

Abstract
A viral entry assay where a beta-lactamase reporter protein fused to the influenza matrix protein-1 (BlaM1) is packaged as a structural component into influenza virus-like particles (VLPs) is described. The Bla reporter is released upon fusion with target cells and can be detected in live cells by flow cytometry, microscopy, or fluorometric plate reader for utility in high-throughput screening approaches. The production of BlaM1 VLPs and subsequent transfer of Bla activity to target cells requires the presence of influenza hemagglutinin (HA) and neuraminidase (NA). In addition, transfer of Bla by the VLPs can be blocked by an influenza neutralizing antibody, is impeded by a chemical inhibitor of influenza virus entry, and requires HA that is cleaved by a protease specific for its cleavage site. An analogous VLP system also was developed for Ebola (EBOV) and Marburg (MARV) viruses, demonstrating that this straightforward assay has broad application for studying the entry steps of enveloped viruses, especially those that require high levels of biosafety containment.
Keywords: Virus entry, Influenza virus, Ebola virus, Marburg virus, Flow cytometry, Microscopy
1. Introduction
Experimentation with highly pathogenic viruses poses myriad practical challenges due to laboratory safety issues. As an alternative, non-infectious viral entry assays provide surrogate systems to characterize mechanistically the early events in the viral life cycle, while circumventing the obstacles of working with authentic, live viruses. Many of these approaches, however, have their share of disadvantages. Fusion assays based on cell surface expression of the viral glycoprotein may not mimic virus-cell fusion events, in either the lipid compositions of the target membranes or the distribution of glycoproteins within those membranes. Furthermore, while pseudotyped retrovirus entry assays depend on fusion of the viral envelope with the cell membrane, these particles may not possess the characteristics of authentic virions.
Influenza viruses claim the lives of approximately 500,000 people worldwide each year and four times in recent history, in 1890, 1918, 1957, and 1968, global influenza pandemics have emerged in the human population with a significant number of fatalities (Smith et al., 2004; Webby and Webster, 2003). Influenza A virus, a member of the family Orthomyxoviridae, is an enveloped virus with a negative-sense, RNA genome, consisting of 8 single-stranded RNA segments and encoding 11 proteins (reviewed in Palese and Shaw, 2007). The genome is encapsidated in a lipid bilayer harboring 3 integral membrane proteins – hemagglutinin (HA), which provides the viral receptor binding and fusogenic functions, neuraminidase (NA), a sialidase that facilitates the release of nascent viral particles from the host cell membrane, and matrix protein-2 (M2) that oligomerizes to form an ion channel necessary for viral uncoating. The HA and NA cytoplasmic tails promote membrane and lipid raft microdomain association of the matrix protein-1 (M1) protein, which lies beneath the lipid bilayer and contacts the viral ribonucleoprotein complexes (Ali et al., 1996; Avalos et al., 2000; Enami and Enami, 1996; Zhang et al., 2000). To initiate infection, the influenza virus HA binds to sialic acid containing-receptors on the target cell surface, triggering receptor-mediated endocytosis and subsequent fusion within the low pH environment of the endosome.
Previous reports have demonstrated that only HA and NA are required for the formation of virus-like particles (VLPs); expression of these 2 proteins alone in mammalian cells generates particles which resemble complete influenza virions (Chen et al., 2007). Beta-lactamase (Bla), a sensitive reporter with a variety of detection methods, has been used previously to tag the HIV-Vpr protein for the examination of HIV and Ebola virus glycoprotein-mediated entry (Cavrois et al., 2002; Yonezawa et al., 2005). Likewise, it was reasoned that if influenza M1 were tagged with Bla (BlaM1) it could be incorporated specifically into VLPs bearing HA and NA on their surfaces, and delivered into target cells following VLP entry. This technique improves on enveloped virus entry/fusion assays described previously. Specifically, the BlaM1 VLPs contain only influenza virus proteins, HA, NA, and M1, which should mimic closely influenza virus particles in their budding, morphology, and entry processes. In addition, influenza VLP reporter activity does not require downstream replication events, but simply the release of the BlaM1 fusion protein into the cytoplasm of the target cell.
In this report the generation of BlaM1 VLPs, the detection of Bla activity by both flow cytometry and microscopy after VLP entry into target cells, and validation of this VLP system as a method to study influenza virus entry are described. An analogous Bla VLP system for the examination of Ebola (EBOV) and Marburg (MARV) virus entry is also described, as has been reported recently (Manicassamy and Rong, 2009). This technique has potential as a universal approach to study the entry pathways of enveloped viruses that have similar structural organization to influenza.
2. Materials and Methods
2.1 Cells and reagents
Madin-Darby canine kidney (MDCK) cells were maintained in minimum essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). 293T cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS. Media and reagents for cell culture were purchased from Gibco-BRL (Invitrogen Inc., Carlsbad, CA, USA). TPCK-treated trypsin, NA from C. perfringens, Proteinase K, soybean trypsin inhibitor, rottlerin, and bafilomycin A1 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Porcine pancreatic elastase was purchased from Worthington Biochemical (Lakewood, NJ, USA). CCF2-AM reagent and loading kit were purchased from Invitrogen (Carlsbad, CA, USA).
