.40. [DOI] [PubMed] [Google Scholar]
- [32].Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- [33].Teixeira MR, Pandis N, Bardi G, Andersen JA, Heim S. Karyotypic comparisons of multiple tumorous and macroscopically normal surrounding tissue samples from patients with breast cancer. Cancer Res. 1996;56:855–859. [PubMed] [Google Scholar]
- [34].Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat. 2007;102:143–155. doi: 10.1007/s10549-006-9317-6. [DOI] [PubMed] [Google Scholar]
- [35].Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998;58:23–28. [PubMed] [Google Scholar]
- [36].Alvarado C, Beitel LK, Sircar K, Aprikian A, Trifiro M, Gottlieb B. Somatic mosaicism and cancer: a micro-genetic examination into the role of the androgen receptor gene in prostate cancer. Cancer Res. 2005;65:8514–8518. doi: 10.1158/0008-5472.CAN-05-0399. [DOI] [PubMed] [Google Scholar]
- [37].Konishi N, Hiasa Y, Matsuda H, Tao M, Tsuzuki T, Hayashi I, Kitahori Y, Shiraishi T, Yatani R, Shimazaki J, et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol. 1995;147:1112–1122. [PMC free article] [PubMed] [Google Scholar]
- [38].Gonzalez-Garcia I, Sole RV, Costa J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci U S A. 2002;99:13085–13089. doi: 10.1073/pnas.202139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Samowitz WS, Slattery ML. Regional reproducibility of microsatellite instability in sporadic colorectal cancer. Genes Chromosomes Cancer. 1999;26:106–114. [PubMed] [Google Scholar]
- [40].Giaretti W, Monaco R, Pujic N, Rapallo A, Nigro S, Geido E. Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: association with degree of DNA aneuploidy. Am J Pathol. 1996;149:237–245. [PMC free article] [PubMed] [Google Scholar]
- [41].Coons SW, Johnson PC, Shapiro JR. Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade human glioma. Cancer Res. 1995;55:1569–1577. [PubMed] [Google Scholar]
- [42].Mora J, Cheung NK, Gerald WL. Genetic heterogeneity and clonal evolution in neuroblastoma. Br J Cancer. 2001;85:182–189. doi: 10.1054/bjoc.2001.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- [44].Sauter G, Moch H, Gasser TC, Mihatsch MJ, Waldman FM. Heterogeneity of chromosome 17 and erbB-2 gene copy number in primary and metastatic bladder cancer. Cytometry. 1995;21:40–46. doi: 10.1002/cyto.990210109. [DOI] [PubMed] [Google Scholar]
- [45].Fujii H, Yoshida M, Gong ZX, Matsumoto T, Hamano Y, Fukunaga M, Hruban RH, Gabrielson E, Shirai T. Frequent genetic heterogeneity in the clonal evolution of gynecological carcinosarcoma and its influence on phenotypic diversity. Cancer Res. 2000;60:114–120. [PubMed] [Google Scholar]
- [46].Horvai AE, Devries S, Roy R, O'Donnell RJ, Waldman F. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol. 2009 doi: 10.1038/modpathol.2009.119. [DOI] [PubMed] [Google Scholar]
- [47].Pantou D, Rizou H, Tsarouha H, Pouli A, Papanastasiou K, Stamatellou M, Trangas T, Pandis N, Bardi G. Cytogenetic manifestations of multiple myeloma heterogeneity. Genes Chromosomes Cancer. 2005;42:44–57. doi: 10.1002/gcc.20114. [DOI] [PubMed] [Google Scholar]
- [48].Tomlinson IP, Lambros MB, Roylance RR. Loss of heterozygosity analysis: practically and conceptually flawed? Genes Chromosomes Cancer. 2002;34:349–353. doi: 10.1002/gcc.10085. [DOI] [PubMed] [Google Scholar]
- [49].Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- [52].Siegmund KD, Marjoram P, Woo YJ, Tavare S, Shibata D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natl Acad Sci U S A. 2009;106:4828–4833. doi: 10.1073/pnas.0810276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Janeway CA. In: Immunobiology : the immune system in health and disease. 6th ed ed. Janeway Charles A., Jr., et al., editors. [Google Scholar]
