

Abstract
Purpose
Transforming growth factor β(TGFβ) is an important cytokine in corneal development and wound healing. Transgenic mice that express an active form of human TGF β1 driven by a lens-specific promoter were used in the current study to determine the biological effects of lens-derived TGFβ1 on postnatal corneal development and homeostasis.
Methods
The postnatal corneal changes in the TGFβ1 transgenic mice were examined by fluorescein labeling and histology. Epithelial/endothelial-to-mesenchymal transition (E/EnMT) in the transgenic mouse cornea was demonstrated by immunostaining for α-smooth muscle actin (α-SMA) and cadherin-11. Expression of E- and N-cadherin in the corneal epithelial and endothelial cells, respectively, was analyzed by in situ hybridization.
Results
Among the established TGF β1 transgenic lines, mice from line OVE853 and OVE917 had normal-sized eyeballs but developed a corneal haze after eyelid opening. Histological examination showed that prenatal corneal development appeared to be normal. However, after postnatal day 7 (P7), the corneal endothelial cells in transgenic line OVE853 began to lose normal cell-cell contact and basement membrane structure. The endothelial layer was eventually absent in the inner surface of the transgenic mouse cornea. The morphological changes in the cornea correlated with abnormal expression of α-SMA, a molecular marker of EMT, and stress fiber formation in myofibroblast-like cells, which initially appeared in the corneal endothelial layer and subsequently in the corneal epithelial and stromal layers. The E/EnMT in the transgenic mouse cornea was further demonstrated by loss of E- and N-cadherin expression in the corneal epithelial and endothelial cells respectively, and meanwhile increasing expression of cadherin-11 in both corneal epithelium and stroma.
Conclusions
Elevated levels of active TGF β1 in the anterior chamber can lead to myofibroblast formation in the corneal endothelial layer and subsequently in the corneal epithelial and stromal layers. Our data suggest that the levels of biologically active TGFβ in the aqueous humor must be under tight control to maintain corneal homeostasis. TGF β1 is the major cytokine during wound healing. Therefore, our findings also suggest a potential mechanism to explain the loss of corneal endothelial barrier and corneal opacification after intraocular surgery or trauma.
1. Introduction
Transforming growth factor β (TGF β) is a multifunctional cytokine that controls a diverse set of cell processes including production of extracellular matrix (ECM), cell proliferation, migration, differentiation, and apoptosis [1; 2; 3; 4; 5]. TGFβ has emerged as one of the most important ligands involved in tissue development, homeostasis and wound healing in the eye [6; 7; 8; 9]. Three mammalian isoforms of TGFβ (TGFβ1, TGFβ2 and TGFβ3) have been identified [10]. While all are expressed in the eye, TGFβ2 is expressed a much higher level than TGFβ1 or TGFβ3 [11; 12; 13; 14; 15]. TGFβ2 knockout mice exhibit multiple defects in anterior segment structures, including thinning of the corneal stroma and an absence of the corneal endothelial layer and anterior chamber [6; 16; 17]. Additional ocular defects in the TGFβ2-null mouse embryos include immature retina and vitreous hypercellularity [16; 18]. The abnormalities found in the eye of the TGFβ2-null mouse are mostly due to the impaired immigration of neural crest cells [6]. Deletion of TGFβ1 and β3 did not cause any visible abnormalities in the cornea or in the eye [19; 20; 21; 22].
TGFβ also plays an important role in corneal maintenance and wound healing [23; 24; 25]. In contrast to the developmental role of TGFβ2 in the eye, TGFβ1 is believed to be the principle isoform during corneal wound healing [25]. In the normal cornea, TGFβ1 and TGFβ2 localize to both corneal epithelium and stroma, and both are present in the tear fluid [26; 27; 28]. Additionally, TGFβ3 is present at a very low level in the cornea [29]. Interestingly, after a corneal wound, the levels and spatial locations of the TGFβ isoforms are altered in the cornea [30]. For instance, TGFβ1 increases dramatically in the tear fluid [31]. Previous studies suggested that increase of TGFβ1 in the cornea may induce scar formation on the ocular surface [32; 33], a process that is also seen in other internal organs such as liver and kidney [34; 35; 36]. As a result of stromal fibrosis and scaring, the transparency of the cornea is reduced, ultimately leading to the impairment of the patients’ vision.
TGFβ signals are relayed through type I and II serine/threonine kinase receptors (TGFβRI and TGFβRII) [37; 38]. Among the three TGFβ isoforms, TGFβ1 and TGFβ3 can independently bind to TGFβRII and then recruit TGFβRI to form an RI-RII-ligand complex. TGFβRII must phosphorylate TGFβRI to transfer the signal to the cell interior. TGFβ2 binds to a type III receptor (TGFβRIII), also known as β-glycan, which then presents TGFβ2 to TGFβRII and phosphorylates TGFβRI [39]. The phosphorylated (active) TGFβRI transduces the signal by phosphorylating the carboxyl terminal serines of the receptor-regulated Smad (R-Smad) proteins, Smad2 and Smad3 [40]. The activated R-Smads form a complex with the common Smad (co-Smad), Smad4. The R-Smad/Co-Smad complex translocates into the nucleus and regulates target gene transcription in conjunction with other nuclear factors. TGFβ receptor activity can be negatively regulated by two inhibitory Smads (I-Smads), Smad6 and Smad7, which compete with R-Smads, to attenuate the receptor signaling activity.
TGFβRI and TGFβRII are located in epithelial, stromal and endothelial layers of the cornea [41; 42; 43]. The non-signaling type III receptor (β-glycan receptor) has been located on both the epithelium and endothelium in vivo, but appears to be absent in keratocytes in vivo. The presence of all three types of receptors in the epithelial and endothelial layers suggests that these corneal cells can respond to any of the three TGFβ isoforms.