2.2 Construction of pCAGGS-BlaM1, VLP production, and VLP infections
The A/BrevigMission/1/18 (1918) M1 sequence was PCR amplified from an M1 expression vector and inserted into the pCAGGS vector (Niwa et al., 1991). The bla gene was PCR amplified from pcDNA3.1 and fused N-terminally to M1 within pCAGGS to create a modified Bla fusion. In the modified Bla the first 24 amino acids are excluded to remove a secretion signal and His 24 was substituted with Asp to create a Kozak consensus sequence. A short linker sequence, encoding GSGGGSAS, was added in frame with the downstream M1 sequence. The pcDNA3.1-BlaVP40 construct has been described (Manicassamy and Rong, 2009).
To generate influenza 1918 BlaM1 VLPs, 293T cells were transfected using Lipofectamine 2000 (Invitrogen) with pCAGGS-BlaM1 (2.5 μg), pCAGGS-HA (A/New York/1/18) and pCAGGS-NA (A/BrevigMission/1/18) (0.5 μg each) (Glaser et al., 2005; Tumpey et al., 2002). WSN BlaM1 VLPs were created by transfection of pCAGGS-BlaM1 in conjunction with pCAGGS-HA (A/WSN/33) and pCAGGS-NA (A/WSN/33) (Neumann et al., 1999). Transfected cells were maintained in OptiMEM for 48-72 h. Following incubation, the BlaM1 VLP-containing 293T supernatants were harvested and centrifuged to remove debris. Supernatants were treated with 2-3 μg/ml TPCK-trypsin for 30 min at 37 °C to activate the HA protein on the VLPs. For antibody experiments, after incubation with TPCK-trypsin, soybean trypsin inhibitor (3.5 μg/ml) and 39E4 (3.0 μg/ml), a neutralizing antibody against 1918 influenza virus, were added to the VLPs, and incubated for an additional 15 min at 37 °C. For infection of MDCK cells in 12-well plates, cells were washed twice with PBS and VLPs were added in a total volume of 400-800 μl. For infection of cells in 4-well chamber slides, 200 μl of VLPs was used. Cells were centrifuged at 1.5 k rpm, for 90 min at 4 °C to synchronize the infection. The cells were transferred to the incubator at 37 °C and further incubated for 3-4 h.
The HA-E mutant was created in the manner described in Stech et al. (2005). Briefly, the pCAGGS-WSN HA vector was used as a template for Quikchange mutagenesis (Stratagene, La Jolla, California, USA) substituting Arg 343 (H1 numbering), adjacent to the trypsin cleavage site, to Val. HA-E containing VLPs were produced in 293T cells as above, by cotransfection of BlaM1, HA-E, and NA expression plasmids. HA-E on the VLPs was activated using 10 μg/ml porcine pancreatic elastase and incubated with MDCK cells as above.
To generate EBOV and MARV BlaVP40 VLPs, 293T cells were transfected using Lipofectamine 2000 (Invitrogen) with pcDNA3.1-BlaVP40 and plasmids expressing EBOV (strain Zaire) or MARV (strain Musoke) GP. Supernatants were harvested and used to infect MDCK cells as described above for influenza VLPs.
2.3 Detection of Bla activity in live cells
For detection of Bla activity by flow cytometry, MDCK cells were detached with trypsin and loaded with the CCF2-AM substrate (Invitrogen) for 45 min - 1 h at 37 °C according to the manufacturer’s instructions using the alternative substrate loading protocol. Following substrate loading, cells were washed with PBS and transferred to FACS tubes. Cells were analyzed at the Mount Sinai Flow Cytometry Shared Resource Facility on an LSRII flow cytometer (Becton Dickinson, Miami, FL, USA), typically collecting 10,000 events. Samples were gated on live cells and analyzed for their cleavage of CCF2 using FlowJo 8.5.2 software (Tree Star Inc., Ashland, OR, USA). For fluorescence microscopy, the CCF2-AM loading solution was added to cells within the chamber slide and stained as above. After washing, cells were maintained in PBS supplemented with bovine serum albumin and analyzed on a Leica SP5 DMI inverted confocal microscope (Leica Microsystems, Bannockburn, IL, USA).
2.4 Hemagglutination (HA) and hemagglutination inhibition (HAI) assays
HA and HAI assays were performed using standard protocols (World Health Organization, 1982). Briefly, for the HA assay, supernatants were harvested, serially diluted 2-fold in PBS, and incubated with 0.5% chicken red blood cells for 30 min at 4 °C. For the HAI assay, VLPs, equivalent to 4-8 HA units/well, were pre-incubated with 2-fold serial dilutions of antibody. Chicken red blood cells were added to a final concentration of 0.5% and samples were incubated for 30 min at 4 °C.
