

Abstract
Considerable success has been achieved in the treatment of HIV-1 infection, and more than two-dozen antiretroviral drugs are available targeting several distinct steps in the viral replication cycle. However, resistance to these compounds emerges readily, even in the context of combination therapy. Drug toxicity, adverse drug-drug interactions, and accompanying poor patient adherence can also lead to treatment failure. These considerations make continued development of novel antiretroviral therapeutics necessary. In this article, we highlight a number of steps in the HIV-1 replication cycle that represent promising targets for drug discovery. These include lipid raft microdomains, the RNase H activity of the viral enzyme reverse transcriptase, uncoating of the viral core, host cell machinery involved in the integration of the viral DNA into host cell chromatin, virus assembly, maturation, and budding, and the functions of several viral accessory proteins. We discuss the relevant molecular and cell biology, and describe progress to date in developing inhibitors against these novel targets.
Keywords: virus entry, virus assembly, retrovirus, drug resistance, HIV-1 drug therapy
A. Introduction
A1. Overview of HIV-1 Replication
The replication cycle of human immunodeficiency virus type 1 (HIV-1) is a complex multi-step process that depends on both viral and host cell factors (Figs. 1 and 2) (Freed, 2007). Replication begins with viral entry into the target cell. Entry proceeds by fusion of the viral lipid envelope and the cellular plasma membrane (Doms, 2000; Melikyan, 2008). The viral component that mediates fusion is the envelope (Env) glycoprotein spike, which is composed of a trimeric, non-covalently associated complex of the surface glycoprotein gp120 and the transmembrane glycoprotein gp41 (Roux and Taylor, 2007). Fusion is initiated by binding of gp120 to the cellular receptor CD4 and a subsequent interaction with the CCR5 or CXCR4 coreceptor (Berger, Murphy, and Farber, 1999; Doms, 2000). Coreceptor binding triggers a series of conformation changes in both gp120 and gp41 that mediate membrane fusion (Doms, 2000; Melikyan, 2008). Fusion delivers the viral core into the cytoplasm of the target cell. The viral core is composed of a capsid (CA) protein shell that encapsidates the single-stranded, dimeric viral RNA genome in complex with the viral nucleocapsid (NC) protein and the viral enzymes reverse transcriptase (RT) and integrase (IN) (Adamson and Freed, 2007; Ganser-Pornillos, Yeager, and Sundquist, 2008). The core uncoats (Warrilow and Harrich, 2007) and RT copies the RNA genome into a double-stranded DNA copy (Sarafianos et al., 2009), which is transported into the nucleus where IN stably integrates it into the host cell genome (Delelis et al., 2008; Suzuki and Craigie, 2007; Vandegraaff and Engelman, 2007). The host cell cofactor LEDGF/p75 (lens epithelium-derived growth factor/transcriptional co-activator 75) plays an important role in the integration process by tethering IN to chromatin (Poeschla, 2008).
Fig. 1.
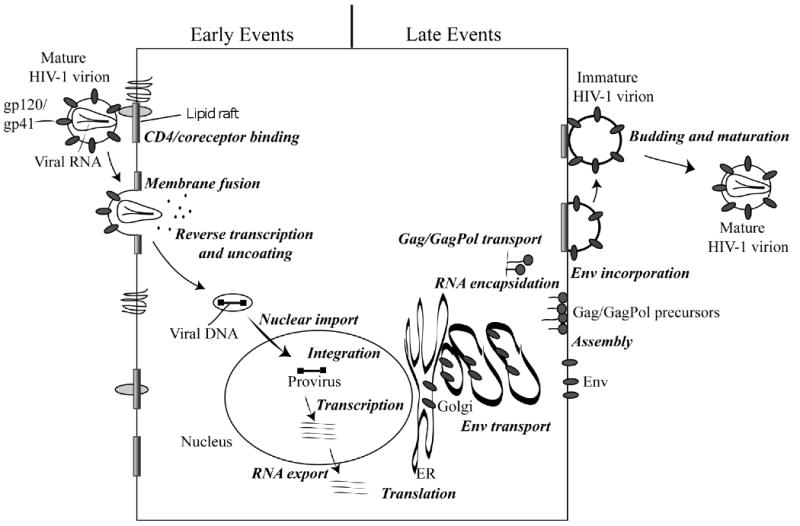
Schematic representation of the HIV-1 replication cycle. Details are provided in the text. Reprinted with permission from Elsevier (Freed, 2004).
Fig. 2.

Organization of the HIV-1 genome. The gene products encoded by HIV-1 include the Gag proteins matrix (MA), capsid (CA), nucleocapsid (NC) and p6 and spacer peptides SP1 and SP2; the Pol proteins protease (PR), reverse transcriptase (RT) and integrase (IN); the surface Env glycoprotein gp120 and the transmembrane Env glycoprotein gp41; the regulatory proteins Tat and Rev; and the accessory proteins Vif, Vpr, Vpu, and Nef. Also shown are the 5′ and 3′ long terminal repeats (LTRs).