Transgenic mice that express a self-activating human TGFβ1 in the ocular lens were generated several years ago to test the biological effects of elevated levels of TGFβ1 on ocular tissue morphogenesis and homeostasis [18; 44]. The pathological consequences of TGFβ1 overexpression on the lens and ocular vascular development have been examined in detail and reported previously [18; 44]. In some of these studies, corneal and anterior segment defects were documented but not investigated in detail [44]. Among the three established transgenic lines, OVE920A had the highest expression level for the transgene, whereas OVE853 and OVE917 had relatively lower expression levels [18]. The developmental abnormalities in the eyes of the OVE920A mice were similar to transgenic mice in which TGFβ1 overexpression was driven by the stronger lens-promoter: chicken βB1-crystallin promoter [45]. In these high expression transgenic lines, differentiation of the corneal mesenchymal cells was disrupted, leading to severe defects in the anterior segment structures. In contrast to OVE920A or βB1-crystallin-TGFβ1 transgenic mice, the development of the cornea and anterior segment in OVE853 and OVE917 transgenic lines appeared to be normal during the prenatal period, but corneal defects were found postnatally [44].
In this study, we show that active TGFβ1 released from the lens of the transgenic mouse has a negative effect on corneal integrity. Data are consistent with corneal cells undergoing classical TGFβ1-induced epithelial-to-mesenchymal transition (EMT), including 1) detachment of the corneal endothelial cells from the basement membrane, 2) induction of α-smooth muscle actin (αSMA) expression and myofibroblast formation in the corneal endothelial and epithelial layers, and later in the corneal stroma, and 3) down-regulation of E- and N-cadherin expression in the corneal epithelial and endothelial cells respectively, and increased expression of cadherin-11, a mesenchymal cell cadherin. Our results suggest that activation of TGFβ1 in the aqueous humor of the postnatal mouse eye can disrupt the maintenance of corneal tissue homeostasis. The findings from this transgenic mouse model may also have clinical implications since increased activation of TGFβ1 after injury, intraocular surgery or in diabetes may potentially lead to pathological changes in the cornea.
2. Materials and Methods
2.1. Transgenic mice
Generation of transgenic mice has been described previously [44]. Briefly, the mouse αA-crystallin promoter was used to drive expression of a secreted and constitutively active form of human TGFβ1 in the lens. All the animals were used in accordance to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and all experimental procedures were approved by the Animal Care and Use Committee at the University of Missouri (MU) and University of Oklahoma Health Science Center (OUHSC).
2.2. Fluorescein labeling of the cornea surface
To evaluate the smoothness of the cornea surface, 3 mice at postnatal day 16 (P16) were anesthetized with ketamine and xylazine, and fluorescein eye drops (Fluress, Akorn Inc.) were applied to the cornea surface. The mouse eyes were examined under blue light using a fluorescent stereomicroscope (SZX12, Olympus).
2.3. Histology
Mouse eyes were fixed in 10% formalin overnight at room temperature, rinsed twice with phosphate buffered saline (PBS), dehydrated by emersion in an ethanol series, cleared with xylene, and embedded in paraffin. Sections were cut at 5 µm and stained with hematoxylin and eosin (H&E). To label the cell nuclei, sections were incubated with DAPI for 5 min and then rinsed twice with PBS before analysis by fluorescence microscopy. Periodic Acid Schiff (PAS) reaction was used to reveal the basement membrane structures in mouse cornea.
2.4. Immunohistochemistry and immunofluorescence
For immunohistochemical staining of TGFβ receptors in mouse cornea, tissue sections were pretreated with 10% methanol and 3% H2O2 in PBS to block endogenous peroxidase. Antigen retrieval was done by heating the slides to 100°C in 10 mM citrate buffer (pH 6.0) for 10 min using a microwave oven. After blocking with 5% normal serum in PBS, sections were incubated with primary antibody overnight at 4°C. The anti-TGFβRI (sc-398) and anti-TGFβRII (sc-220) primary antibodies were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). After washing, the sections were incubated with biotinylated secondary antibody (Vector Laboratory Inc. Burlingame, CA) for 1 h at room temperature. Immune complexes were visualized by using the avidin-biotin peroxidase complex method with diaminobenzidine as the substrate [46]. Sections were counterstained with hematoxylin. For Cadherin-11 immunofluorescence, after blocking, tissue sections were labeled with anti-cadherin-11 antibody (Cat#71-7600, Invitrogen, Carlsbad, CA) overnight at 4°C, and then with Alexa Fluor 568 anti-rabbit secondary antibody for 1 hour at room temperature. Images were captured under Leica fluorescence microscope.
2.5. In situ hybridization
In situ hybridization was performed as described previously [47]. Tissue sections were incubated with radioactive 35S-labeled riboprobes specific for E- and N-cadherin overnight at 50°C. After stringent washing, sections were coated with Kodak NTB-2 emulsion and exposed for 1 week at 4°C before developing. Sections were counterstained with hematoxylin.
2.6. Whole-mount immunofluorescence staining
For whole mount immunofluorescence staining of corneas, mice were killed by CO2 asphyxiation and the eyes were removed and fixed overnight in 4% paraformaldehyde. The following day, corneas were dissected and rinsed three times in PBS. Corneas were then incubated in PBS containing 10% horse serum and 1% Triton for 2 hours to permeabilize tissues and block non-specific binding. Corneas were then incubated with FITC or Cy3 conjugated anti-alpha smooth muscle actin (monoclonal antibody, Sigma-Aldrich) diluted 1:200 in PBS containing 10% horse serum and 1% triton. Following a 16 hour incubation at 4°C corneas were washed three times in PBS with 1% triton, then flat mounted unto glass slides and coverslipped in PBS containing 10% glycerol. Stained corneas were imaged on an Olympus Flowview confocal microscope. Optical secions were obtained using identical detector settings. Maximum projections of image stacks were obtained to demonstrate flat perspective, or rotated in the Z-plane to produce Z-rotated images. Whole corneas were also imaged using epi-fluorescence illumination on a Nikon E800 microscope.
3. Results
3.1. Lens-specific expression of biologically active TGFβ1 causes corneal defects
Among the three previously reported TGFβ1 transgenic mouse lines [18; 44], mouse from line OVE853 and OVE917 share a similar corneal phenotype, this study focused on line OVE853. When OVE853 transgenic mice opened their eyes by P14, the cornea had already lost its transparency and developed haze (Fig. 1A). The defect of the corneal surface in these transgenic mice was also demonstrated by fluorescein binding. Exposure of wild-type corneas to fluorescein dye revealed a smooth surface (Fig. 1B), whereas the TGFβ1 transgenic corneas retained strong fluorescent staining, indicating the roughness of the corneal surface and defects in the epithelial layer (Fig. 1C).
Figure 1.