2.5 VLP purification and immunoblotting
VLPs within transfected cell supernatants were purified over a 30% sucrose cushion by centrifugation at 25 k rpm for 2 h at 4 °C. The pellet was resuspended in PBS and lysed in SDS-lysis buffer. Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride membrane (PVDF) (Millipore, Bedford, MA, USA), and detected using an anti-WSN polyclonal antibody, followed by a horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (GE Healthcare UK Ltd, Little Chalfont, UK). Detection was performed with the Western Lightning Enhanced Chemiluminescence Substrate (Perkin Elmer, Waltham, MA, USA), according to the manufacturer’s instructions.
2.6 Protease protection assay
Supernatants from transfected cells were left untreated, were treated with 30 μg/ml Proteinase K, or with 30 μg/ml Proteinase K and 1% Triton X-100 for 30 min at 37 °C. Following treatment, 1 mM phenylmethylsulphonyl fluoride (PMSF) was added and the samples were lysed in SDS lysis buffer. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane, and immunoblotted as above with an anti-beta-lactamase antibody (Millipore), followed by an HRP-conjugated anti-rabbit secondary antibody (GE Healthcare UK Ltd).
2.7 Rottlerin and bafilomycin A1 inhibitor experiments
For experiments using rottlerin, MDCK cells were pretreated with 1.25 μM rottlerin or DMSO and infected with influenza or EBOV VLPs in the presence of rottlerin or DMSO. MDCK cytotoxicity is not observed at this concentration of rottlerin (H. Hoffmann, personal communication). For bafilomycin experiments, MDCK cells were pretreated with 100 nM bafilomycin A1 or DMSO and infected with influenza VLPs in the presence of bafilomycin or DMSO. Following infection, cells were loaded with CCF2-AM and analyzed by flow cytometry.
3. Results
3.1 Generation and validation of influenza BlaM1 VLPs
Bla enzymatic activity in live cells can be measured by flow cytometry, microscopy, or fluorometric plate reader after loading cells with the commercially-available, fluorogenic substrate CCF2, which contains a beta-lactam ring that is cleavable by Bla (Fig. 1A). The substrate precursor, CCF2-AM, is taken up by cells and retained in the cytoplasm after cleavage by cellular esterases generating CCF2; a quencher present in the loading solution prevents fluorescence from dye that does not enter the cell, thus minimizing background fluorescence. If CCF2 within the cell remains uncleaved it emits a green fluorescence at 520 nm after excitation at 409 nm (Fig. 1A). However, if Bla is present, cleavage of the substrate results in a color change detectable by a shift in emission from 520 to 447 nm, creating blue fluorescence.
Fig. 1. A new VLP assay to study enveloped virus entry.

(A) Schematic illustration of the BlaM1 VLP assay. The fluorogenic substrate CCF2-AM is taken up by cells and cleaved by cellular esterases to form CCF2, which is retained in the cytoplasm. CCF2 is composed of a coumarin (blue) and fluorescein (green) fluorophore joined by a Bla cleavable linker (thick, black line). When Bla is not present (left), the coumarin component of CCF2 is excited at 409 nm and undergoes fluorescence resonance energy transfer (FRET) with the fluorescein moiety resulting in emission at 520 nm (green). When Bla is delivered to the cell by the BlaM1 VLP (right), Bla is released following fusion of the particle and can cleave CCF2, disrupting FRET, resulting in CCF2 emission at 447 nm (blue). (B) Detection of BlaM1 VLPs in the supernatants of transfected cells. 293T cells were transfected with 1918 HA, 1918 NA, and BlaM1 (BlaM1-HA/NA), with 1918 HA and BlaM1 (BlaM1-HA), with BlaM1 alone (BlaM1), or with HA and NA alone (HA/NA). At 72 h post transfection, supernatants were harvested, serially diluted 2-fold in PBS, and incubated with chicken red blood cells for 30 min at 4 °C. The hemagglutinating units per 50 μl of supernatant (HAU) are indicated. PBS is included as a negative control. (C) NA activity is required for the release of influenza VLPs. Cells were transfected as in (B) with the indicated constructs and were left either untreated (left panel) or were maintained in the presence of exogenous NA from Clostridium perfringens (100 mU/ml, right panel). Following incubation, the HAU within the transfected cell supernatants were determined as in (B). (D) Protease treatment of VLPs. Supernatants from BlaM1 alone (no env) or BlaM1-HA/NA transfected cells were left untreated (untr) or were incubated with Proteinase K only (+P), or with both Proteinase K and Triton X-100 (+P, +Tx-100). Samples were lysed in SDS sample buffer and separated by SDS-PAGE (4-15% gradient). Proteins were detected by immunoblotting using an anti-beta-lactamase antibody. (E) Analysis of purified VLPs. Supernatants from BlaM1 alone (no env) or BlaM1-HA/NA transfected cells were purified over a 30% sucrose cushion. BlaM1-HA/NA-transfected cell supernatants were left untreated or incubated with 3 μg/ml TPCK-trypsin for 30 min at 37 °C. The samples were lysed in SDS sample buffer and separated by SDS-PAGE (4-15% gradient). Proteins were detected by immunoblotting with an anti-WSN polyclonal antibody.
