

Abstract
Purpose
Nepafenac is a potent NSAID that rapidly penetrates the eye following topical ocular administration. In the eye, nepafenac is converted to amfenac, which has unique time-dependent inhibitory properties for COX-1 and COX-2. The purpose of the present study was to investigate the ability of amfenac to inhibit discrete aspects of the angiogenic cascade in vitro, and to test the efficacy of amfenac and nepafenac in vivo, using the rat OIR model.
Methods
Müller cells were treated with amfenac, celecoxib (COX-2), or SC-560 (COX-1), and hypoxia-induced VEGF and PGE2 assessed. Endothelial cells were treated with amfenac, celecoxib, or SC-560, and VEGF-induced proliferation and tube formation assessed. Rat pups were subjected to OIR, received intravitreal injections of amfenac, celecoxib, or SC-560, and neovascularization (NV), prostanoid production, and VEGF assessed. Other OIR-exposed pups were treated with topical nepafenac, ketorolac, or diclofenac, and inhibition of NV assessed.
Results
Amfenac treatment failed to inhibit hypoxia-induced VEGF production. Amfenac treatment significantly inhibited VEGF-induced tube formation and proliferation by EC. Amfenac treatment significantly reduced retinal prostanoid production and NV in OIR. Nepafenac treatment significantly reduced retinal NV in OIR; ketorolac and diclofenac had no effect.
Conclusions
Nepafenac and amfenac inhibit OIR more effectively than the commercially available topical and injectable NSAIDs used in this study. Our data suggests there are COX-dependent and COX-independent mechanisms by which amfenac inhibits OIR. Because it is bioavailable to the posterior segment following topical delivery, nepafenac appears to be a promising advancement in the development of therapies for neovascular eye diseases.
Keywords: Retinal angiogenesis, Müller cells, endothelial cells, oxygen-induced retinopathy, COX-2, NSAID, therapeutics
Introduction
Pathological ocular angiogenesis, or ocular neovascularization (NV), is a pivotal pathologic feature of several prevalent, sight-threatening eye diseases. In developed countries, retinopathy of prematurity (ROP), proliferative diabetic retinopathy (PDR), and age-related macular degeneration (AMD) are the leading causes of irreversible blindness in infants, working-age adults, and the elderly, respectively.1–3 Clinical and experimental evidence suggests that ischemia-induced hypoxia is a central etiological factor in retinal NV.4,5 For example, several retinal cell types respond to hypoxia by up-regulating production of vascular endothelial growth factor (VEGF), the principal growth factor promoting retinal NV.6–8 Among these retinal cells, Müller cells exhibit the most consistent and dramatic increase in VEGF synthesis and secretion when subjected to experimental hypoxia.7–9 VEGF binds with high affinity to VEGF receptors (VEGFR-1 and VEGFR-2) expressed on the surface of endothelial cells, initiating signal transduction cascades that lead to angiogenic endothelial cell behaviors.10–13
Cyclooxygenase (COX) enzymes are responsible for the biosynthesis of prostanoids [prostaglandins (PG) and thromboxanes] from arachidonic acid. Studies suggest that COX-1, the constitutively active isoform of COX, plays a role in angiogenic cell behaviors and carcinogenesis.14–19 Additionally, evidence suggests that the inducible isoform of COX, COX-2, plays a key role in regulating angiogenesis through the induction of prostanoid synthesis. Prostanoids subsequently induce the expression of pro-angiogenic factors such as VEGF and bFGF in many cell types,20–21 and several prostanoids have been shown to induce angiogenesis in in vitro and in vivo assays of human angiogenesis and cancer.22–26 A subset of prostanoids, under some conditions, have been shown to be deleterious to the retinal vasculature in ways other than promoting growth factor production. Prostanoid levels are higher in the retinas of infants than in the retinas of adults.27,28 Prostanoids are involved in maintaining retinal and choroidal blood flow.29,30 Specifically, the infant’s retinal prostanoid complement, coupled with their age-dependent responses to the prostanoids, leads to increased retinal vascular relaxation and dilation.28,31–32 This effect is particularly harmful to premature infants on oxygen therapy who do not yet have the ability to auto-regulate retinal and choroidal blood flow; prostanoids serve to enhance oxygen delivery to already-saturated retinal tissue, which is known to worsen the pathology of ROP.29,33 COX-2-dependent production of TXA2 can lead to endothelial cell cytotoxicity, worsening the retinal microvascular degeneration in ischemic retinopathies.34,35 Prostanoid signaling through the EP3, EP4, DP, TP, and IP receptor have all been implicated in mediating discrete cell behaviors that are implicated in the development or pathology of ischemic retinopathies.36–40 Selective inhibition of COX-2 also prevents pathological angiogenesis in the cornea, retina, and experimentally-induced tumors.41–45 Therefore, non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit the activity of the COX enzymes may be viable pharmacologic agents for the treatment of retinal neovascularization (NV).
In 2005, the U.S. Food and Drug Administration (FDA) approved the topical NSAID, NEVANAC® (nepafenac; 0.1% ophthalmic suspension), for the treatment of pain and inflammation associated with cataract surgery.46–49 The active ingredient in NEVANAC® is nepafenac, a potent, reversible COX-1 and COX-2 inhibitor (Kulmacz RJ, et al. 2007: EVER E-Abstract e473). Nepafenac is a pro-drug with superior penetration of cornea and scleral tissues.50 It is quickly metabolized in vivo by amidases in the iris/cilliary body and retina/choroid to form amfenac.46 Amfenac is an NSAID with antipyretic and analgesic properties, and it inhibits both COX-1 and COX-2 activity.47 Amfenac, like nepafenac, is a reversible inhibitor of both COX-1 and COX-2, but unlike nepafenac, amfenac has unique time-dependent inhibitory properties for both COX-1 and COX-2, implying that with time, amfenac irreversibly binds the enzymes, accounting for amfenac’s prolonged activity (Kulmacz RJ, et al. 2007: EVER E-Abstract e473).
Topical ocular administration of nepafenac inhibits posterior segment NV in mouse models of oxygen-induced retinopathy (OIR) and laser-induced choroidal NV (LCNV), and it inhibits the functional abnormalities and retinal vasculopathy observed in rats with streptozotocin-induced diabetes.51,52 Topical ocular administration of nepafenac reduced retinal VEGF expression in the mouse model of OIR.51 This observation is similar to the reported findings demonstrating the anti-VEGF effects of COX-2 inhibitors in tumor angiogenesis models.53
In order to better understand its bioactivity, we used in vitro assays of angiogenic cell behaviors to determine the capacity of amfenac to inhibit discrete aspects of the angiogenic cascade in the retina. We evaluated the effect of amfenac on hypoxia-induced VEGF production by Müller cells. Then, we looked at the effect of amfenac on VEGF-induced angiogenic cell behaviors in retinal endothelial cells. To further investigate the therapeutic potential of nepafenac for human use, we tested the efficacy of amfenac and nepafenac in vivo, using the rat model of OIR developed in our laboratory. This model produces a pattern of pathological pre-retinal NV mimicking that of premature infants with ROP.54 The results of these studies more fully define the mechanism(s) by which nepafenac mediates its anti-angiogenic effect, as well as demonstrate where COX enzymes appear to exert their influence during pathologic retinal angiogenesis.
Materials and Methods
Materials
Nepafenac (NEVANAC®, 0.1% ophthalmic solution), amfenac, and vehicle were synthesized and provided by Alcon Laboratories, Inc. Ketorolac tromethamine (Acular®, 0.5% ophthalmic solution; Allergan, Inc.), diclofenac sodium (Voltaren®, 0.1% ophthalmic solution; Novartis), and celecoxib (Celebrex®; Pfizer) were obtained from commercial sources. SC-560 was purchased from Cayman Chemical (Ann Arbor, MI).
In Vitro Methods
Isolation and Culture of Primary Rat Retinal Müller Cells
Primary rat retinal Müller cell cultures were established from postnatal day (P)7 Long Evans rat pups according to well-established methods.55 Briefly, enucleated eyes were placed in soaking medium, Dulbecco’s Modified Eagle Medium Low Glucose (DMEM; HyClone; Logan, UT) supplemented with 1X Antibiotic/Antimycotic Solution (Sigma; St. Louis, MO), overnight. The following day, eyes were incubated in digestion buffer, comprised of the soaking medium plus 0.1% trypsin and 70 U/ml collagenase, for 60 minutes at 37°C. Retinas were then dissected, triturated, plated, and grown in DMEM supplemented with 10% fetal bovine serum and 1X Antibiotic/Antimycotic Solution. Cultures were maintained at 37°C in a 5% CO2/95% air (20.9% oxygen) atmosphere (normoxia) in a humidified incubator (NuAire; Plymouth, MN). Müller cells were identified by immunocytochemical staining for cellular retinaldehyde binding protein (CRALBP; Abcam; Cambridge, MA). Passages three to six were used for experiments. For treatment of Müller cells with hypoxia, a CO2-enriched environment was generated with a BBL™ GasPak Pouch system (Becton-Dickinson; Sparks, MD).
Quantitative Real Time RT-PCR of VEGF in Rat Müller Cells
Primary rat Müller cells were seeded in 10-cm Petri dishes at equal density and maintained in normoxia. At 80% confluency, the cells were treated with vehicle (0.1% DMSO) or increasing concentrations of amfenac (0.1 to 10 μM) and placed in hypoxia for 24 hours. Total RNA was isolated from the cells using Trizol reagent (Invitrogen Corporation; Carlsbad, CA). Each RNA sample was quality-controlled for DNA and protein contamination. For VEGF amplification, cDNAs were reverse transcribed using the High-Capacity cDNA Archive Kit (Applied Biosystems; Foster City, CA) according to manufacturer’s instructions. Quantitative real-time RT-PCR was performed in duplicate by co-amplification of rat VEGF vs. β-actin (endogenous normalization control) in separate tubes, using gene-specific TaqMan Gene Expression Assays according to the manufacturer’s instructions (Applied Biosystems; primer and probe sequences used in this assay are proprietary).