The integrated proviral DNA is transcribed to generate full-length progeny viral RNA and a number of spliced mRNA transcripts that are translated in the cytoplasm (Rabson and Graves, 1997; Swanstrom and Wills, 1997). Transcription and translation, performed by cellular machinery (Bolinger and Boris-Lawrie, 2009; Nekhai and Jeang, 2006), result in the synthesis of several major structural proteins: (i) the Gag polyprotein precursor, which is composed of four domains - matrix (MA), CA, NC and p6 - and two spacer peptides, SP1 and SP2, (ii) the Gag-Pol polyprotein precursor, which is produced via a -1 ribosomal frameshift during gag translation and encodes the viral enzymes protease (PR), RT and IN, and (iii) the Env glycoprotein precursor, gp160, which is cleaved into the gp120 and gp41 subunits by a host protease during trafficking through the Golgi apparatus (Swanstrom and Wills, 1997). These protein components, together with full-length viral genomic RNA, are each transported to the site of virus particle assembly at the plasma membrane (Adamson and Freed, 2007). Assembly is directed by Gag, which coordinates the incorporation of each of the viral components, together with a number of host cell factors, into the assembling particle (Adamson and Freed, 2007). Virus particle production is completed upon budding of the nascent virion from the plasma membrane (Adamson and Freed, 2007). To facilitate virus release, the p6 domain of Gag hijacks components of the cellular endosomal sorting machinery, which normally function to promote the budding of vesicles into late endosome to form multivesicular bodies (MVBs) (Bieniasz, 2009; Demirov and Freed, 2004; Fujii, Hurley, and Freed, 2007; Morita and Sundquist, 2004). Concomitant with virus release, PR cleaves the Gag and Gag-Pol precursors into their respective protein domains (Swanstrom and Wills, 1997). Gag and Gag-Pol cleavage leads to virion maturation, a reassembly event that produces mature particles containing the condensed, conical core (Adamson and Freed, 2007; Ganser-Pornillos, Yeager, and Sundquist, 2008).
In addition to the structural proteins listed above, HIV-1 encodes two regulatory gene products - Tat and Rev - and several accessory proteins: Vif (viral infectivity factor), Vpu (viral protein U), Nef (negative factor), and Vpr (viral protein R) (Fig. 2). Tat transactivates transcription from the HIV long terminal repeat (LTR) by binding to an RNA element (the transactivation response region, or TAR) at the 5′ end of all viral mRNAs. Rev promotes the export of unspliced mRNAs from the nucleus by binding to the Rev responsive element (RRE) in the viral RNA (Freed, 2007). Vif significantly enhances virus infectivity; Vpu stimulates the release of budded particles from the plasma membrane and induces CD4 degradation. Nef, which is an important determinant of viral pathogenesis and disease progression in vivo, downregulates surface expression of CD4 and major histocompatibility complex I (MHC-I), modulates cell activation pathways, and enhances particle infectivity. Vpr induces cell-cycle arrest, stimulates transcription from some cellular promoters, influences virus-induced apoptosis, and has been reported to promote nuclear import of the preintegration complex (PIC) following reverse transcription in the newly infected target cell (Freed, 2007).
At virtually every step in its life cycle, HIV-1 takes advantage of host cell factors and pathways to promote successful replication. However, it has become clear in recent years that the host cell has set up antiretroviral barriers in the form of restriction factors that markedly impair specific steps in the replication cycle. For example, tripartite motif protein 5α (TRIM5α) acts at a post-entry step by interacting with CA on the incoming viral core leading to its premature degradation. In some cases, retroviruses have responded by evolving counter-defense mechanisms to overcome these restriction factors. For example, Vif counteracts the cytosine deaminase APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G) by inducing its proteasomal degradation, and Vpu counteracts an interferon-induced host protein, variously known as CD317, BST-2, or tetherin, which prevents virus release by “tethering” particles to the plasma membrane.
In this review, we explore a number of unexploited targets for antiretroviral therapy. Many of the approaches discussed here remain hypothetical but may provide future opportunities for drug development. We will cover targets ranging from lipid rafts, RNase H, and LEDGF to budding and maturation, as well as the possibility that information derived from the study of host innate immunity could be applied to the development of novel therapeutics. We will not discuss Tat or Rev as targets as this topic has been treated elsewhere (Baba, 2004; Bannwarth and Gatignol, 2005; Richter and Palu, 2006). We will also not deal with Vpr, as it remains unclear whether this accessory protein is a viable target for drug development.