To generate the BlaM1 construct, which upon expression would be incorporated as a structural component into influenza VLPs, Bla was fused to the N terminus of the M1 protein from the pandemic 1918 (A/BrevigMission/1/18) influenza virus. BlaM1 was cotransfected with 1918 HA and 1918 NA expression constructs into 293T cells. Following transfection, Bla activity in the 293T cells was evident and their supernatants contained an appreciable hemagglutination (HA) titer (data not shown and Fig. 1B), suggesting the generation of BlaM1 VLPs. There was no detectable HA titer in the supernatants of the BlaM1 and HA-transfected cells (Fig. 1B). However, when exogenous bacterial neuraminidase was added during the transfection, HA titer within the cell supernatants was recovered, albeit to a lesser extent than when both HA and NA were present (Fig. 1C). These data indicate a requirement for NA enzymatic activity in releasing the VLPs, but also suggest that the influenza NA protein may contribute to VLP budding. Finally, despite there being no HA or NA in the BlaM1 alone control transfections, BlaM1 protein was detectable in the supernatants of these cells, although it was sensitive to Proteinase K treatment (Fig. 1D). These data suggest that BlaM1 is nonspecifically secreted into the supernatant following its overexpression in 293T cells, rather than incorporated into a VLP containing BlaM1 alone. In contrast, a significant proportion of BlaM1 protein within the supernatants from BlaM1, HA, and NA-containing transfections was protected from protease treatment, presumably by a lipid membrane, unless detergent was also present (Fig. 1D).
It was next examined using flow cytometry whether BlaM1-containing VLPs could transfer Bla activity to naïve MDCK cells. A prerequisite for influenza virus membrane fusion is the proteolytic cleavage of the HA precursor HA0 into HA1 and HA2, liberating the hydrophobic fusion peptide located at the amino terminus of HA2 (Klenk et al., 1975; Lazarowitz et al., 1973). Here this phenomenon was mimicked by the addition of TPCK-trypsin to the VLP preparation. Examination of purified VLPs by immunoblotting supported the incorporation of BlaM1, HA and NA into particles and the cleavage of HA after trypsin addition (Fig. 1E). Only cells that had been incubated with TPCK-trypsin-treated BlaM1 VLPs bearing both HA and NA (influenza VLPs) demonstrated Bla activity in MDCK cells, indicated by the shift in fluorescence from the green to the blue channel (Fig. 2A). Serial dilution of the VLP inoculum resulted in a linear decrease of Bla-positive cells (Fig. 2B). Low levels of background Bla activity, similar to that from mock-treated cells, were observed after incubation of MDCK cells with supernatants from cells transfected with BlaM1 alone or cells transfected with only HA and BlaM1, indicating a requirement for HA and NA in the transfer of BlaM1 to naïve cells (Fig. 2A). Variable levels of VLP-mediated BlaM1 delivery were observed between experiments, most likely due to the multiplicity of infection or differential incorporation of BlaM1 into the VLPs from preparation to preparation. Influenza VLPs containing the HA and NA from A/PuertoRico/8 and A/WSN/33 (WSN) H1N1 viruses demonstrated similar properties, except that for WSN VLPs, TPCK-trypsin was not required for BlaM1 transfer, consistent with the trypsin-independent activation of the WSN HA in the presence of the WSN NA (Choppin, 1969; Goto and Kawaoka, 1998).
Fig. 2. VLP-mediated transfer of Bla activity.
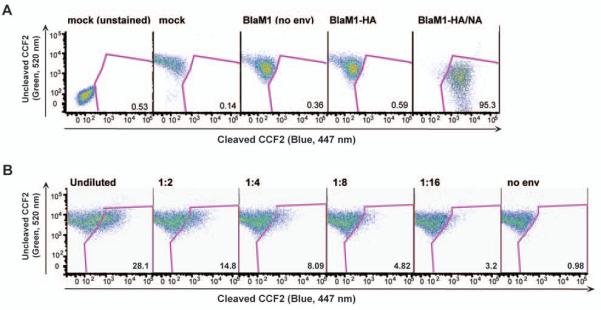
(A) 293T cells were mock transfected, transfected with BlaM1 alone (no env), 1918 HA and BlaM1, or 1918 HA/NA, and BlaM1. MDCK cells were incubated with the transfected 293T cell supernatants, loaded with CCF2, and Bla activity was measured by flow cytometry. The percentage of Bla-positive cells is indicated in the lower right corner of each panel. (B) Serial dilution of influenza VLPs. MDCK cells were infected with 2-fold serial dilutions of influenza VLPs. Following infection, MDCK cells were loaded with CCF2 and Bla activity was measured by flow cytometry. The percentage of Bla-positive cells is indicated in the lower right corner of each panel.