Quantification of Rat Müller Cell-derived VEGF and PGE2 Levels
Primary rat Müller cells were seeded in 12-well plates at equal density and maintained in normoxia. At 80% confluency, cells were treated with vehicle (0.1% DMSO) or 10 μM amfenac, celecoxib, or SC-560, and then maintained in normoxia or hypoxia for 24 hours. Culture medium from cells was collected and assayed for VEGF and PGE2 concentration with colorimetric sandwich ELISA kits (R&D Systems; Minneapolis, MN) according to the manufacturer’s instructions. Cells were washed with CMF-PBS (Invitrogen), lysed with cold lysis buffer (Promega; Madison, WI), and protein concentration was determined with the bicinchoninic acid assay (BCA; Pierce; Rockford, IL). The amount of VEGF and PGE2 (pg/ml) in the culture medium was normalized to total protein concentration (mg/ml) of cell lysates.
Culture of Human Retinal Microvascular Endothelial Cells (HRMEC)
Primary human retinal microvascular endothelial cells (HRMEC; Cell Systems; Kirkland, WA) were seeded in tissue culture flasks coated with attachment factor (Cell Signaling; Danvers, MA) and cultured with endothelial basal medium (EBM; Cambrex; East Rutherford, NJ) supplemented with 10% FBS and EGM single quots (Cambrex). When experimental conditions required serum free (SF) medium, MCDB 131 medium (Sigma) containing 1X Antibiotic/Antimycotic Solution was used. Cultures were maintained at 37°C in a 5% CO2/95% air (20.9% oxygen) atmosphere (normoxia) in a humidified incubator.
HRMEC Tube Formation Assay
In vitro tube formation by HRMEC was carried out in 12-well plates coated with growth factor-reduced Matrigel® matrix (Becton-Dickinson). HRMEC were seeded at 3×104 cells per Matrigel-coated well in complete culture medium. After 4.5 hours, the culture medium was removed and the cells were treated with SF medium alone or SF medium containing 25 ng/ml VEGF (R&D Systems) in the presence or absence of amfenac, celecoxib, or SC-560 (0.01 to 1 μM). Twenty-four hours later, three images of tubes per well were captured using a DMC digitizing camera (Polaroid; Cambridge, MA) mounted on an IMT-2 inverted microscope (Olympus; Melville, NY). Capillary-like structures were measured using Image J software (NIH; Bethesda, MD), and the mean tube length per area of the field was calculated for each well.
HRMEC Cell Proliferation Assay
VEGF-induced HRMEC proliferation was measured using a modified MTT assay. Each well of a 96-well plate was coated with a fibronectin/hyaluronic acid (HA) matrix and seeded with 3×104 cells. Complete medium was added and the cells were incubated for two days. The medium was then aspirated, and the cells were incubated with SF medium overnight. The following day, culture medium was removed and the cells were treated with SF medium alone or SF medium containing 25 ng/ml VEGF (R&D Systems) in the presence or absence of amfenac, celecoxib, or SC-560 (0.01 to 10 μM). Twenty-four hours later, 25 μL of a 5 mg/ml solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Molecular Probes; Eugene, OR) was added to each well and incubated for 4 hours under normal growth conditions. One hundred microliters of lysis buffer (20% SDS in 50:50 dimethylformamide (DMF) and H2O with 2.0% acetic acid and 0.05% HCl) was then added to each well, and the plates were incubated overnight at 37°C and read (Spectramax 190; Molecular Devices; Sunnyvale, CA) at 570 nm. Absorbance values were translated to cell number using standard curves consisting of six cell densities assayed in quadruplicate. The data obtained from the MTT assay and cell counts using a hemocytometer in the presence of trypan blue (Sigma) were found to be highly correlated (r2=0.933, data not shown). A standard curve of absorbance at 570 nm vs. HRMEC number was then produced.
In Vivo Methods
Oxygen-induced Retinopathy (OIR) in the Rat
All animal procedures used in this study were approved by the Vanderbilt University Institutional Animal Care and Use Committee and were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Litters of Sprague-Dawley rat pups and their mothers (Charles River Laboratories; Wilmington, MA) were transferred within four hours after birth to oxygen exposure chambers where they were subjected to alternating 24 hour periods of 50% oxygen and 10% oxygen for 14 days. Control rats were raised simultaneously in room air. On postnatal day (P)14, the oxygen-exposed rats were returned to room air.
Quantification of Retinal Prostanoids
On P14, following removal from the oxygen chambers, rats were administered amfenac (0.05 μg; 40 μM) or vehicle by single intravitreal injection, according to a well-established procedure.56 One day later, on P15, retinas were harvested and homogenized. The lipid soluble prostaglandin compounds were extracted with a Sep-Pak C18 column (Waters; Milford, MA) and were nitrogen-evaporated. O-methoxyamine derivatives were formed by treatment with 2% methoxyamine-HCl in water at room temperature for 30 minutes. Compounds were extracted with ethyl acetate and subsequently converted to pentaflurobenzyl esters. The compounds were chromatographed on TLC plates with ethyl acetate/methanol. The compounds were then converted to trimethylsilyl ether derivatives and analyzed by negative ion chemical ionization mass spectrometry coupled with a gas chromatography system (Agilent Technologies; Palo Alto, CA).
Quantification of Retinal VEGF Levels
On P14, following removal from the oxygen chambers, rats were administered amfenac (0.05 μg; 40 μM) or vehicle by a single intravitreal injection, according to a well-established procedure.56 Because there is a peak in retinal VEGF two days post-oxygen exposure in this model,57 rats were sacrificed on P16 and retinas were harvested and subjected to lysis by homogenization. The total protein concentration of samples was measured by BCA. Retinal VEGF levels were measured with a VEGF colorimetric sandwich ELISA kit (R&D Systems) according to the manufacturer’s instructions. The final mass of retinal VEGF was standardized to total retinal protein.
Quantification of Retinal Neovascularization (NV)
Using commercially available formulations and drop-tainers, nepafenac (0.03%, 0.1%), ketorolac (0.5%), diclofenac (0.1%) or vehicle was dropped directly onto the cornea two or four times a day, depending upon experiment. Topical dosing was performed between P14 and P19. A separate group of oxygen-exposed rat pups received a single intravitreal injection of amfenac (0.05 μg; 40 μM), celecoxib (0.075 μg; 40 μM), SC-560 (0.07 μg; 40 μM), or vehicle (0.1% DMSO) at P14, after return to room air. Our estimations of vitreous volume indicate that these concentrations of injected NSAIDs lead to vitreous concentrations that fall within the middle range of the concentrations used for in vitro assays. Regardless of pharmacologic treatment, all oxygen-exposed rats were sacrificed on P20, 6 days following return to room air. The eyes were enucleated, and retinas were dissected and placed in 10% neutral buffered formalin [CMF-PBS (Invitrogen) with 37% formaldehyde solution (Fisher Scientific; Fair Lawn, NJ)] overnight at 4°C. The retinal vasculature was stained for adenosine diphosphatase (ADPase) activity, according to well-established procedures.58 Images of ADPase-stained retinas were digitized, captured, and displayed at 20X magnification. The total retinal area and the retinal area containing vasculature were independently measured. For each retinal image, pre-retinal vessel tufts were outlined, the pixels within an encircled area were counted, and the total number of pixels from all areas were summed and converted to square millimeters.
Statistical Analysis
Data were analyzed with commercial software (JMP; SAS Institute; Cary, NC). Analysis of variance (ANOVA) with appropriate post-hoc analyses were used to analyze data.
Results
Intravitreally-injected NSAID Efficacy in Rat OIR
The effect of 0.05 μg amfenac on OIR-induced retinal NV was compared to two other NSAIDs, 0.075 μg celecoxib (COX-2 inhibitor) and 0.07 μg SC-560 (COX-1 inhibitor). This concentration of amfenac (40 μM) was empirically chosen using the rat model of OIR; the concentrations of celecoxib and SC-560 were matched to this concentration, to standardize treatment. Because amfenac does not possess the tissue-penetration characteristics of its pro-drug, nepafenac, and because celecoxib and SC-560 are not topically formulated, they were delivered directly to the target tissue with a single intravitreal injection. Oxygen-exposed rats received a single intravitreal injection of amfenac, celecoxib, or SC-560 on P14 and were sacrificed on P20. Amfenac significantly (p ≤ 0.005) reduced the mean area of pre-retinal NV, compared to vehicle-treated eyes (Figure 1). Celecoxib and SC-560 failed to inhibit OIR-induced retinal NV at the doses tested.
Figure 1.
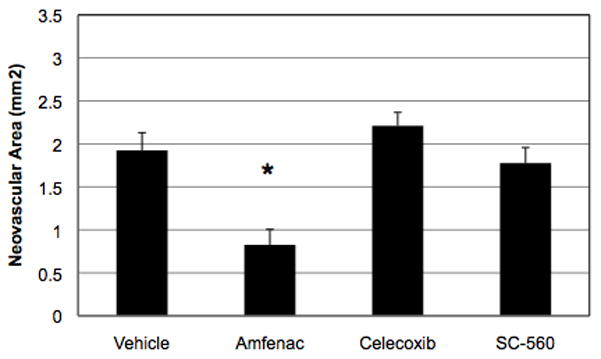
The effect of intravitreally-injected amfenac (40 μM), celecoxib, and SC-560 on the severity of OIR in the rat. Amfenac significantly reduced (* p ≤ 0.005) OIR-induced retinal NV, but neither celecoxib nor SC-560 demonstrated an effect at the concentrations tested. Each bar represents the mean ± SEM.
Effect of OIR on Retinal Prostanoids with and without Amfenac
Amfenac effectively inhibited NV in the rat OIR model, in contrast to celecoxib and SC-560. Thus, we sought to determine, more specifically, the way(s) in which the bioactive metabolite of nepafenac, amfenac, inhibited pathological angiogenesis. The effects of the OIR model and amfenac treatment on retinal prostanoid levels were surveyed. On P14, oxygen-exposed rats received a single intravitreal injection of vehicle or amfenac (0.05 μg; 40 μM). One day later, on P15, the retinas were harvested and retinal prostanoid levels were measured. Compared to room air control retinas, the retinas of oxygen-exposed rats demonstrated increased levels of each of the five prostanoids. Intravitreal amfenac treatment significantly reduced levels of PGE2, PGF2, TxB2, and 6-keto-PGF (p ≤ 0.001) (Figure 2). This data demonstrates that amfenac inhibits COX and prostanoid production as expected, suggesting a possible explanation for the observed anti-angiogenic effect of amfenac in Figure 1.