A2. Clinically Approved Antiretroviral Drugs
The complex, multi-step HIV-1 replication cycle outlined above offers numerous opportunities for pharmacological intervention. To date, more than 20 antiretroviral drugs have been approved for clinical use. These drugs can be divided into six different mechanistic classes that target distinct steps in the HIV-1 replication cycle. Two drugs have been developed that inhibit virus entry. T20 (enfuvirtide) blocks viral fusion by targeting gp41 and maraviroc acts as a CCR5 antagonist, making it the only currently approved antiretroviral drug that targets a host cell factor. Entry inhibitors are discussed by Doms et al. in this issue (Doms, 2010). The remaining four classes of approved drugs target each of the viral enzymes: RT, PR and IN. Inhibitors targeting RT and PR are the most numerous and successful antiretroviral drugs and combinations of these drugs are the standard initial treatment strategy. RT inhibitors fall into two classes based on their mode of action: the nucleoside-analog RT inhibitors (NRTIs) and non-nucleoside-analog RT inhibitors (NNTRIs), which are discussed by Cihlar et al. (Cihlar, 2010) and de Bethune et al. (de bethune, 2010), respectively, in this issue. PR inhibitors (PIs) target the catalytic action of this enzyme [see article by Nijhuis et al in this issue (Nijhuis, 2010)]. The newest antiretroviral drug to achieve widespread clinical use is the IN inhibitor, raltegravir, approved in 2007 [see article by McColl et al. in this issue {McCool, 2010 #384}].
The antiretroviral drugs mentioned above have significantly extended patient survival (Richman et al., 2009; Simon, Ho, and Abdool Karim, 2006). Therapy typically consists of a combination of three to four drugs in therapeutic regimens known as highly active antiretroviral therapy (HARRT) (Chen, Hoy, and Lewin, 2007; Simon, Ho, and Abdool Karim, 2006). The simultaneous use of multiple drugs is required because of the ease with which HIV-1 can acquire drug resistance to any single inhibitor (Emini and Fan, 1997; Simon, Ho, and Abdool Karim, 2006; Temesgen et al., 2006). Resistance arises due to the high degree of HIV-1 genetic diversity within an individual patient, a consequence of a rapid rate of viral replication combined with the error-prone nature of RT and frequent recombination events (Hu et al., 2003; Simon, Ho, and Abdool Karim, 2006; Svarovskaia et al., 2003). Despite the positive impact of HAART on patient survival, drug resistance can still emerge even in the face of this multi-drug treatment (Perno et al., 2008; Simon, Ho, and Abdool Karim, 2006; Temesgen et al., 2006). Drug toxicity, combined with poor patient adherence, can contribute to drug resistance and treatment failure. The serious clinical consequences of multi-drug resistance require the use of alternative treatment regimens, known as salvage therapy (Perno et al., 2008; Temesgen et al., 2006). Salvage therapy is most likely to be effective if new drugs targeting novel sites of action are available (Greene et al., 2008; Perno et al., 2008). As current antiretroviral drugs do not eradicate the virus, patients are required to use HAART on a life-long basis to suppress viral replication (Marsden and Zack, 2009; Richman et al., 2009). Until a cure for HIV infection is achieved, sustained successful treatment of HIV-1-infected patients with antiretroviral drugs may require the development of a continuous pipeline of new drugs. To this end, an intensive research effort into understanding the basic mechanisms governing HIV-1 replication has led to the identification of an array of new therapeutic targets, which have significant potential for future antiretroviral drug development.
B. Novel Therapeutic Approaches
B1. Lipid microdomains as a target for antiviral therapy
The plasma membrane of mammalian cells is composed of a variety of microdomains with specific lipid and protein compositions. Lipid rafts are a particularly well-studied class of plasma membrane microdomain, characterized by a high concentration of saturated lipids and cholesterol (Brown and London, 1998). While the function, and even the existence, of lipid rafts has been controversial (Munro, 2003), there is now general agreement that cholesterol- and saturated lipid-enriched microdomains do exist and serve a variety of functions in cell signaling, cell motility and polarization, intercellular synapse formation, and protein trafficking. Membrane rafts have been defined as “small (10–200 nm), heterogeneous, highly dynamic, sterol- and sphingolipid-enriched domains that compartmentalize cellular processes. Small rafts can sometimes be stabilized to form larger platforms through protein-protein and protein-lipid interactions” (Pike, 2006).