To validate the influenza VLP system as a model for the study of influenza virus entry, it was demonstrated that BlaM1 VLPs utilize an HA-mediated entry pathway, similar to authentic influenza virions. The VLPs were pre-incubated with a neutralizing antibody against 1918 influenza virus (39E4) at a concentration corresponding to 10 hemagglutination inhibition (HAI) units, as measured by classical HAI assay. Antibody-treated and untreated VLPs were used subsequently to infect MDCK cells. Flow cytometry analysis following infection revealed that the antibody treatment had reduced the percentage of Bla-positive cells by greater than 90% compared to untreated VLP infections, to a level comparable to the negative control, BlaM1 alone samples (Fig. 3A). These data demonstrate that BlaM1 VLPs can be bound by and neutralized with an antibody against authentic influenza virus. Furthermore, to show that BlaM1 VLPs require cleaved HA for entry and transfer of Bla to the target cell, a mutant HA (HA-E), which is activated by elastase instead of trypsin, was created by changing one amino acid in the cleavage site of HA, as has been described previously (Stech et al., 2005). Elastase-treated VLPs harboring HA-E readily transferred Bla to target cells (Fig. 3B). Alternatively, trypsin-treated HA-E VLPs had background levels of Bla activity (Fig. 3B). These data establish that treatment of HA with a protease specific for its cleavage site is required for influenza VLP entry.
Fig. 3. Validation of the influenza VLP assay.
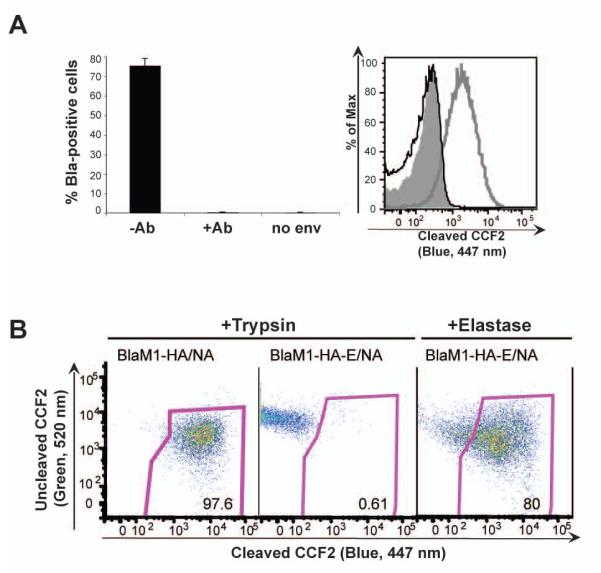
(A) VLP-mediated transfer of Bla activity can be blocked by a neutralizing antibody against influenza virus. 1918 VLPs were left untreated (−Ab) or were treated with a 1918 neutralizing antibody (+Ab) prior to incubation with MDCK cells. Cells were loaded with CCF2 substrate and analyzed by flow cytometry. The percentage of positive cells from triplicate samples is shown (left); error bars indicate standard deviations. The level of cleaved CCF2 in untreated (gray line) or antibody-treated (black line) cells incubated with 1918 or no env (shaded area) VLPs is shown as a histogram (right). (B) Influenza VLP entry requires pretreatment with a protease specific for its HA cleavage site. BlaM1 VLPs were generated as above with WSN HA and NA or HA-E and NA. HA-E is a mutant WSN HA that is activated by elastase instead of trypsin. VLPs were treated with either elastase or trypsin, and used to infect MDCK cells. Cells were loaded with CCF2 substrate and analyzed by flow cytometry.
3.2 Adaptation of Bla VLP system to EBOV and MARV
The VLP system was also adapted to EBOV and MARV viruses, highly pathogenic members of the family Filoviridae. The development of VLP-based entry assays for these viruses is desirable given that experiments with live EBOV and MARV require the use of biosafety level 4 conditions. The filoviruses have similar structural organization to influenza. Enveloped particles contain the viral glycoprotein (GP), which mediates both viral receptor binding and membrane fusion; lying beneath the lipid bilayer is the viral matrix protein VP40, which drives the budding of EBOV particles (Bavari et al., 2002; Jasenosky et al., 2001; Timmins et al., 2001). Therefore, EBOV and MARV VLPs were generated in an analogous manner to influenza VLPs, by tagging the EBOV matrix protein VP40 with Bla (BlaVP40) and cotransfecting with either EBOV or MARV GP, as recently described (Manicassamy and Rong, 2009). Bla activity was detectable in cells by flow cytometry after incubation with the EBOV and MARV VLPs, but not in those incubated with no envelope protein (BlaVP40 alone) VLPs (Fig. 4A). It was also observed that the EBOV inoculum could be diluted to achieve a linear range of infectivity (Fig. 4B). Hence, as the BlaM1 VLPs for influenza, the BlaVP40 VLPs allow detection of filovirus GP-mediated entry.
Fig. 4. Generation of EBOV and MARV VLPs.