- [54].van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, van Krieken JH, Droese J, Gonzalez D, Bastard C, White HE, Spaargaren M, Gonzalez M, Parreira A, Smith JL, Morgan GJ, Kneba M, Macintyre EA. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- [55].Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Panzer-Grumayer ER, Cazzaniga G, van der Velden VH, del Giudice L, Peham M, Mann G, Eckert C, Schrauder A, Germano G, Harbott J, Basso G, Biondi A, van Dongen JJ, Gadner H, Haas OA. Immunogenotype changes prevail in relapses of young children with TEL-AML1-positive acute lymphoblastic leukemia and derive mainly from clonal selection. Clin Cancer Res. 2005;11:7720–7727. doi: 10.1158/1078-0432.CCR-05-1239. [DOI] [PubMed] [Google Scholar]
- [57].Peham M, Konrad M, Harbott J, Konig M, Haas OA, Panzer-Grumayer ER. Clonal variation of the immunogenotype in relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia indicates subclone formation during early stages of leukemia development. Genes Chromosomes Cancer. 2004;39:156–160. doi: 10.1002/gcc.10305. [DOI] [PubMed] [Google Scholar]
- [58].Zuna J, Ford AM, Peham M, Patel N, Saha V, Eckert C, Kochling J, Panzer-Grumayer R, Trka J, Greaves M. TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2004;10:5355–5360. doi: 10.1158/1078-0432.CCR-04-0584. [DOI] [PubMed] [Google Scholar]
- [59].Young RM, Turner BC, Refaeli Y. B-cell receptor signaling in the genesis and maintenance of B-cell lymphoma. Future Oncol. 2008;4:591–594. doi: 10.2217/14796694.4.5.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pandis N, Jin Y, Gorunova L, Petersson C, Bardi G, Idvall I, Johansson B, Ingvar C, Mandahl N, Mitelman F, et al. Chromosome analysis of 97 primary breast carcinomas: identification of eight karyotypic subgroups. Genes Chromosomes Cancer. 1995;12:173–185. doi: 10.1002/gcc.2870120304. [DOI] [PubMed] [Google Scholar]
- [61].Teixeira MR, Pandis N, Bardi G, Andersen JA, Mitelman F, Heim S. Clonal heterogeneity in breast cancer: karyotypic comparisons of multiple intra- and extra-tumorous samples from 3 patients. Int J Cancer. 1995;63:63–68. doi: 10.1002/ijc.2910630113. [DOI] [PubMed] [Google Scholar]
- [62].Kallioniemi OP. Comparison of fresh and paraffin-embedded tissue as starting material for DNA flow cytometry and evaluation of intratumor heterogeneity. Cytometry. 1988;9:164–169. doi: 10.1002/cyto.990090211. [DOI] [PubMed] [Google Scholar]
- [63].Klein CA, Schmidt-Kittler O, Schardt JA, Pantel K, Speicher MR, Riethmuller G. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci U S A. 1999;96:4494–4499. doi: 10.1073/pnas.96.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nakamura T, Kuwai T, Kitadai Y, Sasaki T, Fan D, Coombes KR, Kim SJ, Fidler IJ. Zonal heterogeneity for gene expression in human pancreatic carcinoma. Cancer Res. 2007;67:7597–7604. doi: 10.1158/0008-5472.CAN-07-0874. [DOI] [PubMed] [Google Scholar]
- [65].Leith JT, Michelson S, Faulkner LE, Bliven SF. Growth properties of artificial heterogeneous human colon tumors. Cancer Res. 1987;47:1045–1051. [PubMed] [Google Scholar]
- [66].Hughes S, Lasken R. Whole genome amplification. Scion; Bloxham: 2005. [Google Scholar]
- [67].Lage JM, Leamon JH, Pejovic T, Hamann S, Lacey M, Dillon D, Segraves R, Vossbrinck B, Gonzalez A, Pinkel D, Albertson DG, Costa J, Lizardi PM. Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res. 2003;13:294–307. doi: 10.1101/gr.377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Fialkow PJ. Clonal origin of human tumors. Annu Rev Med. 1979;30:135–143. doi: 10.1146/annurev.me.30.020179.001031. [DOI] [PubMed] [Google Scholar]