Corneal defects in OVE853 TGFβ1 transgenic mice. (A) Eyes from P14 OVE853 transgenic (Tg) and wild-type (Wt) mice. Arrow points to the cloudy corneal surface of the transgenic eye. (B and C) Fluorescein staining of mouse corneas. The wild-type cornea (B) did not retain the fluorescent dye, whereas the transgenic cornea (C) was fluorescein positive (marked with an asterisk).
The progression of the corneal abnormalities in transgenic mice was examined by histology (Fig. 2 and Fig. 3). At P7 (Fig. 2A, B), the wild type cornea contained a single layer of endothelial cells that formed a barrier at the inner surface of the cornea. The corneal endothelial layer in the transgenic mice began to lose its normal cell contact and to detach from the inner surface. The stroma of the transgenic mouse cornea was thicker, probably resulting from the loss of barrier function of the endothelial layer. At P14, the inner surface of the transgenic mouse cornea lacked the distinctive corneal endothelial layer (Fig. 2D, F). Additionally, the corneal epithelial cells were poorly differentiated, for example, the stratified layers and the super basal layers of the corneal epithelium were not seen in transgenic cornea. The mouse corneas were stained with PAS to identify the basement membrane structures (Fig. 3). At P21, the Descemet’s membrane, synthesized by the corneal endothelial cells, can be seen in the wild type cornea (Fig. 3A, indicated by white arrows), but not in either the hemizygous (Fig. 3B) or homozygous (Fig. 3C) transgenic mouse corneas. Loss of corneal endothelial layer and Descemet’s membrane caused the lens and iris to adhere to the cornea in the transgenic eye (Fig.3C and data not shown).
Figure 2.

Corneal histology of wild-type and TGFβ1 transgenic mice. Wild-type (WT) and transgenic (TG) corneas were stained with H&E (A–D) and DAPI (E, F). WT corneas (A, C and E) had three distinctive cell layers at P7 and P14: the corneal epithelium (Epi), the stroma (Str) and the endothelium (End). At P7, the endothelial cells of the TG corneas began to detach from the inner surface (arrows in B). By P14, the endothelial layer was absent from the TG cornea (D and indicated by arrows in F). Additionally, the epithelial layer of the TG cornea was thinner and less differentiated (indicated by arrowheads in D) than the WT cornea (C). The stroma was thicker in the TG cornea, which is indicative of corneal edema. Scale bar represents 100 βm.
Figure 3.

PAS staining. In P21 WT cornea (A), the corneal endothelial cells synthesize a distinctive basement membrane structure, Descemet’s membrane, shown in red purple color (pointed by arrows). In either hemizygous (B) or homozygous (C) transgenic mouse cornea, the Descemet’s membrane was not apparent.
To determine whether corneal cells during the postnatal stage can directly respond to TGFβ1 stimulation, we examined the expression of TGFβRI and TGFβRII in wild-type corneas from P7 mice. Figure 4 shows that both TGFβRI and TGFβRII were expressed in the corneal epithelial, stromal and endothelial cells. The downstream signaling protein Smad 4 was also expressed in all the corneal layers (data not shown). Therefore, it is likely that the corneal phenotype of the TGFβ1 transgenic mice is a direct response to the active TGFβ1 secreted from the lens.
Figure 4.

Immunohistochemical staining for TGFβRI and TGFβRII in the wild-type mouse cornea at P7. Both TGFβRI (upper panel) and TGFβRII (lower panel) were expressed in the corneal epithelial (Epi), stroma (Str) and endothelial (End) cells, suggesting that all the corneal cells can directly respond to TGFβ1 stimulation. Scale bar represents 100 µm.
TGFβ-signaling is known to play a role in EMT [4; 48]. One hallmark feature of EMT is the expression of α-smooth muscle actin (α-SMA). At P16, α-SMA-positive cells were found in both corneal endothelial and epithelial layers (Fig. 5). Although most of the cells still retain their epithelial and endothelial cell morphology, a few cells at the center of the cornea in the transgenic eye experienced morphological changes, and transformed to myofibroblast like cells (Fig. 5B, C).
Figure 5.

Abnormal expression of α-SMA in TGFβ1 transgenic mouse cornea at postnatal day 16. Whole-mount immunofluorescent staining for α-SMA and images were analyzed by using confocal microscopy. α-SMA-expressing cells were present in both the corneal endothelial (A–C) and epithelial (D) layers of the TGFβ1 transgenic mouse. Cells with a myofibroblast shape and a greater level of α-SMA were found in the corneal endothelial layer (as indicated by the arrows in B and C).
To follow the progression of En/EMT in the TGFβ1 transgenic cornea, α-SMA expression was examined at different postnatal ages (Fig. 6). α-SMA expression in the corneal endothelial layer was first detected as early as P2 (Fig. 6B), and the expression level (as indicated by the number of α-SMA-positive cells) increased with age (Fig. 6C–F). At P16, the α-SMA-positive cells had a proto-myofibroblast phenotype but the stain was still prominent in the cytoplasm. Between P21 to P30, corneal endothelial cells had increased α-SMA staining and formed well-organized stress fibers, which are typical of a myofibroblast phenotype.
Figure 6.

Increase of αSMA expression along with the progression of myofibroblast formation in the postnatal TGFβ1 transgenic cornea. α-SMA expression was not detected in the transgenic cornea at postnatal day (P) 0 (A). A few α-SMA-positive cells were found at P2 (B) and the number of α–SMA-expressing cells increased significantly between P6 to P30 (C–F). The myofibroblast-like morphology became visible in the transgenic cornea at P16 (D). By P30 almost all of the αSMA-positive cells in the transgenic cornea were transformed into myofibroblast phenotypes including irregular shape, and the presence of stress fibers.
The distribution of α-SMA-positive cells in different corneal layers was monitored through the confocal x–z imaging stacking technique. α-SMA-expressing cells were initially identified in the corneal endothelial and epithelial layers, and subsequently in the corneal stroma (Fig. 7), suggesting that corneal endothelial and epithelial cells are more susceptible to TGFβ1-induced En/EMT than the keratocytes in the stroma.
Figure 7.

Distribution patterns of αSMA-expressing cells in the cornea of the TGFβ1 transgenic mice. Confocal x–z images of whole-mount-stained corneas shown at different postnatal ages. At P7 (A) and P16 (B), most of the αSMA-expressing cells were localized in the corneal epithelial and endothelial layers. By P30 (C), strong α-SMA staining was distributed in all corneal cell layers. Cornea edema along with the enhanced expression of α-SMA progressed in the transgenic corneas from P7–P30.