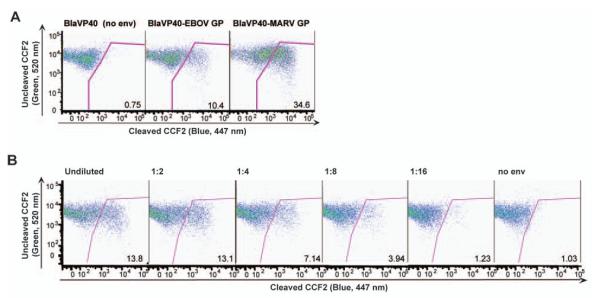
(A) 293T cells were transfected with BlaVP40 alone (no env), EBOV GP and BlaVP40, or MARV GP and BlaVP40. Supernatants were harvested and incubated with MDCK cells. Cells were loaded with CCF2 substrate and analyzed by flow cytometry. The percentage of Bla-positive cells is indicated in the lower right corner of each panel. (B) Serial dilution of EBOV VLPs. MDCK cells were infected with 2-fold serial dilutions of EBOV VLPs. Following infection, MDCK cells were loaded with CCF2 and Bla activity was measured by flow cytometry. The percentage of Bla-positive cells is indicated in the lower right corner of each panel.
3.3 Fluorescence microscopy detection of Bla activity in live cells
Bla activity in live cells can also be detected using microscopy, imaging a shift in CCF2 fluorescence emission from 520 nm (green) to 447 nm (blue) after excitation at 409 nm. VLP-producing 293T cells that had been transfected with BlaM1 or BlaVP40 constructs were examined using an inverted confocal microscope. Fluorescence was observed in the blue channel from BlaM1 or BlaVP40-transfected cells regardless of cotransfection with 1918 HA/NA or EBOV GP, as expected (Fig. 5A). MDCK cells were also examined after incubation with BlaM1 (influenza) or BlaVP40 (EBOV or MARV) VLPs. In accord with the data obtained using flow cytometry, blue cells were observed after infection with influenza VLPs harboring HA and NA from either 1918 or WSN strains, as well as with EBOV and MARV VLPs (Fig. 5B). By comparison, cells that were treated with supernatants from cells expressing either BlaM1 or BlaVP40 alone remained green, again demonstrating that Bla activity could not be transferred to naïve cells in the absence of the viral glycoprotein (Fig. 5B).
Fig. 5. Microscopic detection of Bla activity.

(A) Bla enzymatic activity in VLP producer cells. 293T cells were transfected with BlaM1 or BlaVP40 alone (no env), BlaM1 and 1918 HA/NA (1918), BlaVP40 and EBOV GP (EBOV), or mock-transfected, loaded with CCF2 substrate and imaged under the fluorescent microscope (20x magnification). (B) Bla enzymatic activity in MDCK target cells. MDCK cells were incubated with the indicated VLPs, loaded with CCF2, and imaged under the fluorescent microscope (20x magnification).
3.4 Inhibition of VLP entry
Next it was examined if the influenza and EBOV VLPs could be useful for identifying factors that modulate virion entry. Rottlerin, an inhibitor of protein kinase C (PKC) activity and a known inhibitor of influenza virus replication (Hoffmann et al., 2008), was tested for its effect on influenza and EBOV VLP infection. MDCK cells were incubated with rottlerin and subsequently infected with either influenza or EBOV VLPs in the presence of the compound. At 5 h post infection, flow cytometry analysis indicated that the entry of both influenza and EBOV VLPs was inhibited by rottlerin, the former showing a ~99% reduction compared to DMSO-treated cells (Fig. 6A). A block at the level of virus entry due to rottlerin inhibition of PKC, which regulates a number of cellular processes including membrane trafficking and endocytosis (Alvi et al., 2007), is consistent with the entry pathways of influenza and EBO viruses (Palese and Shaw, 2007; Yonezawa et al., 2005). Additionally, pretreatment of cells with bafilomycin A1, a chemical inhibitor of endosomal acidification, inhibited influenza VLP entry (Fig. 6B). These data suggest that influenza and EBOV VLPs may be useful in the identification, verification, and characterization of potential antiviral compounds that inhibit viral entry.
Fig. 6. Inhibition of VLP entry.

(A) Rottlerin prevents influenza and EBOV VLP entry. MDCK cells were pretreated with DMSO or 1.25 μM rottlerin and subsequently infected with influenza or EBOV VLPs. Values are expressed relative to the percentage of the Bla-positive cells in the DMSO control samples for either the influenza (black bars) or EBOV (white bars) VLP infections. (B) Bafilomycin A1 prevents influenza VLP entry. MDCK cells were pretreated with DMSO or 100 nM Bafilomycin A1 and infected with influenza or BlaM1 (no env) VLPs. Values are expressed relative to the percentage of the Bla-positive cells in the DMSO control samples for either the influenza (black bars) or no env (white bars) VLP infections.