- [69].Weinberg RA. In: The biology of cancer. Weinberg Robert A., editor. [Google Scholar]
- [70].Novelli MR, Williamson JA, Tomlinson IP, Elia G, Hodgson SV, Talbot IC, Bodmer WF, Wright NA. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science. 1996;272:1187–1190. doi: 10.1126/science.272.5265.1187. [DOI] [PubMed] [Google Scholar]
- [71].Leroi AM, Koufopanou V, Burt A. Cancer selection. Nat Rev Cancer. 2003;3:226–231. doi: 10.1038/nrc1016. [DOI] [PubMed] [Google Scholar]
- [72].Gatenby RA, Vincent TL. Application of quantitative models from population biology and evolutionary game theory to tumor therapeutic strategies. Mol Cancer Ther. 2003;2:919–927. [PubMed] [Google Scholar]
- [73].Sieber OM, Tomlinson SR, Tomlinson IP. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer. 2005;5:649–655. doi: 10.1038/nrc1674. [DOI] [PubMed] [Google Scholar]
- [74].Kimura M. In: The neutral theory of molecular evolution. Kimura Motoo., editor. [DOI] [PubMed] [Google Scholar]
- [75].Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- [76].Nowak MA. In: Evolutionary dynamics : exploring the equations of life. Nowak Martin A., editor. 2006. [Google Scholar]
- [77].Priyanga A. Competitive coexistence in spatially structured environments: a synthesis. Ecology Letters. 2003;6:1109–1122. [Google Scholar]
- [78].Chesson P. MECHANISMS OF MAINTENANCE OF SPECIES DIVERSITY. Annual Review of Ecology and Systematics. 2000;31:343–366. [Google Scholar]
- [79].Rainey PB, Travisano M. Adaptive radiation in a heterogeneous environment. Nature. 1998;394:69–72. doi: 10.1038/27900. [DOI] [PubMed] [Google Scholar]
- [80].Korona R, Nakatsu CH, Forney LJ, Lenski RE. Evidence for multiple adaptive peaks from populations of bacteria evolving in a structured habitat. Proc Natl Acad Sci U S A. 1994;91:9037–9041. doi: 10.1073/pnas.91.19.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gatenby RA, Vincent TL. An evolutionary model of carcinogenesis. Cancer Res. 2003;63:6212–6220. [PubMed] [Google Scholar]
- [82].Greaves M. Cancer: The Evolutionary Legacy. 1 edition ed. Oxford University Press; 2002. [Google Scholar]
- [83].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- [84].Bissig H, Richter J, Desper R, Meier V, Schraml P, Schaffer AA, Sauter G, Mihatsch MJ, Moch H. Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am J Pathol. 1999;155:267–274. doi: 10.1016/S0002-9440(10)65120-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cheng L, Bostwick DG, Li G, Wang Q, Hu N, Vortmeyer AO, Zhuang Z. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer. 1999;85:2017–2022. doi: 10.1002/(sici)1097-0142(19990501)85:9<2017::aid-cncr20>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- [86].Kuukasjarvi T, Karhu R, Tanner M, Kahkonen M, Schaffer A, Nupponen N, Pennanen S, Kallioniemi A, Kallioniemi OP, Isola J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–1604. [PubMed] [Google Scholar]
- [87].Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- [88].Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A. 2003;100:15901–15905. doi: 10.1073/pnas.2634067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, Glas AM, Perou CM, Van't Veer LJ. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005;65:9155–9158. doi: 10.1158/0008-5472.CAN-05-2553. [DOI] [PubMed] [Google Scholar]