Studies from many laboratories have demonstrated that a number of viruses, including HIV-1, use lipid rafts as platforms for both viral entry and particle assembly and release (Ono and Freed, 2005; Waheed and Freed, 2009). Lipid rafts can be analyzed biochemically based on their resistance, relative to non-raft membrane, to solubilization in cold, non-ionic detergents (Brown and Rose, 1992). HIV-1 Gag and Env have been reported to associate with such detergent-resistant membrane (DRM), providing support for the hypothesis that HIV-1 assembly takes place in lipid raft microdomains (Nguyen and Hildreth, 2000; Ono and Freed, 2001). Microscopy-based approaches confirm that Gag and Env colocalize with lipid raft markers (Ono and Freed, 2005) and disruption of lipid rafts with cholesterol-depleting agents inhibits both virus release (Ono and Freed, 2001; Pickl, Pimentel-Muinos, and Seed, 2001) and virion infectivity (Campbell et al., 2004; Campbell, Crowe, and Mak, 2002; Graham et al., 2003; Guyader et al., 2002; Liao et al., 2001; Manes et al., 2000; Nguyen and Taub, 2002; Popik, Alce, and Au, 2002). Cholesterol depletion interferes with virus particle production by inhibiting the ability of Gag to bind the plasma membrane (Ono, Waheed, and Freed, 2007). Further support of a raft origin for the assembled particle derives from the finding that the lipid composition of HIV-1 virions is high in raft components, specifically cholesterol and saturated lipids (Aloia, Curtain, and Jensen, 1992; Aloia, Tian, and Jensen, 1993; Brugger et al., 2006; Chan et al., 2008). Taken together, these findings demonstrate that both virus entry and particle egress take place in cholesterol-enriched membrane microdomains. Furthermore, cell-cell transmission of HIV-1 occurs at a “virological synapse” that exhibits a concentration of lipid raft markers (Jolly and Sattentau, 2005).
The use of lipid rafts by HIV-1 at multiple steps in the virus replication cycle opens up the possibility that lipid rafts could in some way be targeted as an antiviral strategy. One report suggested that 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) inhibitors (statin drugs), widely used to treat high cholesterol in vivo, could lower viral loads in HIV-1-infected patients (del Real et al., 2004). However, subsequent studies failed to reproduce these findings (Moncunill et al., 2005; Probasco et al., 2008; Sklar et al., 2005). Systemic treatment with lipid-raft disrupting agents would likely be associated with significant toxicity, making topical treatment in the context of chemoprevention a more realistic approach. Indeed, in a humanized severe combined immunodeficient (SCID) mouse model system, treatment of the vaginal mucosa with a cholesterol-depleting cyclodextrin reduced virus transmission resulting from inoculation with HIV-1-infected human peripheral blood lymphocytes (PBLs) (Khanna et al., 2002). Inhibitors of glycosphingolipid synthesis have also been reported to inhibit HIV-1 infection in culture (for reviews see (Puri and Blumenthal, 2008; Waheed and Freed, 2009).
Another approach to interfering with HIV-1 replication by targeting lipid rafts entails the use of cholesterol-binding agents that associate with the virion lipid bilayer. One such compound, amphotericin B methyl ester (AME), has been shown to inhibit both virus particle production and virion infectivity (Waheed et al., 2006; Waheed et al., 2008). The infectivity of HIV-1 virions bearing heterologous Env glycoproteins (e.g., from murine leukemia virus or vesicular stomatitis virus) or truncated forms of HIV-1 gp41 is not affected by AME, demonstrating that the long gp41 cytoplasmic tail is required for AME-imposed inhibition of infectivity (Waheed et al., 2006). Consistent with this hypothesis, long-term culture of HIV-1 in the presence of AME led to the emergence of AME-resistant variants that acquired mutations in the gp41 cytoplasmic tail (Waheed et al., 2006). Remarkably, the mechanism by which these gp41 mutations conferred resistance to AME involved the cleavage of the gp41 cytoplasmic tail by the viral PR after Env incorporation into the virion (Waheed et al., 2007). Although amphotericin B is used clinically to treat fungal infections it is highly toxic. AME is reportedly less toxic than its parent compound (Parmegiani et al., 1987); however, long-term systemic treatment is unlikely to be well tolerated. Again, use of AME as a chemopreventive agent warrants consideration.
B2. Post–Entry
B2.1 Uncoating of the viral core as a potential antiviral target
Fusion of the viral and cellular membranes delivers the core of the mature virus particle into the cytoplasm of the target cell (Fig. 1). Following entry, (a) the core partially disassembles (uncoats) to form the reverse transcription complex (RTC), (b) the viral RNA genome is reverse transcribed into a double-stranded DNA copy, (c) the PIC, which contains the viral DNA, translocates through the nuclear pore, and (d) the viral DNA integrates into the host genome to establish the provirus (Delelis et al., 2008; Freed, 2007; Sarafianos et al., 2009; Suzuki and Craigie, 2007; Warrilow and Harrich, 2007).