Figure 2.
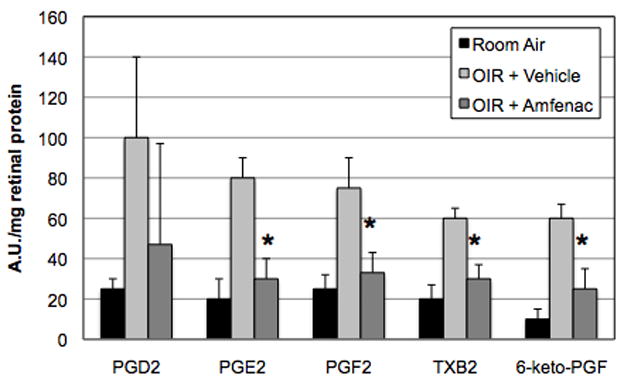
The effect of OIR and amfenac on retinal prostanoid levels. Compared to room air control retinas, the retinas of OIR rats demonstrated increased levels of each of the five prostanoids. Intravitreal amfenac treatment (40 μM) significantly reduced levels of PGE2, PGF2, TxB2, and 6-keto-PGF (* p ≤ 0.001) in OIR rat retinas. Each bar represents the mean ± SD.
Effect of Amfenac on Rat Müller Cell VEGF Expression
Since amfenac decreased retinal prostanoid levels and reduced NV in oxygen-exposed rats, the effect of amfenac on specific angiogenic cell behaviors was studied using in vitro methods. In order to determine whether or not amfenac inhibited hypoxia-induced VEGF production, rat Müller cells were treated with increasing doses of amfenac (0.1 to 10 μM), and placed in hypoxia for 24 hours. Quantitative RT-PCR analysis of VEGF revealed that amfenac exhibited no effect on hypoxia-induced VEGF mRNA expression in rat Müller cells (Figure 3).
Figure 3.
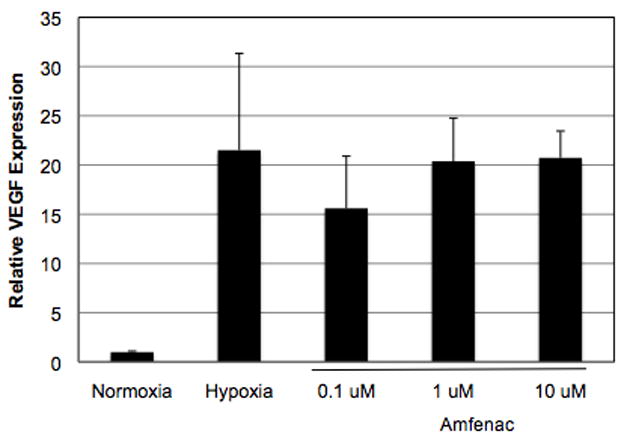
The effect of amfenac on hypoxia-induced VEGF expression in Müller cells. Quantitative RT-PCR analysis of VEGF revealed that amfenac exhibited no effect on hypoxia-induced VEGF mRNA expression in rat retinal Müller cells. Each bar represents the mean ± SD.
Effect of NSAIDs on VEGF and PGE2 Production in Rat Müller cells
It is important to note that a change in VEGF mRNA does not always correlate with a change in VEGF protein (Wang FE, et al. IOVS 2004;45:ARVO E-Abstract 3711). Because the production, secretion, and turnover of VEGF protein directly contributes to the pathology observed in the rat OIR model, we determined the effect of amfenac treatment on VEGF protein in hypoxic rat Müller cells. Rat Müller cells were treated with 1 μM amfenac, celecoxib, or SC-560 and placed in hypoxia for 24 hours. Amfenac, celecoxib, and SC-560 had no significant effect on hypoxia-induced VEGF production in rat Müller cells (Figure 4A). However, amfenac, celecoxib, and SC-560 treatment profoundly and significantly reduced PGE2 levels in these cells (p ≤ 0.001), implying that hypoxia-induced VEGF expression in rat Müller cells is not affected by pharmacologic manipulation of the COX-2 enzyme (Figure 4B). Since inhibition of COX did not reduce pro-angiogenic VEGF production in rat Müller cells, it cannot explain the inhibition of retinal NV by amfenac.
Figure 4.
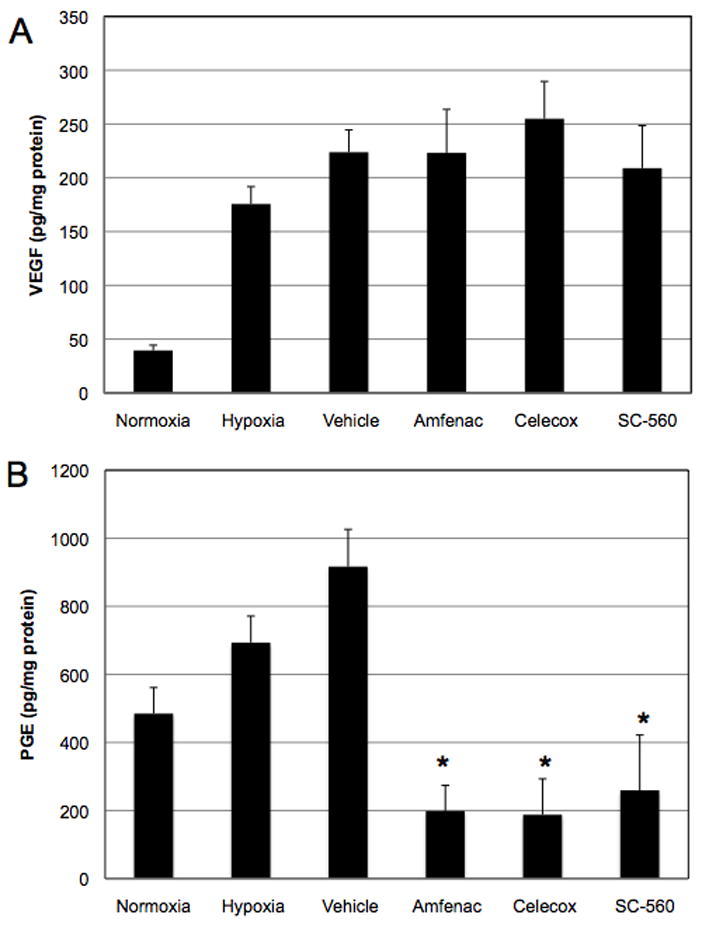
A and B. The effect of amfenac, celecoxib, and SC-560 on Müller cell production of VEGF and PGE2. (A) 1 μM amfenac, celecoxib, and SC-560 had no effect on hypoxia-induced VEGF production in rat retinal Müller cells. (B) However, amfenac, celecoxib, and SC-560 treatment significantly reduced PGE2 levels in these cells (* p ≤ 0.001). Each bar represents the mean ± SD.
Effect of Amfenac on Retinal VEGF Production
To complement our in vitro data (Figure 4A), we returned to the OIR model in order to determine whether amfenac inhibited retina-wide, as opposed to Müller cell-derived, VEGF production. Amfenac (0.05 μg; 40 μM) was administered by a single intravitreal injection to oxygen-exposed rats upon return to room air. Two days later, retinas were harvested and retinal VEGF levels were measured. As expected, oxygen-exposed rats experienced a 4-fold increase in retinal VEGF compared to room air controls (Figure 5). However, and in agreement with our in vitro findings, amfenac treatment demonstrated no significant effect on retinal VEGF in oxygen-exposed rats. Although a modest (20%) reduction in mean VEGF level was observed following amfenac treatment, this result was not statistically significant. Retinal VEGF levels following topical nepafenac treatment were also assayed to determine whether the natural in vivo metabolism of amfenac was required in order to achieve a VEGF response, and again no effect was observed (data not shown).
Figure 5.
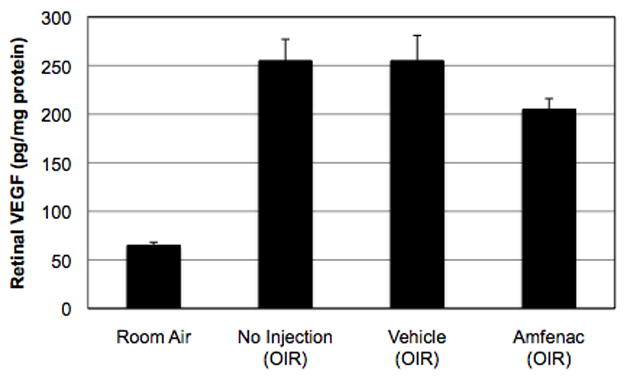
The effect of intravitreally-injected amfenac on retinal VEGF production. In agreement with the in vitro findings, 40 μM amfenac treatment demonstrated no significant effect on retinal VEGF levels in OIR rats. Each bar represents the mean ± SEM.
Effect of Amfenac on VEGF-induced HRMEC Behaviors
Because nepafenac proved ineffective in preliminary in vitro assays, we used its bioactive metabolite, amfenac, to determine the effect of the drug on angiogenic endothelial cell behaviors. The effects of amfenac, celecoxib and SC-560 on VEGF-induced tube formation and proliferation were examined. VEGF-induced (25 ng/ml) HRMEC tube formation (as determined by mean tube length) was significantly inhibited by amfenac (p ≤ 0.001), the COX-2-selective celecoxib (* p ≤ 0.001; † p ≤ 0.006), and, at higher concentrations, the COX-1 selective SC-560 (* p ≤ 0.001; ‡ p ≤ 0.01) (Figure 6). Amfenac also lead to a significant reduction (32.5%; 10.00 ± 2.12 in VEGF-treated HRMEC vs. 6.75 ± 2.45 in 0.01 μM amfenac-treated HRMEC) in the number of HRMEC branch points in this assay (p ≤ 0.0233; data not shown). VEGF-induced HRMEC proliferation was significantly inhibited by amfenac and celecoxib (p ≤ 0.001, respectively) in a dose-dependent manner, whereas SC-560 (p ≤ 0.001) was only inhibitory at the highest concentration tested (Figure 7). Although 10 μM SC-560 significantly inhibited VEGF-induced HRMEC proliferation, this concentration is known to inhibit both COX-1 and COX-2, and to exert COX-independent effects. These experiments suggest that amfenac, likely through COX inhibition, affects discrete aspects of the angiogenic cascade downstream of VEGFR-2 activation.