Additional features of EMT include changes in cadherin expression [49; 50]. Cadherins belong to a family of calcium-dependent adhesion molecules that have been shown to play critical roles in tissue morphogenesis and homeostasis [51; 52]. Previous studies have documented that E-cadherin is expressed in corneal epithelial cells whereas N-cadherin is expressed in corneal endothelial cells [47]. Therefore, E- and N-cadherin can serve as molecular markers for cornea epithelial and endothelial cells, respectively. In situ hybridization showed that at P0, the expression patterns of E- and N-cadherin in the cornea of the transgenic mice appeared similar to their expression patterns in wild-type mice, suggesting that prenatal development of these cell layers were not affected in the OVE853 mice (Fig. 8 A–D). However, by P7, the expression levels of E- and N-cadherin in the transgenic cornea were downregulated significantly, a finding consistent with the observation that En/EMT occurred in these two corneal cell layers during the postnatal development.
Figure 8.

Decrease in cadherin expression in the cornea of the TGFβ1 transgenic mouse. At P0 (A–D), E-cadherin was expressed in the corneal epithelial cells (arrowheads) and N-cadherin was in the corneal endothelial cells (arrows). E-cadherin expression was also detected in the epithelial cells of the eyelid and lens, and N-cadherin in the lens epithelial cells. Similar E- and N-cadherin expression patterns were found for the wild-type and the TGFβ1 transgenic mice, suggesting that prenatal development of the cornea and lens were not affected in the OVE853 transgenic mice. At P7 (E–H), E- and N-cadherin expression decreased in the corneal epithelial (arrowhead in F) and endothelial layers (arrows in H), respectively, in the TGFβ1 transgenic mouse, a feature consistent with EnMT and EMT in the cornea of the transgenic mouse. Scale bar represents 100 µm.
Cadherin-11 is a non-classical cadherin found in mesenchymal cells [53]. Its expression was detected in the corneal myofibroblasts in a cell culture model [54]. We compared the cadherin-11 expression patterns in the wild type and transgenic mouse corneas (Fig. 9). In the wild type eye, cadherin-11 was expressed at a low level in the corneal epithelial layer and in the cells associated with the vasculature systems (e.g. the blood vessel in the iris in Fig. 9A). In the transgenic eye, cadherin-11 expression was increased in both corneal epithelial and stromal layers, and also in a group of lens epithelial cells that detached from the lens capsule (arrow in Fig. 9B). Increase of cadherin-11 expression, along with αSMA expression, confirmed the myofibroblast phenotype in the cornea of TGFβ1 transgenic mouse.
Figure 9.
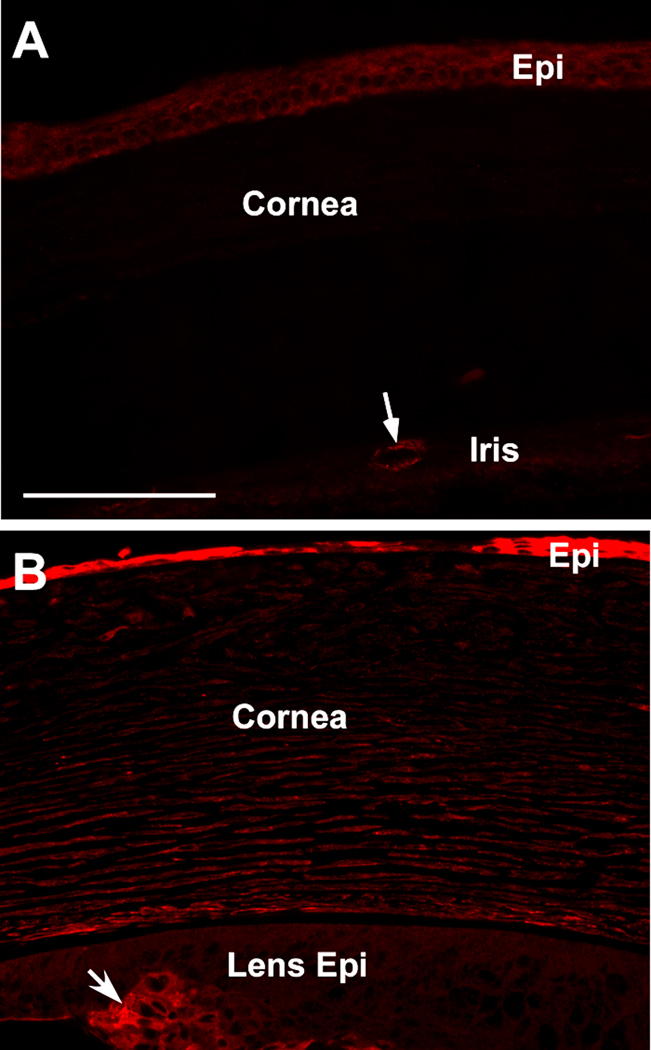
Cadherin-11 immunofluorescence in P21 mouse cornea. Cadherin-11 staining was found in the vescular tissue of the iris (arrow in A). A low level of cadherin-11 expression was also detected in the WT corneal epithelial layer (A). The intensity of cadherin-11 staining was increased in the transgenic mouse cornea, particularly in the corneal epithelial layer (B). On the same section, cadherin-11 was also shown in the group of lens epithelial cells (arrow in B) which were positive for αSMA and had transformed into myofibroblasts.