- [90].Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, Chen L, Ewing CM, Eisenberger MA, Carducci MA, Nelson WG, Yegnasubramanian S, Luo J, Wang Y, Xu J, Isaacs WB, Visakorpi T, Bova GS. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ruijter ET, van de Kaa CA, Schalken JA, Debruyne FM, Ruiter DJ. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol. 1996;180:295–299. doi: 10.1002/(SICI)1096-9896(199611)180:3<295::AID-PATH663>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [92].Aihara M, Wheeler TM, Ohori M, Scardino PT. Heterogeneity of prostate cancer in radical prostatectomy specimens. Urology. 1994;43:60–66. doi: 10.1016/s0090-4295(94)80264-5. discussion 66–67. [DOI] [PubMed] [Google Scholar]
- [93].Schlimok G, Funke I, Holzmann B, Gottlinger G, Schmidt G, Hauser H, Swierkot S, Warnecke HH, Schneider B, Koprowski H, et al. Micrometastatic cancer cells in bone marrow: in vitro detection with anti-cytokeratin and in vivo labeling with anti-17-1A monoclonal antibodies. Proc Natl Acad Sci U S A. 1987;84:8672–8676. doi: 10.1073/pnas.84.23.8672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Passlick B, Izbicki JR, Kubuschok B, Nathrath W, Thetter O, Pichlmeier U, Schweiberer L, Riethmuller G, Pantel K. Immunohistochemical assessment of individual tumor cells in lymph nodes of patients with non-small-cell lung cancer. J Clin Oncol. 1994;12:1827–1832. doi: 10.1200/JCO.1994.12.9.1827. [DOI] [PubMed] [Google Scholar]
- [95].Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- [96].Klein CA, Blankenstein TJ, Schmidt-Kittler O, Petronio M, Polzer B, Stoecklein NH, Riethmuller G. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002;360:683–689. doi: 10.1016/S0140-6736(02)09838-0. [DOI] [PubMed] [Google Scholar]
- [97].Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, Riethmuller G, Eils R, Klein CA. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bedenne L, Michel P, Bouche O, Milan C, Mariette C, Conroy T, Pezet D, Roullet B, Seitz JF, Herr JP, Paillot B, Arveux P, Bonnetain F, Binquet C. Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol. 2007;25:1160–1168. doi: 10.1200/JCO.2005.04.7118. [DOI] [PubMed] [Google Scholar]
- [99].Stoecklein NH, Hosch SB, Bezler M, Stern F, Hartmann CH, Vay C, Siegmund A, Scheunemann P, Schurr P, Knoefel WT, Verde PE, Reichelt U, Erbersdobler A, Grau R, Ullrich A, Izbicki JR, Klein CA. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell. 2008;13:441–453. doi: 10.1016/j.ccr.2008.04.005. [DOI] [PubMed] [Google Scholar]
- [100].Tutt A, Ashworth A. Close relatives or distant cousins? Lancet Oncol. 2003;4:593–594. doi: 10.1016/s1470-2045(03)01217-8. [DOI] [PubMed] [Google Scholar]
- [101].Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and Propagation of Untransformed Mouse Mammary Cells in the Lung. Science. 2008 doi: 10.1126/science.1161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Gadi VK, Nelson JL. Fetal microchimerism in women with breast cancer. Cancer Res. 2007;67:9035–9038. doi: 10.1158/0008-5472.CAN-06-4209. [DOI] [PubMed] [Google Scholar]
- [103].Solakoglu O, Maierhofer C, Lahr G, Breit E, Scheunemann P, Heumos I, Pichlmeier U, Schlimok G, Oberneder R, Kollermann MW, Kollermann J, Speicher MR, Pantel K. Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc Natl Acad Sci U S A. 2002;99:2246–2251. doi: 10.1073/pnas.042372199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gangnus R, Langer S, Breit E, Pantel K, Speicher MR. Genomic profiling of viable and proliferative micrometastatic cells from early-stage breast cancer patients. Clin Cancer Res. 2004;10:3457–3464. doi: 10.1158/1078-0432.CCR-03-0818. [DOI] [PubMed] [Google Scholar]
- [105].Abbruzzese JL, Abbruzzese MC, Hess KR, Raber MN, Lenzi R, Frost P. Unknown primary carcinoma: natural history and prognostic factors in 657 consecutive patients. J Clin Oncol. 1994;12:1272–1280. doi: 10.1200/JCO.1994.12.6.1272. [DOI] [PubMed] [Google Scholar]