Figure 6.
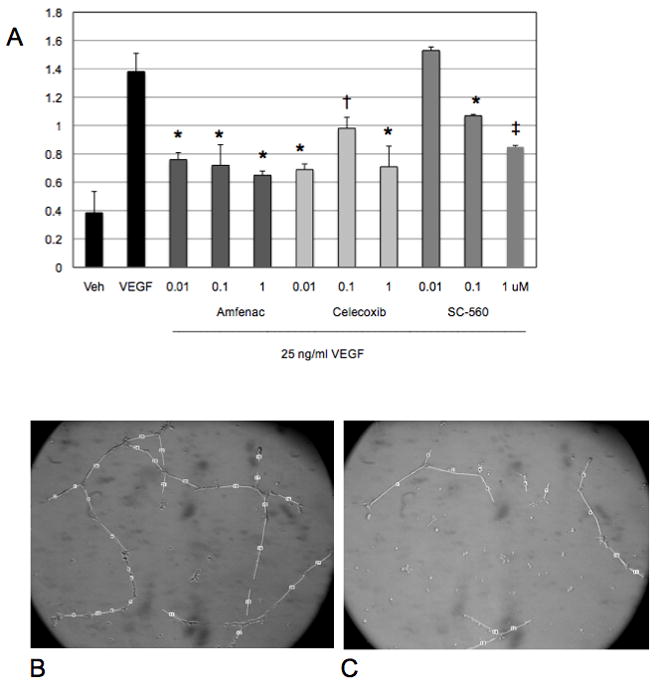
A–C. The effect of amfenac, celecoxib, and SC-560 on VEGF-induced HRMEC tube formation. (A) HRMEC tube formation was induced by 25 ng/ml VEGF, and this induction was significantly inhibited by amfenac (* p ≤ 0.001), the COX-2-selective NSAID celecoxib (* p ≤ 0.001; † p ≤ 0.006), and, at higher concentrations, the COX-1 selective NSAID SC-560 (* p ≤ 0.001; ‡ p ≤ 0.01). Each bar represents the mean ± SD. (B) A representative image of tube formation in VEGF-stimulated HRMEC. (C) A representative image of tube formation in VEGF-stimulated HRMEC treated with 1 μM amfenac. Amfenac-treated HRMEC demonstrate reduced tube formation.
Figure 7.
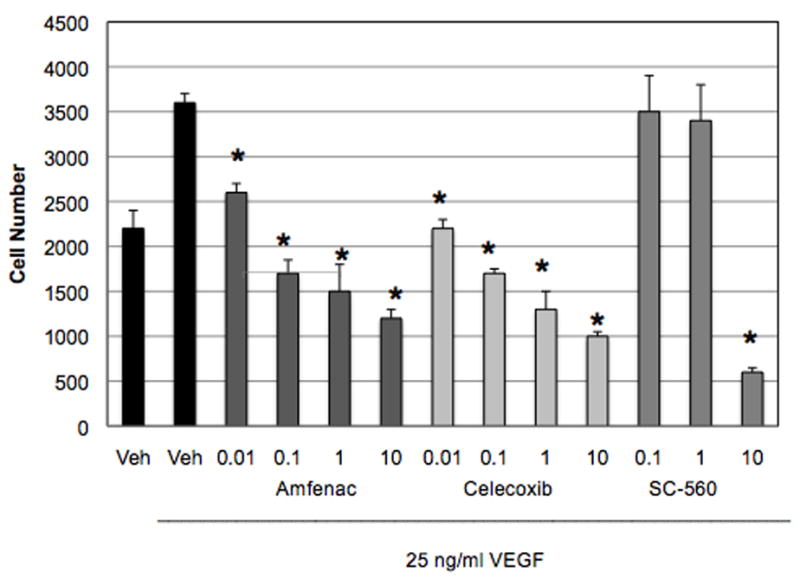
The effect of amfenac, celecoxib, and SC-560 on VEGF-induced HRMEC proliferation. VEGF-induced HRMEC proliferation was significantly inhibited by amfenac and celecoxib (* p ≤ 0.001) in a dose-dependent manner, and by SC-560 (* p ≤ 0.001) at the highest concentration tested. The effect of SC-560 at the highest concentration may be attributed to lethality. Each bar represents the mean ± SD.
Topical Nepafenac Efficacy in Rat OIR
Topical administration of a drug that has the capacity to substantially reduce retinal NV would be a promising advancement in the development of therapies for neovascular eye diseases. Thus, we tested the capacity of topical nepafenac to inhibit retinal NV in the rat model of OIR. Oxygen-exposed rats were treated with topical nepafenac four times daily (QID) or twice daily (BID) from P14 through P19 and were sacrificed on P20. Nepafenac (0.1%) delivered QID or BID significantly reduced (p ≤ 0.001) the amount of pre-retinal NV in an apparent dose-dependent manner (Figure 8). Like nepafenac, ketorolac and diclofenac are labeled for the treatment of pain and inflammation following cataract surgery. We compared the effect of topical nepafenac (0.1%, QID) to these commercially available, topically formulated NSAIDs, ketorolac (0.5%, QID) and diclofenac (0.1%, QID), on OIR-induced retinal NV. Nepafenac significantly reduced the mean area of pre-retinal NV by 59.3% (p ≤ 0.007), but neither ketorolac nor diclofenac demonstrated an effect at the tested doses (Figure 9).
Figure 8.
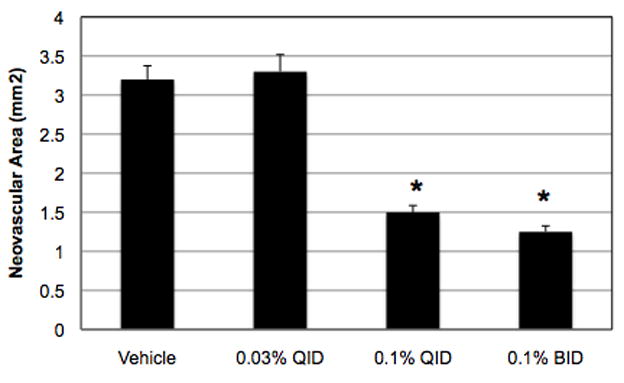
The effect of topical nepafenac on the severity of OIR in the rat. 0.1% nepafenac, given QID or BID from P14-P19, significantly reduced (* p ≤ 0.001) OIR-induced retinal NV. Each bar represents the mean ± SEM.
Figure 9.
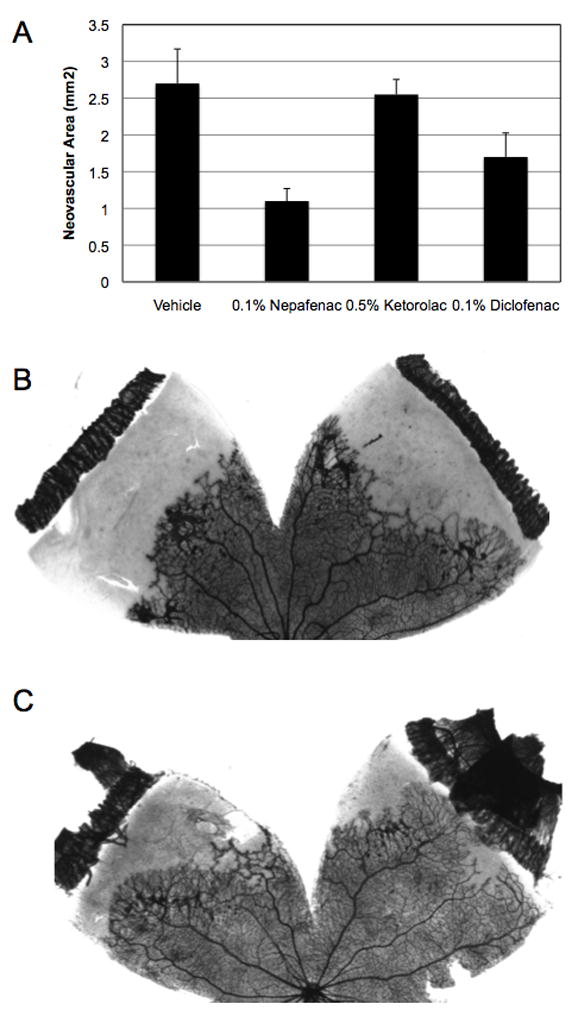
A–C. The effect of topical nepafenac, ketorolac, and diclofenac on the severity of OIR in the rat. (A) Drugs were administered topically, QID, from P14-P19. Nepafenac significantly reduced (* p ≤ 0.007) OIR-induced retinal NV. Ketorolac and diclofenac failed to demonstrate an effect. Each bar represents the mean ± SEM. (B) A representative image of NV in vehicle-treated eyes. (C) A representative image of NV in nepafenac-treated eyes. As demonstrated by representative ADPase-stained retinal flat mounts, nepafenac significantly reduced retinal NV.
Discussion
The goal of this study was two-fold. First, we used three in vitro assays to determine the capacity of amfenac to inhibit discrete aspects of retinal angiogenesis. Using these model systems, we were better able to determine where in the angiogenic cascade COX isoforms exert their influence. Second, in order to further investigate the therapeutic potential of nepafenac as an angiostatic agent for human ocular use, we tested the efficacy of nepafenac in vivo, using the rat model of OIR developed in our laboratory.
In 2005, the FDA approved nepafenac for the treatment of pain and inflammation associated with cataract surgery.46–49 In the eye, nepafenac is converted to an active metabolite, amfenac, which like nepafenac is a reversible inhibitor of both COX-1 and COX-2, but unlike nepafenac has unique time-dependent inhibitory properties for both COX-1 and COX-2 (Kulmacz RJ, et al. 2007: EVER E-Abstract e473). Thus, we wanted to determine if, and more specifically how, amfenac inhibited pathological angiogenesis. Amfenac inhibits COX activity and COX-dependent prostanoid production. The cancer literature has shown that COX-2 and the prostanoids are involved in the angiogenesis that occurs during tumor growth.25,26,59–61 Moreover, recent studies have shown that COX inhibitors, including topical nepafenac, ameliorate various experimental pathologies in the posterior segment of the eye.45,48,51,52,62–67 We tested the hypothesis that amfenac, by virtue of its capacity to inhibit COX activity, would inhibit pathological angiogenesis. This was done using three NSAIDs with varying selectivities for COX-1 and COX-2. Amfenac is a relatively non-selective NSAID, inhibiting both COX-1 (IC50 = 0.25 μM) and COX-2 (IC50 = 0.15 μM).47 Celecoxib is highly COX-2-selective (COX-2 IC50 = 0.06 μM, COX-1 IC50 = 19 μM).68 SC-560 is highly COX-1-selective (COX-1 IC50 = 0.009 μM, COX-2 IC50 = 6.3 μM).69 Only amfenac inhibited NV in the rat OIR model (Figure 1).