4. Discussion
Earlier studies have shown that TGFβ1 overexpression can alter the development patterns of the anterior segments and the ocular vasculature in the eye [18; 44; 45]. TGFβ1 overexpression can also induce EMT in the lens [44; 55; 56]. In this study, we found that the two transgenic lines OVE853 and OVE917 that have relatively lower levels of TGFβ1 transgene expression had normal prenatal development of the anterior segment (Figure 8). The postnatal corneal defects were investigated in detail in this study using line OVE853. Histological examination revealed that the corneal endothelial layer started to deteriorate by P7. Initially the endothelial cells lost both normal cell contact and adhesion to the basement membrane. By P14, the resulting edema suggested that the corneal endothelial barrier was lost in the eye of the transgenic mice. At P21, the distinctive basement membrane structure of the corneal endothelium, the Descemet’s membrane, was absent in the transgenic mouse cornea (Fig. 3B, C). Changes in the corneal endothelial layer in transgenic mice is consistent with endothelial-to-mesenchymal transformation (EnMT), as demonstrated by the expression of myofibroblast marker α-SMA, and loss of N-cadherin expression combined with the formation of stress fibers in the inner surface of the cornea. Following EnMT, the fate of endothelial cells is not certain. It is possible that they die or detach from the basement membrane or they migrate as myofibroblast into the stroma, as suggested by the loss of Descemet’s membrane (Fig. 3B, C) and the appearance of α-SMA adjacent to the endothelial layer. Following the loss of corneal endothelial barrier function, EMT occurred in the corneal epithelium and later in the stromal layer. Our data suggest that balancing the biological activity of TGFβ1 in the eye is crucial not only for normal anterior segment development, but also for maintaining the cornea integrity and homeostasis during the postnatal period.
TGFβ1 is known to induce EMT in many tissues. The transgenic mice we used in this study develop anterior subcapsular cataracts (ASC) by P21 [44]. Previous studies showed that the underlying mechanism of ASC is through TGFβ1-induced EMT in the lens epithelial cells [57; 58]. Despite the lens defects in the OVE853 transgenic mice, we conclude that EnMT and EMT in the corneal endothelial and epithelial layers respectively are a direct response to the TGFβ1 released from the lens, not a secondary effect resulting from EMT in the lens epithelial cells. In our study, α-SMA expression in the cornea occurred earlier than that in the lens. When the eyelids open on P14, the cornea in the OVE853 transgenic mice looked hazy and the EMT marker, α-SMA, was expressed in the corneal endothelial and epithelial layers before P14. In contrast, the lens of OVE853 transgenic mice looked normal and no ASC was found at P14 [44]. The expression of α-SMA in the lens epithelial cells was detected at P21. During mouse eye development, both type I and the type II TGFβ receptors are highly expressed in the anterior corneal mesenchymal cells, from which the corneal endothelial and stromal keratocytes differentiate. The type II TGFβ receptor was not expressed at a detectable level in the lens epithelial cells before P3, but significant expression of both type I and type II receptors occurred by age P21 [59]. These results together suggest that the corneal cells are susceptible to TGFβ1 stimulation at an earlier age than the lens.
Our data indicate that active TGFβ1 from the lens can directly induce epithelial/endothelial-to-myofibroblast transformation in the cornea. However, we can not rule out the possibility that other factors, such as the growth factors in the aqueous humor and tears, also play a role in the progression of EnMT and EMT in the cornea [60; 61; 62]. A recent report showed that epidermal growth factor (EGF), which is normally present in tears, synergizes with TGFβ1 in the induction of differentiation of keratocytes to myofibroblasts [63]. Growth factors, including PDGF, IGF-1 and FGFs, are present in the anterior chamber. Some of these, such as PDGF, were shown to synergistically induce keratocyte-to-myofibroblast differentiation with TGFβ in a cell culture system [64; 65]. The initial loss of the barrier function of the corneal endothelial layer in the transgenic eye can result in the exposure of stromal keratocytes and the corneal epithelial cells to various growth factors in the aqueous humor. Therefore, it is likely that the increase of α-SMA expression and stress fiber formation in the cornea of the transgenic mice are the combined effects of TGFβ1 and other growth factors.
It is noteworthy to mention that while our data demonstrate that TGFβ1 can induce EnMT and EMT in the cornea, they do not demonstrate that TGFβ1 is responsible for this activity in injured eyes. While TGFβ1 levels increases during normal corneal wound healing [25; 66], TGFβ2 is the predominant form of TGF in normal eyes, and TGFβ2 is present at a relatively high level in the aqueous humor [67; 68]. TGFβ2 expression is found in the ciliary and iris epithelial cells as well as in the equatorial region of lens cells in mice [15]. These findings raise a question as to why a high level of endogenous TGFβ2 does not induce the same EMT response in normal eyes. One possibility is that the endogenous TGFβ2 in the anterior chamber is normally present in the inactive form [67]. TGFβ proteins are secreted as inactive complexes that contain the mature TGFβ homodimer tightly associated with dimers of the amino-terminal domain, which is referred as the latency-associated peptide (LAP) [69]. The complexes are termed “latent” because they do not bind to TGFβ receptors. The release of the active TGFβ form from the latent complex is thought to be regulated by specific proteases in vivo. The protease activity is tightly controlled under the normal physiological conditions to keep the levels of active TGFβ2 in the aqueous humor low. Another possibility is that the co-receptor TGFβRIII is required to present TGFβ2 to TGFβRII [69]. Expression of TGFβRIII in the corneal endothelial layer of the normal rat cornea was not detectable before P10 [23]. Therefore, the developing corneal endothelial cells would not be sensitive to TGFβ2 stimulation until after P10. Another possibility is that inhibitory molecules, which antagonize TGFβ activity, are present in the anterior chamber of the normal eye [70].
In the cornea, TGFβ1 plays a critical role in wound healing [7]. After corneal injury, quiescent keratocytes at the wound periphery become activated and transform into corneal fibroblasts or myofibroblasts. The transformed cells can migrate to the damaged area, proliferate, and deposit a disorganized and fibrotic ECM to repair the wound. Abnormal wound healing can lead to corneal fibrosis as a result of deregulated production of ECM components and contractile elements by corneal fibroblasts and myofibroblasts [71; 72; 73; 74]. In our transgenic model, we found that TGFβ1 from the lens is able to induce corneal endothelial- and epithelial-to-myofibroblast transformation. The transformation in these two cell layers occurred before myofibroblast formation in the stroma. In our transgenic mouse model, TGFβ1 is secreted from the lens, therefore, it is not surprising that the corneal endothelial cells underwent EnMT prior to the stromal cells. After the loss of endothelial barrier, the differential susceptibility to TGFβ1 stimulation in the corneal epithelium and stroma could be a result of different levels of receptor expression. Zieske et al showed that both type I and type II TGFβ receptors were upregulated in the corneal epithelial cells during wound repair [43]. In cultured primary human corneal epithelial cells, EGF and TGFβ1 can induce the expression of TGFβRII. If this is also true in our transgenic mouse model, TGFβ1 overexpression in the lens can upregulate the TGFβRII expression in the corneal epithelial cells, give the cells the potential for enhanced TGFβ1-signaling, and result in an earlier onset of EMT in these cells than in the stromal keratocytes.