4. Discussion
In conclusion, the VLP system presented here is a simple, but powerful approach for the study of virus entry, especially in cases where high levels of biocontainment are necessary for experiments with live virus. The influenza, EBOV, and MARV VLPs capitalize on the inherent budding machinery of these viruses, utilizing their own viral proteins rather than deriving these functions from a heterologous virus, such as a retrovirus. Thus, it is expected that these particles should more faithfully mimic the budding, morphology, and authentic entry process of the corresponding, live virus. Moreover, the Bla protein, fused to the viral matrix protein, either M1 or VP40, appears to be incorporated as a structural component of the VLP. Therefore, Bla reporter expression is uncomplicated by events downstream from entry and can be detected in a sensitive and quantitative manner upon virion fusion with the target cell. We hypothesize that this technique potentially can be applied to any enveloped virus that possesses similar structural organization to influenza or filoviruses.
Generation of influenza VLPs that were competent to transfer Bla to the MDCK target cells required expression of both HA and NA in the 293T producer cells. Current literature suggests that HA provides the budding, attachment, and fusogenic functions for influenza plasmid-derived VLPs, while the role of NA is in the efficient release of particles (Chen et al., 2007). Consistent with this, it was observed that when HA was coexpressed with BlaM1 in the absence of NA, no VLPs were detectable in the supernatants of the producer cells, unless exogenous neuraminidase was added to the transfection. These data also support a possible role of NA in the budding process given that the exogenous neuraminidase treatment did not restore HA titers to BlaM1-HA/NA VLP levels. Alternatively, this observation may simply reflect differences in the efficiencies of the influenza NA versus the bacterial NA in the release of particles. The data presented here also support that subsequent influenza VLP entry into MDCK cells requires a cleaved HA molecule and occurs in a manner that mimics the pathway of authentic influenza virions because Bla transfer could be blocked by a neutralizing antibody, rottlerin, a PKC inhibitor, and bafilomycin, a chemical inhibitor of endosomal acidification. As for the filovirus VLPs, although we did not examine formally the requirements for VLP formation, which is driven by the VP40 matrix protein (Bavari et al., 2002; Janenosky et al., 2001; Timmins et al., 2001), our data is consistent with a GP-dependent mechanism of Bla transfer.
There are a number of methods to detect Bla enzymatic activity in mammalian cells, two of which are presented here. Both flow cytometry and fluorescence microscopy can be used to detect Bla VLP entry on the single cell level. Flow cytometry offers the added opportunity to determine the magnitude of VLP entry by examining the levels of uncleaved (green) versus cleaved (blue) CCF2 within a particular cell. This is more difficult to distinguish under the fluorescent microscope. The Bla VLP assay can also be adapted to a fluorometric plate reader format, allowing its use in high-throughput screening applications. One consideration with any of these techniques is the differential loading and retention time of the CCF2 substrate in different cell types. Some cells require the addition of an anion transport inhibitor to the substrate loading solution, as was done for MDCK cells, which improves the uptake and limits the export of CCF2. Likewise, the optimal time and temperature of substrate loading must also be determined empirically for a specific cell type. Taken together, the availability of approaches for Bla detection suggests that the Bla VLP system can be utilized to answer a variety of experimental questions regarding enveloped virus entry.
Although the virulence of a particular influenza virus strain is determined polygenically, the cleavability of the HA precursor correlates well with the pathogenicity of avian influenza viruses (Garten and Klenk, 1999; Horimoto and Kawaoka, 2001; Steinhauer, 1999). In human influenza virus strains, the correlation between the efficiency of HA cleavage and virulence is less clear. The BlaM1 VLP technique described here can examine directly the protease requirements for HA cleavage, and how combinations of HA and NA from different subtypes, such as may be present in a reassortant virus, may affect particle budding, binding, and fusion. The effects of specific domains in HA, NA, and M1 required for viral budding and entry can also be analyzed quickly by this assay. Similar studies using BlaVP40 VLPs can be envisioned for EBOV/MARV GP and VP40. Perhaps the most attractive use of the Bla VLPs is their utility in identifying new or more efficient antiviral drugs for treatment of these viruses. If adapted to a high-throughput platform, libraries of compounds or antibodies can be screened for their ability to block the virus entry step, an attractive target for the development of an antiviral or therapeutic agent. Surely, further investigation into the entry pathways of these highly pathogenic viruses will allow the development of improved predictive measures, prevention techniques, and treatment strategies.