The reverse transcription and integration steps are relatively well understood at the molecular level (Delelis et al., 2008; Sarafianos et al., 2009). Antiretroviral drugs targeting the RT and IN enzymes have been successfully developed [See the following articles in this issue: {Cihlar, 2010 #381;de bethune, 2010 #382;McCool, 2010 #384}]. New approaches to target reverse transcription and integration are discussed in sections B2.2 and B2.3 below. In contrast, core uncoating, which converts the viral core into the RTC and ultimately the PIC, is poorly understood. The precise order of events and the exact composition, structure, location and transport pathway of the RTC and PIC remain to be defined (Suzuki and Craigie, 2007; Warrilow and Harrich, 2007). This is currently an active and rapidly progressing field of research. While many details await elucidation, it is clear that correct regulation of core uncoating is essential for completion of the early steps of the HIV-1 replication cycle. Mutations in CA that affect core stability but not core formation result in impaired reverse transcription and infection (Brun et al., 2008; Fitzon et al., 2000; Forshey et al., 2002). Furthermore, the vulnerability of core-uncoating has been highlighted by the discovery of the host restriction factor, TRIM5α, which appears to target this early step in retroviral replication (Bieniasz, 2004; Luban, 2007; Ozato et al., 2008; Stremlau et al., 2004; Towers, 2007).
The mechanism by which TRIM5α exerts its antiretroviral effect is not fully understood. However, it is known that TRIM5α targets intact or partially uncoated incoming viral cores via a pattern-recognition function that identifies a structure formed by the CA lattice (Forshey, Shi, and Aiken, 2005; Hatziioannou et al., 2004a; Kar et al., 2008; Langelier et al., 2008; Sebastian and Luban, 2005; Shi and Aiken, 2006; Stremlau et al., 2006) (See section B4.2 for further details of the CA lattice). The block to replication occurs at a post-entry but pre-RT step, such that completion of reverse transcription is inhibited (Besnier, Takeuchi, and Towers, 2002; Cowan et al., 2002; Munk et al., 2002; Shibata et al., 1995; Stremlau et al., 2004; Towers et al., 2000). It has been suggested that TRIM5α blocks retroviral replication either by causing cores to undergo rapid and premature disassembly (Perron et al., 2007; Stremlau et al., 2006) and/or by recruiting cellular proteasomal degradation machinery (Anderson et al., 2006; Campbell et al., 2008; Rold and Aiken, 2008; Wu et al., 2006). Importantly, the antiretroviral effects of TRIM5α are exerted in a species-specific manner (Bieniasz, 2004; Hatziioannou et al., 2004b; Keckesova, Ylinen, and Towers, 2004; Luban, 2007; Ozato et al., 2008; Perron et al., 2004; Stremlau et al., 2004; Towers, 2007; Yap et al., 2004). For example, HIV-1 replication is potently blocked by TRIM5α from the non-human primate rhesus macaque (TRIM5αrh); in contrast, human TRIM5α (TRIM5αhu) is largely ineffective against HIV-1 but does inhibit other retroviruses.
The specificity of retroviral restriction correlates with the ability of TRIM5α to recognize the incoming viral core and has been mapped to a CA recognition domain in TRIM5α (Bieniasz, 2004; Luban, 2007; Ozato et al., 2008; Towers, 2007). TRIM family proteins are composed of three domains (Fig. 3), (i) a RING-finger, (ii) a B-box, and (iii) a coiled-coil domain, which together form the tripartite RBCC domain (Ozato et al., 2008). The α splice variant of TRIM5 (TRIM5α) contains an additional C-terminal PRYSPRY (or B30.2) domain (Reymond et al., 2001). The PRYSPRY domain is the main determinant of core recognition, CA binding, and hence restriction specificity (Kar et al., 2008; Langelier et al., 2008; Li et al., 2006; Perez-Caballero et al., 2005; Sawyer et al., 2005; Stremlau et al., 2004; Stremlau et al., 2006; Stremlau et al., 2005; Yap, Nisole, and Stoye, 2005). The degree of CA binding and restriction potency correlates with amino acid variations in the PRYSPRY domain and, remarkably, a single-amino-acid substitution in the PRYSPRY domain is sufficient to enable TRIM5αhu to restrict HIV-1 (Langelier et al., 2008; Li et al., 2006; Stremlau et al., 2005; Yap, Nisole, and Stoye, 2005). Conversely, mutations in CA give rise to differential susceptibility of incoming cores to TRIM5α restriction (Hatziioannou et al., 2004a; Li et al., 2006; Owens et al., 2004; Stremlau et al., 2006).
Fig. 3.

Domain organization of TRIM5α and TRIM-Cyp. The major domains of TRIM5α – RING, B-box 2, coiled-coil, and PRYSPRY (B30.2) – are indicated. In TRIM-Cyp, the PRYSPRY (B30.2) domain has been replaced with cyclophilin A (Cyp). Details are provided in the text.