In this study, we demonstrated that TGFβ1-induced EMT in corneal endothelial and epithelial layers is associated with loss of N- and E-cadherin expression respectively. Our result supports the hypothesis proposed by Masur et al based on their cell culture model that myofibroblast differentiation requires at least two factors: action of TGFβ and loss of cell contact [75]. In their in vitro system, the low-density culture had more myofibroblasts and active proliferating cells than the high-density culture. Interestingly, the same change in cell proliferation was also observed in our TGFβ1 transgenic mouse and was reported in the previous study [44]. It was shown that BrdU-labeled cells in the basal corneal epithelium were markedly increased in the P21 TGFβ1 transgenic mouse. In contrast to the in vitro cell culture model, it is unclear to us whether TGFβ1-induced EMT in the transgenic mouse cornea resulted in the formation of both fibroblasts and myofibroblasts. Cadherin-11 is a non-classical cadherin which was expressed in cultured corneal myofibroblasts [54]. We found that cadherin-11 expression was upregulated in the corneal epithelium and stroma in TGFβ1 transgenic mouse. Cadherin-11 is a maker for loosely connected and migratory mesenchymal cells. In human cancers, cadherin-11 expression is associated with the most aggressive and metastatic subsets of cancer cells [76; 77]. In our TGFβ1 transgenic mouse model, loss of E- and N-cadherin and increase of cadherin-11 expression in the cornea can potentially increase cell motility and promote the migration/invasion of transformed cells into the stromal layer. Our data raise the question: during corneal fibrosis, can EMT in corneal epithelial and EnMT in endothelial layer also contribute to the myofibroblasts in the corneal stroma? This hypothesis would challenge the original theory that myofibroblast formation during corneal wound healing is derived from mesenchymal cells, i.e. subconjunctival fibroblasts or keratocytes (corneal fibroblasts).
In summary, a threshold level of TGFβ1 appears to be critical for corneal development and homeostasis. As seen in the OVE920A transgenic mice and the TGFβ1 transgenic mice with a βB1-crystallin lens promoter [18; 45], TGFβ1 at high levels acts as a mitogen to the corneal mesenchymal cells and prevents these cells from differentiating into the corneal endothelial cells and keratocytes. In the OVE853 and OVE917 transgenic mice that have lower TGFβ1 expression, TGFβ1 does not interfere with the normal differentiation of the corneal mesenchymal cells. As a result, the corneal endothelial layer is formed properly, as shown by the expression of N-cadherin in the inner corneal surface (Figure 7). However, after P7 the corneal endothelium of the OVE853 strain started to lose its integrity and some of the cells expressed α-SMA, a molecular marker of EMT.
The effect of TGFβ1 on the postnatal cornea could be of clinical relevance. During intraocular injury or post-operative wound healing, the level of TGFβ1 in the ocular media may rise. Our data suggest that such an increase can result in the loss of corneal endothelial cells through the EnMT process. For patients with relatively low corneal endothelial cell density, blocking the increase of TGFβ activity in the aqueous humor after insult or surgery may help to preserve corneal endothelium and prevent corneal complications.
Acknowledgements
The work was supported by NIH/NEI grants R01 EY13146 (LWR), R24 EY14795 (MU Ophthalmology minicore), R01 EY016459 (JDA), P20 RR017703 (OUHSC), P30 EY012190 (OUHSC), and an unrestricted grant from Research to Prevent Blindness (OUHSC and MU Ophthalmology), The University of Missouri Research Board (LWR), and the American Diabetes Association (1-06-RA-05, JDA)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare that they have no competing financial interests.
References
- 1.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akhurst RJ. TGF beta signaling in health and disease. Nat Genet. 2004;36:790–792. doi: 10.1038/ng0804-790. [DOI] [PubMed] [Google Scholar]
- 3.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 5.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 6.Saika S, Liu CY, Azhar M, Sanford LP, Doetschman T, Gendron RL, Kao CW, Kao WW. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- 7.Saika S. TGFbeta pathobiology in the eye. Lab Invest. 2006;86:106–115. doi: 10.1038/labinvest.3700375. [DOI] [PubMed] [Google Scholar]
- 8.Saika S, Yamanaka O, Sumioka T, Miyamoto T, Miyazaki K, Okada Y, Kitano A, Shirai K, Tanaka S, Ikeda K. Fibrotic disorders in the eye: targets of gene therapy. Prog Retin Eye Res. 2008;27:177–196. doi: 10.1016/j.preteyeres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Kao WW, Xia Y, Liu CY, Saika S. Signaling pathways in morphogenesis of cornea and eyelid. Ocul Surf. 2008;6:9–23. [PubMed] [Google Scholar]
- 10.Piek E, Heldin CH, Ten Dijke P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. Faseb J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- 11.Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- 12.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115:1091–1105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lutty GA, Merges C, Threlkeld AB, Crone S, McLeod DS. Heterogeneity in localization of isoforms of TGF-beta in human retina, vitreous, and choroid. Invest Ophthalmol Vis Sci. 1993;34:477–487. [PubMed] [Google Scholar]
- 14.Kurosaka D, Nagamoto T. Inhibitory effect of TGF-beta 2 in human aqueous humor on bovine lens epithelial cell proliferation. Invest Ophthalmol Vis Sci. 1994;35:3408–3412. [PubMed] [Google Scholar]
- 15.Gordon-Thomson C, de Iongh RU, Hales AM, Chamberlain CG, McAvoy JW. Differential cataractogenic potency of TGF-beta1, -beta2, and -beta3 and their expression in the postnatal rat eye. Investigative Ophthalmology & Visual Science. 1998;39:1399–1409. [PubMed] [Google Scholar]