It is possible that amfenac demonstrates superior ocular pharmacokinetics and bioavailability, and/or pharmacodynamic mechanisms than do celecoxib and SC-560. Or, amfenac may exert distinct COX-independent effects that mediate its angiostatic activity. The non-selective nature of amfenac’s COX inhibition may be one possible pharmacodynamic explanation for its superior performance. The importance of inhibiting both COX isoforms during ischemia-induced retinal NV has been suggested by results from studies using COX-1 null and COX-2 null mice.70 Due to its superior performance, we wanted to determine, more specifically, the way(s) in which amfenac inhibited pathological angiogenesis. We surveyed the effects of the oxygen exposure model and amfenac treatment on retinal prostanoid levels in the OIR model (Figure 2). All five of the prostanoids exhibited at least a two-fold increase upon exposure to the OIR protocol, suggesting a potential role in the development of retinal NV. The observed increase in retinal prostanoid production in oxygen-exposed rats could be due to: (1) increased cPLA2 level or activity, which serves to liberate arachidonic acid, the substrate that is converted by COX into prostanoids; (2) increased level or activity of COX-2; (3) increased prostanoid synthase activity; or (4) some combination of these. Regardless of the mechanism by which prostanoids were increased, intravitreal injection of amfenac significantly inhibited the response.
Retinal NV can be studied in vitro by distilling it into two basic components: hypoxia-induced VEGF production by retinal, e.g. Müller, cells and VEGF-induced angiogenic behaviors (proliferation and tube formation) in endothelial cells. We tested the capacity of amfenac to inhibit each of these processes in vitro so that we could more clearly define its mechanism of action in vivo. Amfenac had no effect on hypoxia-induced VEGF expression or production by rat Müller cells (Figure 3 and Figure 4A). This was confirmed in vivo: amfenac did not significantly decrease retinal VEGF levels in OIR rats (Figure 5). These data suggest that amfenac likely does not inhibit retinal NV in the rat model of OIR by reducing hypoxia-induced VEGF production. In accordance with our findings, Kern et al. reported that topically applied nepafenac did not reduce the increased retinal VEGF production found in diabetic rats.52 These results suggest that VEGF inhibition is unlikely to be a major contribution to amfenac’s anti-angiogenic activity. Our results contradict those of Takahashi et al. who showed that topical nepafenac reduced retinal VEGF mRNA in mice exposed to the OIR model.51 The discrepancy between our findings and Takahashi et al.’s findings may be due to: (1) inherent differences between the rat and mouse models of OIR; (2) inherent differences between the two species; or (3) the fact that Takahashi et al. looked at VEGF mRNA, whereas we looked at protein. It is important to note that a change in VEGF mRNA does not always correlate with a change in VEGF protein (Wang FE, et al. IOVS 2004;45:ARVO E-Abstract 3711). Because the production, secretion, and turnover of VEGF protein directly contributes to the pathology observed in the rat OIR model, we chose this endpoint. Point number two brings up an important distinction, because it calls into question the universal capacity of NSAIDs to affect VEGF production.
Since amfenac, a potent COX inhibitor, had no effect on VEGF production in vitro or in vivo, it is unlikely that pharmacologic manipulation of COX-2 affects this process. We tested this hypothesis using three different NSAIDs with varying selectivities for COX-1 and COX-2. The NSAID concentrations used in vitro were chosen because they fall within the range that allows us to distinguish between COX-1 and COX-2 effects. Amfenac, celecoxib, and SC-560 significantly inhibited Müller cell PGE2 production (Figure 4B), indicating that they did, in fact, inhibit COX activity in our cultures. However, the drugs had no effect on hypoxia-induced VEGF production. This demonstrates that hypoxia-induced VEGF production by Müller cells is not diminished by pharmacological inhibition of the COX-2 enzyme, and that the inhibition of pro-angiogenic VEGF production by Müller cells does not appear to be the mechanism by which amfenac inhibits retinal NV. It is possible that COX-dependent prostanoid production may influence VEGF production by other retinal cell types [as Amrite et al. have shown in retinal pigment epithelial (RPE) cells],71 although our in vivo studies suggest that this is not the case in the rat model of OIR (Figure 5). Therefore, COX inhibition by NSAIDs likely influences hypoxia-induced angiogenic cell behavior and OIR by a bioactivity unrelated to VEGF induction.
Although we chose to focus on VEGF, it is possible that COX-dependent prostanoid production influences the production of angiogenic factors other than VEGF. Cheng et al. demonstrated that PGE2 induces bFGF expression in cultured rat Müller cells.20 Others have demonstrated that a different prostanoid, PGF2, induces bFGF expression in rat osteoblasts and endometrial adenocarcinoma explants.72,73 These and other studies demonstrate that there are other angiogenic factors whose production may be prostanoid-dependent, and thus inhibited by amfenac treatment. We did not assess amfenac’s effect on these proteins [namely bFGF, the VEGF receptors, erythropoietin (EPO), adenosine, or insulin-like growth factor (IGF)]. Instead, we chose to look at the effect of amfenac on VEGF-stimulated angiogenic endothelial events because: (1) despite the presence and potential involvement of other, prostanoid-dependent angiogenic factors in the retina, none have been demonstrated to be both necessary and sufficient for the development of retinal NV, as VEGF has; (2) we see increases in VEGF in our model of OIR, but do not see increases in bFGF;74 and (3) HRMEC are exposed and respond to VEGF in human ROP, making it an appropriate means by which to stimulate and manipulate (with amfenac) angiogenic endothelial cell behaviors in vitro.
Next, we sought to determine whether the effect of amfenac on retinal NV was being mediated through the inhibition of VEGF-induced angiogenic behaviors in endothelial cells. VEGF binds and activates high affinity VEGF receptors on retinal endothelial cells.75 Binding of VEGF to VEGFR-2 induces receptor dimerization and tyrosine autophosphorylation, activating complex and incompletely-defined signaling cascades.75 These signal transduction pathways ultimately lead to the induction of various endothelial behaviors necessary for angiogenesis, including proliferation, migration, survival, and the production of nitric oxide that leads to increased permeability. We tested the effect of amfenac, celecoxib, and SC-560 on two of these VEGF-induced behaviors: tube formation and proliferation. Amfenac and celecoxib dose-dependently inhibited both VEGF-induced behaviors. These findings confirm those of Wu et al. who reported that HUVEC demonstrated reduced VEGF-induced proliferation and tube formation when they were treated with NS-398 (a COX-2-selective inhibitor) or with siRNA directed against COX-2.76 In our studies, the COX-1 inhibitor SC-560 was only mildly effective against tube formation alone (Figure 6 and Figure 7). Notably, 10 μM SC-560 significantly inhibited VEGF-induced HRMEC proliferation. This concentration of SC-560 inhibits COX-2 (in addition to inhibiting the COX-1 target enzyme) and exerts COX-independent effects on HRMEC proliferation, suggesting an explanation for its dramatic effect. Notably, amfenac inhibited two measures of tube formation, mean tube length and the number of HRMEC branch points (Figure 6). The effect of COX-2 inhibition on endothelial cell branching has been documented in the literature, and was confirmed by our study.77 Amfenac, likely through inhibition of COX-2, affects discrete aspects of the angiogenic cascade downstream of VEGFR-2 activation. It is known that VEGF-stimulated endothelial cells produce PGs.78 It is also known that PGs stimulate proliferation and tube formation, therefore demonstrating angiogenic effects.79,80 Our data suggests that the capacity of nepafenac to inhibit proliferation and tube formation is dependent on its capacity to inhibit pro-angiogenic PG production by COX-2. This in vitro data suggests that nepafenac’s mechanism of action in ROP is dependent on its capacity to inhibit endothelial cell bioactivities like proliferation and tube formation, two behaviors that are central to the development of pathological ocular NV in ROP.
Safe and effective anti-angiogenic therapies that can be delivered noninvasively remain an unmet need in ophthalmology. Lucentis®, an anti-VEGF antibody fragment (Fab) delivered via intravitreal injection, is the current standard-of-care for neovascular age-related macular degeneration (AMD). During multiple registration studies, intravitreal injections of Lucentis® stabilized vision in over 90% of patients, and improved vision in up to 40% of patients. However, repeated intravitreal injections were necessary for the majority of patients to maintain this level of benefit.81–85 Intravitreal injections require an office visit, are often expensive, can be physically uncomfortable, and they expose the patient to a number of potential vision-threatening complications such as intraocular infection. Topical administration of a drug that has the capacity to substantially reduce retinal NV would be a promising advancement in the development of therapies for neovascular eye diseases. Nepafenac, topically applied to the cornea two or four times daily, significantly inhibited the development of retinal NV in the rat model of OIR (Figure 8). This finding is consistent with those of Takahashi et al. who reported that topical nepafenac inhibited ischemia-induced retinal NV in mice.51 We hypothesized that the anti-angiogenic effect of nepafenac was due to its capacity to inhibit COX and pro-angiogenic prostanoid production. However, it was unexpected that nepafenac proved to be unique in its capacity to significantly inhibit oxygen-induced retinal NV; ketorolac and diclofenac demonstrated no significant effect (Figure 9). This observation cannot be explained by the COX-2 selectivities of the three compounds, because their respective COX-2 IC50’s are within the same range: amfenac = 0.15 μM, ketorolac = 0.086 μM, and diclofenac = 0.038 μM.47,86 A more plausible explanation is that topical nepafenac likely has superior bioavailability to the posterior segment. In early pre-clinical trials, nepafenac exhibited superior corneal penetration and suppressed prostanoid production by the iris/cilliary body and retina/choroid more efficiently and for a longer duration than did diclofenac.46,47 In rabbits, topical administration of 0.1% nepafenac lead to nanomolar concentrations of amfenac in both anterior and posterior segment tissues, were above the COX-2 IC50, indicating sufficient penetration for inhibitory activity (Hariprasad SM, et al. IOVS 2009:50:ARVO E-Abstract 5999). Topical administration of nepafenac provided highest concentrations in the sclera > choroid > retina > vitreous. Pharmacologically relevant concentrations in the posterior segment were achieved through a scleral/choroidal distribution. Together, these data suggest that prostanoid synthesis is an important aspect of oxygen-induced retinal NV and that nepafenac’s inhibitory effect on retinal NV is due, at least in part, to its capacity to efficiently penetrate the cornea/sclera and inhibit COX-dependent prostanoid synthesis in the retina.