Interestingly, in some primate species (e.g., owl monkey) the PRYSPRY domain has been replaced by cyclophilin A (CypA) to generate what is referred to as a “TRIM-Cyp” fusion (Luban, 2007; Nisole et al., 2004; Sayah et al., 2004; Stoye and Yap, 2008). The CypA portion of TRIM-Cyp binds CA (Gamble et al., 1996; Luban et al., 1993) and disruption of this interaction abolishes the restriction activity of TRIM-Cyp (Towers et al., 2003). Indeed, fusion of CypA to the C-termini of non-restricting TRIM proteins (e.g., TRIM1, 18 or 19) generated functional HIV-1 restriction factors (Yap, Dodding, and Stoye, 2006). The CypA portion of TRIM-Cyp therefore replaces the CA binding function of the PRYSPRY domain in TRIM5-mediated retroviral restriction.
CA binding recruits the N-terminal tripartite RBCC domain to the viral core. Each of the RBCC domains contributes to efficient retroviral restriction. TRIM proteins assemble in cells into low molecular weight oligomers (dimers or trimers), as well as large aggregations known as cytoplasmic bodies (Kar et al., 2008; Langelier et al., 2008; Mische et al., 2005; Stremlau et al., 2004). The primary function of the coiled-coil domain is to mediate oligomerization to increase CA binding (Javanbakht et al., 2006; Mische et al., 2005; Rold and Aiken, 2008; Yap et al., 2007). The role of the B-box is the least well understood but it has been implicated in several functions, including the capacity to induce higher-order self-association of TRIM5α oligomers to promote cooperative binding to the multimeric retroviral CA (Li and Sodroski, 2008). The ring domain possesses E3 ubiquitin ligase activity and is thought to recruit cellular proteasomal degradation machinery that plays an important role in TRIM5α restriction (Anderson et al., 2006; Campbell et al., 2008; Rold and Aiken, 2008; Wu et al., 2006; Xu et al., 2003; Yamauchi et al., 2008).
TRIM5α provides a conceptual framework for the development of novel antiretroviral strategies that target the vulnerable incoming viral core and disrupt early events in HIV-1 replication. As TRIM5αhu is ineffective against HIV-1, simple use of interferon to induce its expression (Asaoka et al., 2005; Sakuma, Mael, and Ikeda, 2007) is unlikely to be therapeutically effective. An alternative strategy is to use gene delivery to express an isoform of TRIM5α that potently inhibits HIV-1. As described above, expression of non-human primate TRIM5α's, such as TRIM5αrh, in human cells restricts HIV-1. However, these non-human TRIM5α's are likely to be recognized as foreign in vivo and to provoke an immune response. The restriction specificity of TRIM5α can be manipulated by genetically engineering the CA-recognizing PRYSPRY domain. Chimeric human-rhesus TRIM5α molecules may therefore be more immunologically tolerated for a gene therapy application (Anderson and Akkina, 2008). The gene therapy approach has yielded initial promising results in vitro in a range of human cell types including CD34+ hematopoietic progenitor stem cells that give rise to both T cells and macrophages, and in vivo in SCID-hu mouse-derived thymocytes (Anderson and Akkina, 2005; Anderson and Akkina, 2008; Sakuma et al., 2007). While theoretically possible, a TRIM5α-mediated gene therapy approach is not currently practical for treating HIV-1 infected patients.
Small molecules are a more realistic and cost-effective strategy for antiretroviral therapy. TRIM5α-like molecules that restrict HIV-1 could potentially be developed; however, the requirements for restriction are complex, involving multiple protein domains that function as part of an oligomer. Indeed, a study to design artificial restriction factors concluded that multimerization of a CA-binding domain could be the common minimal design feature for CA-dependent retroviral restriction (Yap et al., 2007). The design of a peptide with restriction activity that is capable of delivery into the cell therefore seems unrealistic. However, it is worth noting that in a recent study the peptide CAI that inhibits virus assembly and maturation by binding to CA (see sections B3.2 and B4.2) was also observed to dismantle CA tubes preassembled in vitro, suggesting that CAI binding to CA may also affect core stability (Barklis et al., 2009). An alternative approach could exploit the fact that a single-amino-acid substitution is sufficient to enable TRIM5αhu to inhibit HIV-1, suggesting that a conformational change can transform TRIM5αhu into an effective restriction factor (Li et al., 2006). Binding of a small molecule to TRIM5αhu could perhaps induce such a conformational change. This approach would present a major challenge, as most drugs are designed to either disrupt an interaction or inhibit an enzymatic activity rather than to elicit a gain-of-function interaction between two binding partners. Also, a suitable high-throughput screen to identify such molecules would need to be developed. Further elucidation of the mechanism of restriction will be required to advance these and other future therapeutic strategies. A significant advance will be provided by high-resolution structures of TRIM5α or TRIM-Cyp bound to the viral core.