- 16.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunker N, Krieglstein K. Reduced programmed cell death in the retina and defects in lens and cornea of Tgfbeta2(−/−) Tgfbeta3(−/−) double-deficient mice. Cell Tissue Res. 2003;313:1–10. doi: 10.1007/s00441-003-0761-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhao S, Overbeek PA. Elevated TGFbeta signaling inhibits ocular vascular development. Dev Biol. 2001;237:45–53. doi: 10.1006/dbio.2001.0360. [DOI] [PubMed] [Google Scholar]
- 19.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 23.Joyce NC, Harris DL, Mello DM. Mechanisms of mitotic inhibition in corneal endothelium: contact inhibition and TGF-beta2. Invest Ophthalmol Vis Sci. 2002;43:2152–2159. [PubMed] [Google Scholar]
- 24.Ittner LM, Wurdak H, Schwerdtfeger K, Kunz T, Ille F, Leveen P, Hjalt TA, Suter U, Karlsson S, Hafezi F, Born W, Sommer L. Compound developmental eye disorders following inactivation of TGFbeta signaling in neural-crest stem cells. J Biol. 2005;4:11. doi: 10.1186/jbiol29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrington LM, Albon J, Anderson I, Kamma C, Boulton M. Differential regulation of key stages in early corneal wound healing by TGF-beta isoforms and their inhibitors. Invest Ophthalmol Vis Sci. 2006;47:1886–1894. doi: 10.1167/iovs.05-0635. [DOI] [PubMed] [Google Scholar]
- 26.Nishida K, Kinoshita S, Yokoi N, Kaneda M, Hashimoto K, Yamamoto S. Immunohistochemical localization of transforming growth factor-beta 1, -beta 2, and -beta 3 latency-associated peptide in human cornea. Invest Ophthalmol Vis Sci. 1994;35:3289–3294. [PubMed] [Google Scholar]
- 27.Pasquale LR, Dorman-Pease ME, Lutty GA, Quigley HA, Jampel HD. Immunolocalization of TGF-beta 1, TGF-beta 2, and TGF-beta 3 in the anterior segment of the human eye. Invest Ophthalmol Vis Sci. 1993;34:23–30. [PubMed] [Google Scholar]
- 28.Gupta A, Monroy D, Ji Z, Yoshino K, Huang A, Pflugfelder SC. Transforming growth factor beta-1 and beta-2 in human tear fluid. Curr Eye Res. 1996;15:605–614. doi: 10.3109/02713689609008900. [DOI] [PubMed] [Google Scholar]
- 29.Chen C, Michelini-Norris B, Stevens S, Rowsey J, Ren X, Goldstein M, Schultz G. Measurement of mRNAs for TGFss and extracellular matrix proteins in corneas of rats after PRK. Invest Ophthalmol Vis Sci. 2000;41:4108–4116. [PubMed] [Google Scholar]
- 30.Kaji Y, Mita T, Obata H, Tsuru T, Soya K, Shirasawa E, Sakai H, Hanyu A, Kato M, Yamashita H. Expression of transforming growth factor beta superfamily and their receptors in the corneal stromal wound healing process after excimer laser keratectomy. Br J Ophthalmol. 1998;82:462–463. doi: 10.1136/bjo.82.4.456g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vesaluoma M, Teppo AM, Gronhagen-Riska C, Tervo T. Release of TGF-beta 1 and VEGF in tears following photorefractive keratectomy. Curr Eye Res. 1997;16:19–25. doi: 10.1076/ceyr.16.1.19.5119. [DOI] [PubMed] [Google Scholar]
- 32.Jester JV, Barry-Lane PA, Petroll WM, Olsen DR, Cavanagh HD. Inhibition of corneal fibrosis by topical application of blocking antibodies to TGF beta in the rabbit. Cornea. 1997;16:177–187. [PubMed] [Google Scholar]
- 33.Mita T, Yamashita H, Kaji Y, Obata H, Hanyu A, Suzuki M, Tobari I. Functional difference of TGF-beta isoforms regulating corneal wound healing after excimer laser keratectomy. Exp Eye Res. 1999;68:513–519. doi: 10.1006/exer.1998.0627. [DOI] [PubMed] [Google Scholar]
- 34.Czaja MJ, Weiner FR, Flanders KC, Giambrone MA, Wind R, Biempica L, Zern MA. In vitro and in vivo association of transforming growth factor-beta 1 with hepatic fibrosis. J Cell Biol. 1989;108:2477–2482. doi: 10.1083/jcb.108.6.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto T, Noble NA, Miller DE, Border WA. Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int. 1994;45:916–927. doi: 10.1038/ki.1994.122. [DOI] [PubMed] [Google Scholar]
- 36.Cutroneo KR. TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound Repair Regen. 2007;15 Suppl 1:S54–S60. doi: 10.1111/j.1524-475X.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- 37.Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 38.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 39.Massague J. TGF-beta signal transduction. Annual Review of Biochemistry. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 40.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 41.Joyce NC, Zieske JD. Transforming growth factor-beta receptor expression in human cornea. Investigative Ophthalmology & Visual Science. 1997;38:1922–1928. [PubMed] [Google Scholar]
- 42.Obata H, Kaji Y, Yamada H, Kato M, Tsuru T, Yamashita H. Expression of transforming growth factor-beta superfamily receptors in rat eyes. Acta Ophthalmol Scand. 1999;77:151–156. doi: 10.1034/j.1600-0420.1999.770207.x. [DOI] [PubMed] [Google Scholar]
- 43.Zieske JD, Hutcheon AE, Guo X, Chung EH, Joyce NC. TGF-beta receptor types I and II are differentially expressed during corneal epithelial wound repair. Invest Ophthalmol Vis Sci. 2001;42:1465–1471. [PubMed] [Google Scholar]
- 44.Srinivasan Y, Lovicu FJ, Overbeek PA. Lens-specific expression of transforming growth factor beta1 in transgenic mice causes anterior subcapsular cataracts. Journal of Clinical Investigation. 1998;101:625–634. doi: 10.1172/JCI1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flugel-Koch C, Ohlmann A, Piatigorsky J, Tamm ER. Disruption of anterior segment development by TGF-beta1 overexpression in the eyes of transgenic mice. Dev Dyn. 2002;225:111–125. doi: 10.1002/dvdy.10144. [DOI] [PubMed] [Google Scholar]
- 46.Fromm L, Shawlot W, Gunning K, Butel JS, Overbeek PA. The retinoblastoma protein-binding region of simian virus 40 large T antigen alters cell cycle regulation in lenses of transgenic mice. Molecular & Cellular Biology. 1994;14:6743–6754. doi: 10.1128/mcb.14.10.6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu L, Overbeek PA, Reneker LW. Systematic analysis of E-, N- and P-cadherin expression in mouse eye development. Experimental Eye Research. 2002;74:753–760. doi: 10.1006/exer.2002.1175. [DOI] [PubMed] [Google Scholar]