The findings that HRMEC treated with amfenac and celecoxib demonstrate reduced VEGF-induced tube formation and proliferation suggest that there are COX-2-dependent mechanisms through which amfenac inhibits oxygen-induced retinal NV. Amfenac and celecoxib may also inhibit VEGF-induced angiogenic cell behaviors through COX-2-independent mechanisms. For example, Amrite et al. reported that choroidal endothelial cells treated with celecoxib demonstrated reduced proliferation, but that the anti-proliferative effect of celecoxib was independent of its COX-2-inhibitory action.87 Nepafenac appears to be a rational therapeutic strategy for the non-invasive treatment of oxygen-induced retinopathies and other neovascular diseases of the eye, and it appears that nepafenac’s mechanism of action is dependent on its capacity to inhibit endothelial cell bioactivities like proliferation and tube formation, two behaviors that are central to the development of pathological ocular NV.
Evidence suggests that oxidative compounds play a role in ROP and other angiogenic diseases of the retina.88–90,52 The retina is particularly susceptible to oxidative damage because it has a high rate of oxygen consumption.91 Furthermore, premature infants have an incompletely developed antioxidant system, leading to a reduced ability to scavenge reactive oxidative species (ROS).92 This may increase their vulnerability to the effects of damaging oxidative species. These findings have led to a large body of basic and clinical research focused on understanding the effect of antioxidant supplementation in ROP. A meta-analysis of clinical studies that tested the effect of vitamin E supplementation on the incidence and severity of ROP development demonstrated that vitamin E supplementation led to a 52% reduction in the development of stage 3 ROP (characterized by NV).93 Vitamin E, superoxide dismutase, and apocynin (an NADPH oxidase inhibitor) have all been shown to prevent the development of pathological features that present in the rat model of ROP.94–97 More recently, Kern et al. have shown that nepafenac demonstrates anti-oxidant activity.52 Nepafenac inhibited diabetes-induced production of superoxide anion (a ROS) in rat retinas. It is known that ROS activates cytosolic phospholipase A2 (cPLA2) and COX-2, which can lead to the production of potentially pro-angiogenic PGs.98,99 cPLA2 is the enzyme responsible for liberating arachidonic acid, a COX substrate, from membrane-derived phospholipids. cPLA2 has been shown to have a pro-angiogenic effect on retinal cell behaviors.100 These may be additional mechanisms of nepafenac’s action: it may prevent the ROS-dependent activation of cPLA2 and/or COX-2 and the resultant PG-induced angiogenic cell behaviors. Although we did not assess nepafenac’s anti-oxidant capacity in our model of ROP, it is feasible that a portion of nepafenac’s mechanism of action may have been related to its capacity to scavenge damaging ROS.
Various stimuli, including COX-derived PGs, stimulate endothelial nitric oxide synthase (eNOS).101 Stimulation of eNOS leads to the production of nitric oxide (NO), a potent signaling molecule. NO plays a role in maintaining blood flow and vascular tone. Increased NO production leads to vasodilation, which can be particularly harmful to the retinas of premature infants on oxygen therapy. In adults, retinal blood flow and choroidal blood flow are tightly regulated; when an adult retina is exposed to hyperoxia, retinal and choroidal blood vessels constrict, limiting excessive oxygen delivery to the retina. This vascular regulation is lacking in infants. Failure of the vasculature to constrict in response to high oxygen, coupled with the vaso-dilatory effect of high NO, means that the infant is particularly sensitive to the deleterious consequences of hyper-oxygenation. This inability to limit oxygen delivery may contribute to the infant’s susceptibility to hyperoxia-induced ROP.102 These findings suggest that inhibiting COX-derived PG production will inhibit deleterious NO production, providing protection against hyperoxia-induced retinopathy. We have shown that amfenac inhibits PG production in vitro and in vivo (Figure 2 and Figure 4). Although we did not assess nepafenac’s capacity to inhibit NO production in our model of ROP, it is possible that a portion of nepafenac’s mechanism of action may have been related to its capacity to prevent PG-mediated NO production and vasodilation. Notably, NO inhibition may also be detrimental to premature infants on supplemental oxygen therapy. eNOS and NO are required for the development of normal lung vasculature. MacRitchie et al. have shown that there is reduced eNOS in the pulmonary circulation and respiratory tract of preterm lambs on oxygen therapy and suggest that reduced eNOS may play a role in the development of chronic lung disease in the lambs.103 Furthermore, eNOS deficient mice exhibit defective lung vascular development and respiratory distress.104 Therefore, if it is found that nepafenac inhibits NO production and has a beneficial effect on the ocular vasculature in models of ROP, it’s effect on the pulmonary vasculature will need to be carefully assessed in clinical trials.
Pharmacologic inhibition of COX-2 did not have a major effect on hypoxia-induced VEGF production in our models. Despite pharmacologic inhibition, residual COX activity may have remained, which could continue to produce pro-angiogenic prostanoids with the capacity to affect VEGF production.20,21 Alternatively, Lukiw et al. reported that hypoxia-induced VEGF production was directly regulated by HIF-1, and only indirectly regulated through NF-κB-mediated COX-2 in choroidal endothelial cells.105 This suggests that HIF-1-dependent VEGF production may have the capacity to overpower the effect of COX-2-inhibition, and could explain the results of our Müller cell studies. In order to more clearly define the role of COX-2 in this process, it is necessary to assess VEGF production in COX-2 knock-out animals and the cells derived from these animals. These studies are on going.
Acknowledgments
Supported by NIH EY07533, NIH EY01826, Alcon Research, Ltd., an Unrestricted Grant from Research to Prevent Blindness, Inc., and a Research to Prevent Blindness Senior Scientific Investigator Award to JSP.
Footnotes
Conflict of Interest Statement
JSP: receives research support from Alcon Research Ltd., and is a paid consultant for Alcon Research Ltd.
DPB: is employed by Alcon Research Ltd.
SEY: no conflict of interest
MLC: no conflict of interest
RY: no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Susan E. Yanni, Email: susan.e.yanni@vanderbilt.edu.
Monika L. Clark, Email: monika8382@yahoo.com.
Rong Yang, Email: rong.yang@vanderbilt.edu.
David P. Bingaman, Email: David.Bingaman@Alconlabs.com.
John S. Penn, Email: john.penn@vanderbilt.edu.
References
- 1.Rahmani B, Tielsch JM, Kat J, et al. The cause-specific prevalence of visual impairment in an urban population. The Baltimore eye survey. Ophthalmology. 1996;103:1721–1726. doi: 10.1016/s0161-6420(96)30435-1. [DOI] [PubMed] [Google Scholar]
- 2.Lee P, Wang CC, Adamis AP. Ocular neovascularization: an epidemiologic review. Surv Opthalmol. 1998;43(3):245–269. doi: 10.1016/s0039-6257(98)00035-6. [DOI] [PubMed] [Google Scholar]
- 3.Steinkuller PG, Du L, Gilbert C, et al. Childhood blindness. J AAPOS. 1999;3:26–32. doi: 10.1016/s1091-8531(99)70091-1. [DOI] [PubMed] [Google Scholar]
- 4.D’Amore PA. Mechanisms of retinal and choroidal angiogenesis. Invest Ophthalmol Vis Sci. 1994;35:3974–3979. [PubMed] [Google Scholar]
- 5.Casey R, Li WW. Factors controlling ocular angiogenesis. Am J Ophthalmol. 1997;124:521–529. doi: 10.1016/s0002-9394(14)70868-2. [DOI] [PubMed] [Google Scholar]
- 6.Aiello LP, Northrup JM, Keyt BA, et al. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalomol. 1995;113:1538–1544. doi: 10.1001/archopht.1995.01100120068012. [DOI] [PubMed] [Google Scholar]
- 7.Pierce EA, Avery RL, Foley ED, et al. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins SG, Conaway JR, Ford BL, et al. Detection of VEGF protein in vascular and non-vascular cells of the normal and oxygen-injured rat retina. Growth Factors. 1997;14:229–241. doi: 10.3109/08977199709021522. [DOI] [PubMed] [Google Scholar]
- 9.Robbins SG, Rajaratnam VS, Penn JS. Evidence for upregulation and redistribution of VEGF receptors flt-1 and flk-1 in the oxygen-injured rat retina. Growth Factors. 1998;16:1–9. doi: 10.3109/08977199809017487. [DOI] [PubMed] [Google Scholar]
- 10.Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortéga N, Hutchings H, Plouët J. Signal relays in the VEGF system. Front Biosci. 1999;4:141–152. doi: 10.2741/A417. [DOI] [PubMed] [Google Scholar]
- 12.Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto T, Claesson-Welsh L. VEGF Receptor Signal Transduction. Sci STKE. 2001;112:RE21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 14.Sano H, Noguchi T, Miyajima A, et al. Anti-angiogenic activity of basic-type, selective cyclooxygenase (COX)-1 inhibitors. Bioorg Med Chem Lett. 2006;16:3068–3072. doi: 10.1016/j.bmcl.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 15.von Rahden BH, Stein HJ, Pühringer F, et al. Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res. 2005;65:5038–5044. doi: 10.1158/0008-5472.CAN-04-1107. [DOI] [PubMed] [Google Scholar]