B2.2 RNase H Inhibitors
RT plays an essential role in HIV-1 replication, as it copies viral RNA into double-stranded DNA for integration into the host cell genome (Freed, 2007). This is achieved through the use of two distinct enzymatic activities (Champoux and Schultz, 2009; Sarafianos et al., 2009; Schultz and Champoux, 2008). First, as a DNA polymerase, RT copies an RNA and then a DNA template to generate minus- and plus-strand viral DNA, respectively. Second, RT possesses an RNase H activity (Fig. 4) that degrades the RNA strand within the RNA-DNA duplex(es) formed during minus-strand DNA synthesis. This activity also serves to create the plus-strand primers required for initiation of plus-strand DNA synthesis, as well as to remove the minus- and plus-strand primers once synthesis is complete. Inhibitors that target RT polymerase activity have been highly successful, with multiple drugs routinely used to treat HIV-1-infected patients [see (Cihlar, 2010; de bethune, 2010) in this issue]. Development of drugs targeting RNase H activity, however, has proven to be more difficult, and no RNase H inhibitors (RNHIs) have been approved for clinical use (Jochmans, 2008; Klumpp and Mirzadegan, 2006; Yu et al., 2008). Nevertheless, RNase H remains an attractive drug target, as its activity is essential for HIV-1 replication (Tisdale et al., 1991).
Fig. 4.
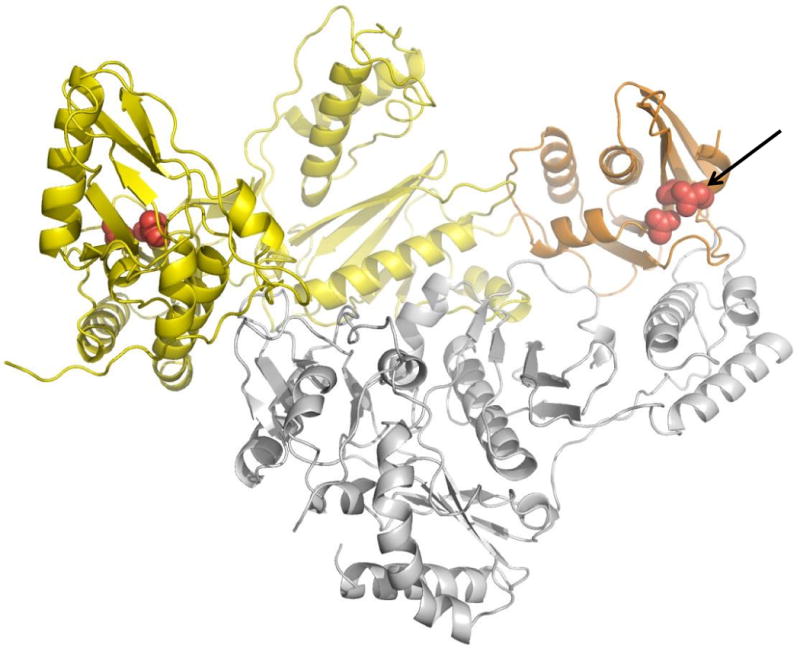
Structure of HIV-1 RT. The polymerase domain is in yellow, RNase H in orange, and p51 subunit in gray. The polymerase and RNase H active sites are highlighted in red and the RNase H active site is also indicated with an arrow (Das et al., 2008). We thank Kalyan Das and Eddy Arnold for providing the figure.
Efforts to develop RNHIs have been hampered by a lack of therapeutic value, with the limited number of promising candidates tested in vitro frequently encountering problems of poor cellular uptake or cytotoxicity or the targeting of activities other than RNase H (Jochmans, 2008; Klumpp and Mirzadegan, 2006; Yu et al., 2008). Despite these setbacks, recent development of high-throughput RNase H assays, as well as advances in our understanding of the mechanisms of RNase H activity, have reinvigorated RNHI development efforts. The RNase H domain was the first fragment of HIV-1 RT for which the structure was solved (Davies et al., 1991). Structures of RNase H's from several sources in complex with RNA/DNA hybrid substrates have provided potential clues for future antiretroviral drug exploration (Nowotny et al., 2005; Nowotny et al., 2007; Sarafianos et al., 2001). Structural information has also contributed to an increased understanding of the molecular mechanisms of RNase H activity (Nowotny et al., 2005; Nowotny et al., 2007; Nowotny and Yang, 2006; Yang, Lee, and Nowotny, 2006). Specifically, it has become clear that HIV-1 RNase H utilizes a two metal ion-dependent mechanism of catalysis, and residues critical for positioning the RNA strand within the RNase H active site, as well as the catalytic residues themselves, have been identified (Cristofaro et al., 2002; Davies et al., 1991; Klumpp et al., 2003).