- [106].Klein CA, Holzel D. Systemic cancer progression and tumor dormancy: mathematical models meet single cell genomics. Cell Cycle. 2006;5:1788–1798. doi: 10.4161/cc.5.16.3097. [DOI] [PubMed] [Google Scholar]
- [107].Arteaga CL, Baselga J. Tyrosine kinase inhibitors: why does the current process of clinical development not apply to them? Cancer Cell. 2004;5:525–531. doi: 10.1016/j.ccr.2004.05.028. [DOI] [PubMed] [Google Scholar]
- [108].Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- [109].Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat Rev Cancer. 2004;4:197–205. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- [110].Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- [111].Hofmann WK, Komor M, Wassmann B, Jones LC, Gschaidmeier H, Hoelzer D, Koeffler HP, Ottmann OG. Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood. 2003;102:659–661. doi: 10.1182/blood-2002-06-1756. [DOI] [PubMed] [Google Scholar]
- [112].Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, Fenaux P, Preudhomme C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- [113].Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- [114].Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. 2008;3:557–586. doi: 10.1146/annurev.pathmechdis.3.121806.151538. [DOI] [PubMed] [Google Scholar]
- [115].Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- [116].Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Pierga JY, Reis-Filho JS, Cleator SJ, Dexter T, Mackay A, Simpson P, Fenwick K, Iravani M, Salter J, Hills M, Jones C, Ashworth A, Smith IE, Powles T, Dowsett M. Microarray-based comparative genomic hybridisation of breast cancer patients receiving neoadjuvant chemotherapy. Br J Cancer. 2007;96:341–351. doi: 10.1038/sj.bjc.6603483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Blagosklonny MV. Oncogenic resistance to growth-limiting conditions. Nat Rev Cancer. 2002;2:221–225. doi: 10.1038/nrc743. [DOI] [PubMed] [Google Scholar]
- [119].Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008;18:322–329. doi: 10.1016/j.semcancer.2008.03.019. [DOI] [PubMed] [Google Scholar]
- [120].Thilly WG. Have environmental mutagens caused oncomutations in people? Nat Genet. 2003;34:255–259. doi: 10.1038/ng1205. [DOI] [PubMed] [Google Scholar]
- [121].Haddow A. Cellular inhibition and the origin of cancer. Acta Unio Int Contra Cancrum. 1938:342–352. [Google Scholar]
- [122].Marusyk A, Casás-Selves M, Henry CJ, Zaberezhnyy V, Klawitter J, Christians U, DeGregori J. Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-0604. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Crespi B, Summers K. Evolutionary biology of cancer. Trends Ecol Evol. 2005;20:545–552. doi: 10.1016/j.tree.2005.07.007. [DOI] [PubMed] [Google Scholar]
- [124].Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008;1:158–164. doi: 10.1593/tlo.08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Heppner GH. Cancer cell societies and tumor progression. Stem Cells. 1993;11:199–203. doi: 10.1002/stem.5530110306. [DOI] [PubMed] [Google Scholar]
- [126].Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- [128].Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, Wu H, Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- [130].Marusyk A, DeGregori J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim Biophys Acta. 2008;1785:1–11. doi: 10.1016/j.bbcan.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bilousova G, Marusyk A, Porter CC, Cardiff RD, DeGregori J. Impaired DNA replication within progenitor cell pools promotes leukemogenesis. PLoS Biol. 2005;3:e401. doi: 10.1371/journal.pbio.0030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Laconi S, Pani P, Pillai S, Pasciu D, Sarma DS, Laconi E. A growth-constrained environment drives tumor progression invivo. Proc Natl Acad Sci U S A. 2001;98:7806–7811. doi: 10.1073/pnas.131210498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lensch MW, Rathbun RK, Olson SB, Jones GR, Bagby GC., Jr. Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: a view from the window of Fanconi anemia. Leukemia. 1999;13:1784–1789. doi: 10.1038/sj.leu.2401586. [DOI] [PubMed] [Google Scholar]