- 16.Sales KJ, Katz AA, Howard B, et al. Cyclooxygenase-1 is up-regulated in cervical carcinomas: autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res. 2002;62:424–432. [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta RA, Tejada LV, Tong BJ, et al. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003;63:906–911. [PubMed] [Google Scholar]
- 18.Daikoku T, Wang D, Tranguch S, et al. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005;65:3735–3744. doi: 10.1158/0008-5472.CAN-04-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Xu RJ, Lin ZY, et al. Effects of a cyclooxygenase-1-selective inhibitor in a mouse model of ovarian cancer, administered alone or in combination with ibuprofen, a nonselective cyclooxygenase inhibitor. Med Oncol. 2008 doi: 10.1007/s12032-008-9104-9. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20.Cheng T, Cao W, Wen R, et al. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Muller cells. Invest Ophthalmol Vis Sci. 1998;39(3):581–591. [PubMed] [Google Scholar]
- 21.Pai R, Szabo IL, Soreghan BA. PGE2 stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochem Biophys Res Commun. 2001;286(5):923–928. doi: 10.1006/bbrc.2001.5494. [DOI] [PubMed] [Google Scholar]
- 22.Gullino PM. Prostaglandins and gangliosides of tumor microenvironment: their role in angiogenesis. Acta Oncol. 1995;34:439–441. doi: 10.3109/02841869509094005. [DOI] [PubMed] [Google Scholar]
- 23.Daniel TO, et al. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial cell migration and angiogenesis. Cancer Res. 1999;59:4574–4577. [PubMed] [Google Scholar]
- 24.Pradono P, Tazawa R, Maemondo M, et al. Gene transfer of thromboxane A2 synthase and prostaglandin I2 synthase antithetically altered tumor angiogenesis and tumor growth. Cancer Res. 2002;62:63–66. [PubMed] [Google Scholar]
- 25.Ziche M, Jones J, Gullino PM. Role of prostaglandin E1 and copper in angiogenesis. J Natl Cancer Inst. 1982;69:475–482. [PubMed] [Google Scholar]
- 26.Diaz-Flores L, Gutierrez R, Varela H. Angiogenesis: an update. Histol Histopathol. 1994;9(4):807–843. [PubMed] [Google Scholar]
- 27.Hardy P, Bhatthacharya M, Abran D, et al. Increases in retinovascular prostaglandin receptor functions by cyclooxygenase-1 and -2 inhibition. Invest Ophthalmol Vis Sci. 1998;39:1888–1898. [PubMed] [Google Scholar]
- 28.Abran D, Dumont I, Hardy P, et al. Characterization and regulation of prostaglandin E2 receptor and receptor-coupled functions in the choroidal vasculature of the pig during development. Circ Res. 1997;80:463–472. [PubMed] [Google Scholar]
- 29.Chemtob S, Beharry K, Rex J, et al. Ibuprofen enhances retinal and choroidal blood flow autoregulation in newborn piglets. Invest Ophthalmol Vis Sci. 1991;32:1799–1807. [PubMed] [Google Scholar]
- 30.Hardy P, Abran D, Li DY, et al. Free radicals in retinal and choroidal blood flow autoregulation in the piglet: Interaction with prostaglandins. Invest Ophthalmol Vis Sci. 1994;35:580–591. [PubMed] [Google Scholar]
- 31.Abran D, Varma DR, Chemtob S. Regulation of prostanoid vasomotor effects and receptors in choroidal vessels of newborn pigs. Am J Physiol. 1997;272:R995–R1001. doi: 10.1152/ajpregu.1997.272.3.R995. [DOI] [PubMed] [Google Scholar]
- 32.Abran D, Varma DR, Li D-Y, et al. Reduced responses of retinal vessels of the newborn pig to prostaglandins but not to thromboxane. Can J Physiol Pharmacol. 1994;72:168–173. doi: 10.1139/y94-026. [DOI] [PubMed] [Google Scholar]
- 33.Hardy P, Peri KG, Lahaie I, et al. Increased nitric oxide synthesis and action preclude choroidal vasoconstriciton to hyperoxia in newborn pigs. Circ Res. 1996;79:504–511. doi: 10.1161/01.res.79.3.504. [DOI] [PubMed] [Google Scholar]
- 34.Flower RW, McLeod DS, Wajer SD, et al. Prostaglandins as mediators of vasotonia in the immature retina. Pediatrics. 1984;73:440–444. [PubMed] [Google Scholar]
- 35.Zhu Y, Park TS, Gidday GM. Mechanisms of hyperoxia-induced reductions in retinal blood flow in newborn pig. Exp Eye Res. 1998;67:357–369. doi: 10.1006/exer.1998.0535. [DOI] [PubMed] [Google Scholar]
- 36.Sennlaub F, Valamanesh F, Vazquez-Tello A, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204. doi: 10.1161/01.CIR.0000080735.93327.00. [DOI] [PubMed] [Google Scholar]
- 37.Yanni SE, Barnett JM, Clark ML, et al. PGE2 Receptor EP4 is a Potential Therapeutic Target for the Treatment of Pathological Ocular Angiogenesis. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3652. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satarug S, Wisedpanichkij R, Takeda K, et al. Prostaglandin D2 induces heme oxygenase-1 mRNA expression through the DP2 receptor. Biochem Biophys Res Commun. 2008;377:878–883. doi: 10.1016/j.bbrc.2008.10.094. [DOI] [PubMed] [Google Scholar]
- 39.Quiniou C, Sennlaub F, Beauchamp MH, et al. Dominant role for calpain in thromboxane-induced neuromicrovascular endothelial cytotoxicity. J Pharmacol Exp Ther. 2006;316:618–627. doi: 10.1124/jpet.105.093898. [DOI] [PubMed] [Google Scholar]
- 40.Burnette JO, White RE. PGI2 opens potassium channels in retinal pericytes by cyclic AMP-stimulated, cross-activation of PKG. Exp Eye Res. 2006;83:1359–1365. doi: 10.1016/j.exer.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 41.Masferrer JL, Leahy KM, Koki AT. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000;60(5):1306–1311. [PubMed] [Google Scholar]
- 42.Leahy KM, Ornberg RL, Wang Y. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res. 2002;62(3):625–631. [PubMed] [Google Scholar]
- 43.Yamada M, Kawai M, Kaway Y, et al. The effect of selective cyclooxygenase-2 inhibitor on corneal angiogenesis in the rat. Curr Eye Res. 1999;19(4):300–304. doi: 10.1076/ceyr.19.4.300.5301. [DOI] [PubMed] [Google Scholar]
- 44.Sawaoka H, Tsuji S, Tsuji M, et al. Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab Invest. 1999;79(12):1469–1477. [PubMed] [Google Scholar]
- 45.Wilkinson-Berka JL, Alousis NS, Kelly DJ, et al. COX-2 inhibition and retinal angiogenesis in a mouse model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2003;44(3):974–979. doi: 10.1167/iovs.02-0392. [DOI] [PubMed] [Google Scholar]
- 46.Ke T-L, Graff G, Spellman JM, et al. Nepafenac, a unique nonsteroidal prodrug with potential utility in the treatment of trauma-induced ocular inflammation: II. In vitro bioactivation and permeation of ocular barriers. Inflammation. 2000;24(4):371–384. doi: 10.1023/a:1007001131987. [DOI] [PubMed] [Google Scholar]
- 47.Gamache DA, Graff G, Brady MT, et al. Nepafenac, a unique nonsteroidal prodrug with potential utility in the treatment of trauma-induced ocular inflammation: I. Assessment of anti-inflammatory efficacy. Inflammation. 2000;24(4):357–370. doi: 10.1023/a:1007049015148. [DOI] [PubMed] [Google Scholar]
- 48.Kapin MA, Yanni JM, Brady MT, et al. Inflammation-mediated retinal edema in the rabbit is inhibited by topical nepafenac. Inflammation. 2003;27(5):281–291. doi: 10.1023/a:1026024409826. [DOI] [PubMed] [Google Scholar]
- 49.Lane SS, Modi SS, Lehmann RP, et al. Nepafenac ophthalmic suspension 0.1% for the prevention and treatment of ocular inflammation associated with cataract surgery. J Cataract Refract Surg. 2007;33(1):53–58. doi: 10.1016/j.jcrs.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 50.Walters T, Raizman M, Ernest P, et al. In vivo pharmacokinetics and in vitro pharmacodynamics of nepafenac, amfenac, ketorolac, and bromfenac. J Cataract Refract Surg. 2007;33:1539–1545. doi: 10.1016/j.jcrs.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi K, Saishin Y, Saishin Y, et al. Topical nepafenac inhibits ocular neovascularization. Invest Ophthalmol Vis Sci. 2003;44(1):409–415. doi: 10.1167/iovs.02-0346. [DOI] [PubMed] [Google Scholar]
- 52.Kern TS, Miller CM, Du Y. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes. 2007;56:373–379. doi: 10.2337/db05-1621. [DOI] [PubMed] [Google Scholar]
- 53.Chiarugi V, Magnelli L, Gallo O. Cox-2, iNOS and p53 as play-makers of tumor angiogenesis. Int J Mol Med. 1998;2:715–719. doi: 10.3892/ijmm.2.6.715. [DOI] [PubMed] [Google Scholar]
- 54.Penn JS, Henry MM, Tolman BL. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res. 1994;26(6):724–731. doi: 10.1203/00006450-199412000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Hicks D, Curtois Y. The growth and behaviour of rat retinal Müller cells in vitro. 1. An improved method for isolation and culture. Exp Eye Res. 1990;51:119–129. doi: 10.1016/0014-4835(90)90063-z. [DOI] [PubMed] [Google Scholar]
- 56.Barnett JM, McCollum GW, Fowler JA, et al. Pharmacologic and genetic manipulation of MMP-2 and -9 affects retinal neovascularization in rodent models of OIR. Invest Ophthalmol Vis Sci. 2007;28:907–915. doi: 10.1167/iovs.06-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Werdich XQ, McCollum GW, Rajaratnam VS, Penn JS. Variable oxygen and retinal VEGF levels: correlation with incidence and severity of pathology in a rat model of oxygen-induced retinopathy. Exp Eye Res. 2004;79:623–630. doi: 10.1016/j.exer.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Penn JS, Tolman BL, Henry MM. Oxygen-induced retinopathy in the rat: relationship of retinal nonperfusion to subsequent neovascularization. Invest Ophthalmol Vis Sci. 1994;35:3429–3435. [PubMed] [Google Scholar]
- 59.Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998;58:409–412. [PubMed] [Google Scholar]
- 60.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer and development. Oncogene 1999. 1983;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 61.Form DM, Auerbach R. PGE2 and angiogenesis. Proc Soc Exp Biol Med. 1983;172:214–218. doi: 10.3181/00379727-172-41548. [DOI] [PubMed] [Google Scholar]