5. Acknowledgements
We thank Richard Cadagan and Osman Lizardo for technical support, Chris Basler for the 39E4 antibody, EBOV GP plasmid, and MARV GP plasmid, and Lijun Rong for the EBOV BlaVP40 plasmid. We also thank Osvaldo Martinez for useful discussions. Flow cytometry was performed at the Mount Sinai Flow Cytometry Shared Resource Facility. Microscopy was performed at the Mount Sinai Microscopy Shared Resource Facility, supported with funding from NIH-NCI shared resources grant 5R24 CA095823-04, NSF Major Research Instrumentation grant DBI-9724504 and NIH shared instrumentation grant 1 S10 RR0 9145-01. Research on influenza viruses in the Adolfo García-Sastre lab is partly funded by the Center for Research on Influenza Pathogenesis (CRIP), a National Institute for Allergy and Infectious Diseases (NIAID) sponsored Center of Excellence for Influenza Research and Surveillance HHSN266200700010C, by the Northeast Biodefense Center and by the Center for Investigating Viral Immunity and Antagonism (CIVIA) (NIAID grants U54AI57158 and U19AI62623, respectively), and by NIAID grants U01AI70469 and P01AI58113. Donna Tscherne is funded by NIAID grant 1F32AI081428.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- Ali A, Avalos RT, Ponimaskin E, Nayak DP. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000;74:8709–19. doi: 10.1128/jvi.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvi F, Idkowiak-Baldys J, Baldys A, Raymond JR, Hannun YA. Regulation of membrane trafficking and endocytosis by protein kinase C: emerging role of pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell Mol. Life Sci. 2007;64:263–70. doi: 10.1007/s00018-006-6363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmalijohn C, Schmalijohn A, Aman MJ. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002;195:593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay for detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002;20:1151–4. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Leser GP, Morita E, Lamb RA. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007;81:7111–23. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choppin PW. Replication of influenza virus in a continuous cell line: high yield of infective virus from cells inoculated at high multiplicity. Virology. 1969;39:130–4. doi: 10.1016/0042-6822(69)90354-7. [DOI] [PubMed] [Google Scholar]
- Enami M, Enami K. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J. Virol. 1996;70:6653–7. doi: 10.1128/jvi.70.10.6653-6657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten W, Klenk HD. Understanding influenza virus pathogenicity. Trends Microbiol. 1999;7:99–100. doi: 10.1016/s0966-842x(99)01460-2. [DOI] [PubMed] [Google Scholar]
- Glaser L, Stevens J, Zamarin D, Wilson IA, Garcia-Sastre A, Tumpey TM, Basler CF, Taubenberger JK, Palese P. A single amino acid substitution in the 1918 influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 2005;79:11533–6. doi: 10.1128/JVI.79.17.11533-11536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Kawaoka Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. U.S.A. 1998;95:10224–8. doi: 10.1073/pnas.95.17.10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HH, Palese P, Shaw ML. Modulation of influenza virus replication by alteration of sodium ion transport and protein kinase C activity. Antiviral Res. 2008;80:124–34. doi: 10.1016/j.antiviral.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horimoto T, Kawaoka Y. Pandemic threat posed by avian influenza A viruses. Clin. Microbiol. Rev. 2001;14:129–49. doi: 10.1128/CMR.14.1.129-149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 2001;75:5205–14. doi: 10.1128/JVI.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk HD, Rott R, Orlich M, Blodorn J. Activation of influenza A viruses by trypsin treatment. Virology. 1975;68:426–39. doi: 10.1016/0042-6822(75)90284-6. [DOI] [PubMed] [Google Scholar]
- Lazarowitz SG, Compans RW, Choppin PW. Proteolytic cleavage of the hemagglutinin polypeptide of influenza virus: function of the uncleaved polypeptide HA. Virology. 1973;52:199–212. doi: 10.1016/0042-6822(73)90409-1. [DOI] [PubMed] [Google Scholar]
- Manicassamy B, Rong L. Expression of Ebolavirus glycoprotein on the target cells enhances viral entry. Virol. J. 2009;6:75. doi: 10.1186/1743-422X-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U.S.A. 1999;96:9345–50. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P, Shaw ML. Orthomyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5th edition Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1647–89. [Google Scholar]
- Smith DJ, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305:371–6. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Stech J, Garn H, Wegmann M, Wagner R, Klenk HD. A new approach to an influenza live vaccine: modification of the cleavage site of hemagglutinin. Nat. Med. 2005;1:683–9. doi: 10.1038/nm1256. [DOI] [PubMed] [Google Scholar]
- Steinhauer DA. Role of hemagglutinin cleavage for the pathogencity of influenza virus. Virology. 1999;258:1–20. doi: 10.1006/viro.1999.9716. [DOI] [PubMed] [Google Scholar]
- Timmins J, Scianimanico S, Schoehn G, Weissenhorn W. Vesicular release of Ebola virus matrix protein VP40. Virology. 2001;283:1–6. doi: 10.1006/viro.2001.0860. [DOI] [PubMed] [Google Scholar]
- Tumpey TM, Garcia-Sastre A, Mikulasova A, Taubenberger JK, Swayne DE, Palese P, Basler CF. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13849–54. doi: 10.1073/pnas.212519699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webby RJ, Webster RG. Are we ready for pandemic influenza? Science. 2003;302:1519–22. doi: 10.1126/science.1090350. [DOI] [PubMed] [Google Scholar]
- World Health Organization Collaborating Centers for Reference and Research on Influenza . In: Concepts and Procedures for Laboratory-Based Influenza Surveillance. Kendal AP, Skehel JJ, Pereira MS, editors. Centers for Disease Control and Prevention; 1982. pp. B17–B35. [Google Scholar]
- Yonezawa A, Cavrois M, Greene WC. Studies of Ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J. Virol. 2005;79:918–26. doi: 10.1128/JVI.79.2.918-926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Pekosz A, Lamb RA. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 2000;74:4634–44. doi: 10.1128/jvi.74.10.4634-4644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]