- [134].Begon M, Harper JL, Townsnd CR. Ecology. 3rd edition ed. Blackwell Science; 1998. [Google Scholar]
- [135].Nagy JD. Competition and natural selection in a mathematical model of cancer. Bull Math Biol. 2004;66:663–687. doi: 10.1016/j.bulm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- [136].Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- [137].Miller BE, Miller FR, Wilburn DJ, Heppner GH. Analysis of tumour cell composition in tumours composed of paired mixtures of mammary tumour cell lines. Br J Cancer. 1987;56:561–569. doi: 10.1038/bjc.1987.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Miller BE, Miller FR, Wilburn D, Heppner GH. Dominance of a tumor subpopulation line in mixed heterogeneous mouse mammary tumors. Cancer Res. 1988;48:5747–5753. [PubMed] [Google Scholar]
- [139].Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- [140].O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- [141].Guba M, Cernaianu G, Koehl G, Geissler EK, Jauch KW, Anthuber M, Falk W, Steinbauer M. A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res. 2001;61:5575–5579. [PubMed] [Google Scholar]
- [142].Czaran TL, Hoekstra RF, Pagie L. Chemical warfare between microbes promotes biodiversity. Proc Natl Acad Sci U S A. 2002;99:786–790. doi: 10.1073/pnas.012399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Tomlinson IP. Game-theory models of interactions between tumour cells. Eur J Cancer. 1997;33:1495–1500. doi: 10.1016/s0959-8049(97)00170-6. [DOI] [PubMed] [Google Scholar]
- [144].Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, Reinhardt F, Harris LN, Hylander BL, Repasky EA, Weinberg RA. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Tomlinson IP, Bodmer WF. Modelling the consequences of interactions between tumour cells. Br J Cancer. 1997;75:157–160. doi: 10.1038/bjc.1997.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Yamamoto K, Hanada R, Kikuchi A, Ichikawa M, Aihara T, Oguma E, Moritani T, Shimanuki Y, Tanimura M, Hayashi Y. Spontaneous regression of localized neuroblastoma detected by mass screening. J Clin Oncol. 1998;16:1265–1269. doi: 10.1200/JCO.1998.16.4.1265. [DOI] [PubMed] [Google Scholar]
- [148].Gross K. Positive interactions among competitors can produce species-rich communities. Ecol Lett. 2008;11:929–936. doi: 10.1111/j.1461-0248.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- [149].Axelrod R, Hamilton WD. The evolution of cooperation. Science. 1981;211:1390–1396. doi: 10.1126/science.7466396. [DOI] [PubMed] [Google Scholar]
- [150].Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A. 2006;103:13474–13479. doi: 10.1073/pnas.0606053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lyons JG, Lobo E, Martorana AM, Myerscough MR. Clonal diversity in carcinomas: its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin Exp Metastasis. 2008;25:665–677. doi: 10.1007/s10585-007-9134-2. [DOI] [PubMed] [Google Scholar]
- [152].Maley CC, Reid BJ, Forrest S. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev. 2004;13:1375–1384. [PubMed] [Google Scholar]
- [153].Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69:4894–4903. doi: 10.1158/0008-5472.CAN-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]