- 62.Joussen AM, Poulaki V, Qin W, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 63.Sennlaub F, Valamanesh F, Vazquez-Tello A, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204. doi: 10.1161/01.CIR.0000080735.93327.00. [DOI] [PubMed] [Google Scholar]
- 64.Berkowitz BA, Roberts R, Luan H, et al. Drug intervention can correct subnormal retinal oxygenation response in experimental diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:2954–2960. doi: 10.1167/iovs.05-0132. [DOI] [PubMed] [Google Scholar]
- 65.Ayalasomayajula SP, Kompella UB. Subconjunctivally administered celecoxib-PLGA microparticles sustain retinal drug levels and alleviate diabetes-induced oxidative stress in a rat model. Euro J Pharm. 2005;511:191–198. doi: 10.1016/j.ejphar.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi H, Yanagi Y, Tamaki Y, et al. COX-2-selective inhibitor, etodolac, suppresses choroidal neovascularization in a mice model. Biochem Biophys Res Commun. 2004;325:461–466. doi: 10.1016/j.bbrc.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 67.Hariprasad SM, Callanan D, Gainey S, et al. Cystoid and diabetic macular edema treated with nepafenac 0.1% J Ocul Pharmacol Ther. 2007;23:585–589. doi: 10.1089/jop.2007.0062. [DOI] [PubMed] [Google Scholar]
- 68.Gierse JK, Koboldt CM, Walker MC, et al. Kinetic basis for selective inhibition of cylco-oxygenases. Biochem J. 1999;339:607–614. [PMC free article] [PubMed] [Google Scholar]
- 69.Smith CJ, Zhang Y, Koboldt CM, et al. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cryan LM, Pidgeon GP, Fitzgerald DJ, et al. COX-2 protects against thrombosis of the retinal vasculature in a mouse model of proliferative retinopathy. Mol Vis. 2006;12:405–414. [PubMed] [Google Scholar]
- 71.Amrite AC, Ayalasomayajula SP, Cheruvu NPS, et al. Single periocular injection of celecoxib-PLGA microparticles inhibits diabetes-induced elevations in retinal PGE2, VEGF, and vascular leakage. Invest Ophthalmol Vis Sci. 2006;47:1149–1160. doi: 10.1167/iovs.05-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sabbieti MG, Marchetti L, Gabrielli MG, et al. Prostaglandins differently regulate FGF-2 and FGF receptor expression and induce nuclear translocation in osteoblasts via MAPK kinase. Cell Tissue Res. 2005;319:267–278. doi: 10.1007/s00441-004-0981-8. [DOI] [PubMed] [Google Scholar]
- 73.Sales KJ, Boddy SC, Williams AR, et al. F-prostanoid receptor regulation of fibroblast growth factor 2 signaling in endometrial adenocarcinoma cells. Endocrinology. 2007;148:3635–3644. doi: 10.1210/en.2006-1517. [DOI] [PubMed] [Google Scholar]
- 74.Recchia FM, Xu L, Penn JS, et al. Identification of genes and pathways involved in retinal neovascularization by microarray analysis of two animal models of retinal angiogenesis. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-4006. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thieme H, Aiello LP, Takagi H, et al. Comparative analysis of vascular endothelial growth factor receptors on retinal and aortic vascular endothelial cells. Diabetes. 1995;44:98–103. doi: 10.2337/diab.44.1.98. [DOI] [PubMed] [Google Scholar]
- 76.Wu G, Luo J, Rana JS, et al. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc Res. 2006;69:512–519. doi: 10.1016/j.cardiores.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 77.Wu G, Luo J, Rana JS, et al. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc Res. 2006;69:512–519. doi: 10.1016/j.cardiores.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 78.Akarasereenont PC, Techatraisak K, Thaworn, et al. The expression of COX-2 in VEGF-treated endothelial cells is mediated through protein tyrosine kinase. Mediators Inflamm. 2002;11:17–22. doi: 10.1080/09629350210311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murphy JF, Fitzgerald DJ. Vascular endothelial cell growth factor (VEGF) induces cyclooxygenase (COX)-dependent proliferation of endothelial cells (EC) via the VEGF-2 receptor. FASEB J. 2001;15:1667–1669. doi: 10.1096/fj.00-0757fje. [DOI] [PubMed] [Google Scholar]
- 80.Nie D, Lamberti M, Zacharek A, et al. Thromboxane A2 regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun. 2000;267:245–251. doi: 10.1006/bbrc.1999.1840. [DOI] [PubMed] [Google Scholar]
- 81.Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 82.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 83.Kaiser PK, Blodi BA, Shapiro H, et al. Angiographic and optical coherence tomographic results of the MARINA study of ranibizumab in neovascular age-related macular degeneration. Ophthalmol. 2007;114:1868–1875. doi: 10.1016/j.ophtha.2007.04.030. [DOI] [PubMed] [Google Scholar]
- 84.Regillo CD, Brown DM, Abraham P, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145:239–248. doi: 10.1016/j.ajo.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 85.Fung AE, Lalwani GA, Rosenfeld PJ, et al. An optical coherence tomography-guided, variable dosing regimen with intravitreal ranibizumab (Lucentis) for neovascular age-related macular degeneration. Am J Ophthalmol. 2007;143:566–583. doi: 10.1016/j.ajo.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 86.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid Drug Selectivities for Cyclo-Oxygenase-1 Rather than Cyclo-Oxygenase-2 Are Associated with Human Gastrointestinal Toxicity: A Full in vitro Analysis. PNAS. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amrite AC, Kompella UB. Celecoxib inhibits proliferation of retinal pigment epithelial cells and choroid-retinal endothelial cells by a cyclooxygenase-2-independent mechanism. J Pharmacol Exp Ther. 2008;324:749–758. doi: 10.1124/jpet.107.128918. [DOI] [PubMed] [Google Scholar]
- 88.Katz ML, Robison WG., Jr Autoxidative damage to the retina: potential role in retinopathy of prematurity. Birth Defects Orig Artic Ser. 1988;24:237–248. [PubMed] [Google Scholar]
- 89.Penn JS. Oxygen-induced retinopathy in the rat: possible contribution of peroxidation reactions. Doc Ophthalmol. 1990;74:179–186. doi: 10.1007/BF02482607. [DOI] [PubMed] [Google Scholar]
- 90.Penn JS, Tolman BL, Lowery LA. Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest Ophthalmol Vis Sci. 1993;34:576–585. [PubMed] [Google Scholar]
- 91.Rodieck RW. The Vertebrate Retina: Principles of Structure and Function. San Francisco: WH Freeman; 1973. p. 159. [Google Scholar]
- 92.Asikainen TM, Heikkila P, Kaarteenaho-Wiik R, et al. Cell-specific expression of manganese superoxide dismutase protein in the lungs of patients with respiratory distress syndrome, chronic lung disease, or persistent pulmonary hypertension. Pediatr Pulmonol. 2001;32:193–200. doi: 10.1002/ppul.1108. [DOI] [PubMed] [Google Scholar]
- 93.Raju TN, Langenberg P, Bhutani V, et al. Vitamin E prophylaxis to reduce retinopathy of prematurity: a reappraisal of published trials. J Pediatr. 1997;131:844–850. doi: 10.1016/s0022-3476(97)70031-3. [DOI] [PubMed] [Google Scholar]
- 94.Penn JS, Thum LA, Naash MI. Oxygen-induced retinopathy in the rat. Vitamins C and E as potential therapies. Invest Ophthalmol Vis Sci. 1992;33:1836–1845. [PubMed] [Google Scholar]
- 95.van Rossum GS, Drummen GP, Verkleij AJ, et al. Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: a p42/44(MAPK)-dependent and phosphorylation-independent mechanism. J Biochim Biophys Acta. 2004;1636:183–195. doi: 10.1016/j.bbalip.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 96.Sun Y, Chen J, Rigas B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis. 2009;30:93–100. doi: 10.1093/carcin/bgn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Penn JS, Tolman BL, Bullard LE. Effect of a water-soluble vitamin E analog, trolox C, on retinal vascular development in an animal model of retinopathy of prematurity. Free Radic Biol Med. 1997;22:977–984. doi: 10.1016/s0891-5849(96)00479-0. [DOI] [PubMed] [Google Scholar]
- 98.Niesman MR, Johnson KA, Penn JS. Therapeutic effect of liposomal superoxide dismutase in an animal model of retinopathy of prematurity. Neurochem Res. 1997;22:597–605. doi: 10.1023/a:1022474120512. [DOI] [PubMed] [Google Scholar]
- 99.Saito Y, Geisen P, Uppal A, et al. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853. [PMC free article] [PubMed] [Google Scholar]
- 100.Barnett JM, McCollum G, Penn JS. The Role of Cytosolic Phospholipase A2 in Retinal Neovascularization. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3691. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dumont I, Hardy P, Peri KG, et al. Regulation of endothelial nitric oxide synthase by PGD(2) in the developing choroid. Am J Physiol Heart Circ Physiol. 2000;278:H60–66. doi: 10.1152/ajpheart.2000.278.1.H60. [DOI] [PubMed] [Google Scholar]
- 102.Hardy P, Dumont I, Bhattacharya M, et al. Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis for ischemic retinopathy. Cardiovasc Res. 2000;47:489–509. doi: 10.1016/s0008-6363(00)00084-5. [DOI] [PubMed] [Google Scholar]
- 103.MacRitchie AN, Albertine KH, Sun J, et al. Reduced endothelial nitric oxide synthase in lungs of chronically ventilated preterm lambs. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1011–1020. doi: 10.1152/ajplung.2001.281.4.L1011. [DOI] [PubMed] [Google Scholar]
- 104.Han RN, Babaei S, Robb M, et al. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res. 2004;94:1115–1123. doi: 10.1161/01.RES.0000125624.85852.1E. [DOI] [PubMed] [Google Scholar]
- 105.Lukiw WJ, Ottlecz A, Lambrou G, et al. Coordinate activation of HIF-1 and NF-kappaB DNA binding and COX-2 and VEGF expression in retinal cells by hypoxia. Invest Ophthalmol Vis Sci. 2003;44:4163–4170. doi: 10.1167/iovs.02-0655. [DOI] [PubMed] [Google Scholar]