- 48.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 49.Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23:912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 50.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008;121:727–735. doi: 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- 51.Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes & Development. 2000;14:1169–1180. [PubMed] [Google Scholar]
- 52.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 53.Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, Amann E. Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J Biol Chem. 1994;269:12092–12098. [PubMed] [Google Scholar]
- 54.Masur SK, Conors RJ, Jr., Cheung JK, Antohi S. Matrix adhesion characteristics of corneal myofibroblasts. Invest Ophthalmol Vis Sci. 1999;40:904–910. [PubMed] [Google Scholar]
- 55.Lovicu FJ, Schulz MW, Hales AM, Vincent LN, Overbeek PA, Chamberlain CG, McAvoy JW. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br J Ophthalmol. 2002;86:220–226. doi: 10.1136/bjo.86.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs. 2005;179(1–2):43–55. doi: 10.1159/000084508. 179 (2005) 43–55. [DOI] [PubMed] [Google Scholar]
- 57.Robertson JV, Nathu Z, Najjar A, Dwivedi D, Gauldie J, West-Mays JA. Adenoviral gene transfer of bioactive TGFbeta1 to the rodent eye as a novel model for anterior subcapsular cataract. Mol Vis. 2007;13:457–469. [PMC free article] [PubMed] [Google Scholar]
- 58.Hales AM, Chamberlain CG, McAvoy JW. Cataract induction in lenses cultured with transforming growth factor-beta. Investigative Ophthalmology & Visual Science. 1995;36:1709–1713. [PubMed] [Google Scholar]
- 59.de Iongh RU, Gordon-Thomson C, Chamberlain CG, Hales AM, McAvoy JW. Tgfbeta receptor expression in lens: implications for differentiation and cataractogenesis. Exp Eye Res. 2001;72:649–659. doi: 10.1006/exer.2001.1001. [DOI] [PubMed] [Google Scholar]
- 60.Wilson SE, Chen L, Mohan RR, Liang Q, Liu J. Expression of HGF, KGF, EGF and receptor messenger RNAs following corneal epithelial wounding. Exp Eye Res. 1999;68:377–397. doi: 10.1006/exer.1998.0603. [DOI] [PubMed] [Google Scholar]
- 61.Imanishi J, Kamiyama K, Iguchi I, Kita M, Sotozono C, Kinoshita S. Growth factors: importance in wound healing and maintenance of transparency of the cornea. Prog Retin Eye Res. 2000;19:113–129. doi: 10.1016/s1350-9462(99)00007-5. [DOI] [PubMed] [Google Scholar]
- 62.Lee JG, Kay EP. FGF-2-induced wound healing in corneal endothelial cells requires Cdc42 activation and Rho inactivation through the phosphatidylinositol 3-kinase pathway. Invest Ophthalmol Vis Sci. 2006;47:1376–1386. doi: 10.1167/iovs.05-1223. [DOI] [PubMed] [Google Scholar]
- 63.He J, Bazan HE. Epidermal growth factor synergism with TGF-beta1 via PI-3 kinase activity in corneal keratocyte differentiation. Invest Ophthalmol Vis Sci. 2008;49:2936–2945. doi: 10.1167/iovs.07-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaur H, Chaurasia SS, Agrawal V, Wilson SE. Expression of PDGF receptor-alpha in corneal myofibroblasts in situ. Exp Eye Res. 2009 doi: 10.1016/j.exer.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jester JV, Huang J, Petroll WM, Cavanagh HD. TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Exp Eye Res. 2002;75:645–657. doi: 10.1006/exer.2002.2066. [DOI] [PubMed] [Google Scholar]
- 66.Kaji Y, Soya K, Amano S, Oshika T, Yamashita H. Relation between corneal haze and transforming growth factor-beta1 after photorefractive keratectomy and laser in situ keratomileusis. J Cataract Refract Surg. 2001;27:1840–1846. doi: 10.1016/s0886-3350(01)01141-5. [DOI] [PubMed] [Google Scholar]
- 67.Helbig H, Kittredge KL, Coca-Prados M, Davis J, Palestine AG, Nussenblatt RB. Mammalian ciliary-body epithelial cells in culture produce transforming growth factor-beta. Graefes Arch Clin Exp Ophthalmol. 1991;229:84–87. doi: 10.1007/BF00172268. [DOI] [PubMed] [Google Scholar]
- 68.Jampel HD, Roche N, Stark WJ, Roberts AB. Transforming growth factor-beta in human aqueous humor. Curr Eye Res. 1990;9:963–969. doi: 10.3109/02713689009069932. [DOI] [PubMed] [Google Scholar]
- 69.Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 70.Schulz MW, Chamberlain CG, McAvoy JW. Inhibition of transforming growth factor-beta-induced cataractous changes in lens explants by ocular media and alpha 2-macroglobulin. Investigative Ophthalmology & Visual Science. 1996;37:1509–1519. [PubMed] [Google Scholar]
- 71.Garana RM, Petroll WM, Chen WT, Herman IM, Barry P, Andrews P, Cavanagh HD, Jester JV. Radial keratotomy. II. Role of the myofibroblast in corneal wound contraction. Invest Ophthalmol Vis Sci. 1992;33:3271–3282. [PubMed] [Google Scholar]
- 72.Jester JV, Petroll WM, Barry PA, Cavanagh HD. Expression of alpha-smooth muscle (alpha-SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci. 1995;36:809–819. [PubMed] [Google Scholar]
- 73.Jester JV, Petroll WM, Barry PA, Cavanagh HD. Temporal, 3-dimensional, cellular anatomy of corneal wound tissue. J Anat. 1995;186(Pt 2):301–311. [PMC free article] [PubMed] [Google Scholar]
- 74.Esquenazi S, He J, Bazan NG, Bazan HE. Comparison of corneal wound-healing response in photorefractive keratectomy and laser-assisted subepithelial keratectomy. J Cataract Refract Surg. 2005;31:1632–1639. doi: 10.1016/j.jcrs.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 75.Masur SK, Dewal HS, Dinh TT, Erenburg I, Petridou S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci U S A. 1996;93:4219–4223. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 1999;59:947–952. [PubMed] [Google Scholar]
- 77.Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, Schalken JA. Cadherin switching in human prostate cancer progression. Cancer Res. 2000;60:3650–3654. [PubMed] [Google Scholar]