An important class of RNHIs targets the metal-ion requirement of RNase H. Small molecules that act by this mechanism can be divided into three major groups i) N-hydroxyimides (Hang et al., 2004; Klumpp et al., 2003) ii) diketo acids (Shaw-Reid et al., 2003; Tramontano et al., 2005) and iii) hydroxylated tropolones (Beilhartz et al., 2009; Budihas et al., 2005; Didierjean et al., 2005). Other classes of RNHIs with alternative or undefined mechanisms of action include; i) hydrazones (Borkow et al., 1997; Himmel et al., 2006), i) vinologous ureas (Wendeler et al., 2008), iii) napthoquinones (Min, Miyashiro, and Hattori, 2002) and iv) small nucleic acid fragments (aptamers) (Hannoush et al., 2004; Somasunderam et al., 2005). Cellular uptake and cytotoxicity issues remain challenging; however, a small number of molecules with acceptable levels of cytotoxicity have been shown to exhibit antiviral activity in cells (Borkow et al., 1997; Somasunderam et al., 2005; Tramontano et al., 2005). Moreover, inhibitor screening has recently become more sophisticated, as lead compounds are scored not only according to the degree with which they inhibit retroviral RNase H, but also on whether they inhibit human RNase H (Budihas et al., 2005). Despite recent progress, none of the current RNHIs have entered clinical trials and an outstanding lead candidate whose antiviral properties can be definitively linked to RNase H activity has yet to be identified (Jochmans, 2008; Klumpp and Mirzadegan, 2006; Yu et al., 2008).
B2.3. LEDGF/p75 as a novel target for integration inhibitors
As mentioned above, after reverse transcription and nuclear import of the newly synthesized viral DNA, IN catalyzes the insertion of the viral DNA into the host cell genome. While purified IN can mediate most aspects of the integration reaction in vitro, numerous studies have demonstrated that other proteins of both viral and cellular origin enhance the efficiency of integration and are likely to be important for integration in the context of a viral infection. The host factor that has received the most attention in recent years is LEDGF/p75 [for review see (Engelman and Cherepanov, 2008)]. This chromatin-associated protein is thought to normally function in transcriptional regulation in stress response and apoptosis pathways (Ganapathy, Daniels, and Casiano, 2003). A role for LEDGF/p75 in lentiviral integration was first suggested by studies that identified an interaction between this protein and HIV-1 IN (Cherepanov et al., 2003; Emiliani et al., 2005). Depletion of LEDGF/p75 shifted the localization of exogenously expressed IN from the nucleus to the cytoplasm, implying a functional relevance for this interaction. Initial studies on the effect of LEDGF/p75 depletion on HIV-1 infectivity produced conflicting and inconsistent results. However, more complete knock-downs, or the use of mouse embryo fibroblasts derived from LEDGF/p75 knock-out mice, revealed a significant loss in HIV-1 infectivity in the absence of this host factor (Llano et al., 2006; Shun et al., 2007). The defect imposed by LEDGF/p75 depletion is at the level of integration, and infectivity of retroviruses that do not interact with LEDGF/p75 is not affected by LEDGF/p75 disruption. Furthermore, overexpression of the IN-binding domain (IBD) of LEDGF/p75 also imposes a severe defect in HIV-1 infectivity (De Rijck et al., 2006; Llano et al., 2006). The preferential targeting of HIV-1 integration to actively transcribed regions of the host cell genome (Schroder et al., 2002) is to a large extent eliminated by LEDGF/p75 knock-down (Ciuffi et al., 2005; Marshall et al., 2007; Shun et al., 2007).
LEDGF/p75 is a multidomain protein that not only binds IN via its IBD but also bears several motifs that function in DNA and chromatin binding (Fig. 5). These include a nuclear localization signal (NLS) and two AT-hooks (so named for their preferential binding to AT-rich DNA), and a PWWP motif that binds chromatin [reviewed in (Engelman and Cherepanov, 2008)]. Structural studies have elucidated the basis for the interaction between IN and the IBD of LEDGF/p75. IN is known to form multimers (dimers and tetramers) and an interhelical loop in the LEDGF/p75 IBD fits into a cleft formed by the dimer interface of IN (Cherepanov et al., 2005). While the catalytic core of IN is primarily responsible for binding to LEDGF/p75, the N-terminal domain of IN contributes charge-charge interactions (Hare et al., 2009).
Fig. 5.

Schematic representation of newly synthesized viral DNA tethered to chromatin by LEDGF. The preintegration complex (green) containing a tetramer of IN (yellow) and double-stranded viral DNA (vDNA) is shown. The IN-binding domain (IBD) of LEDGF is shown in association with the IN tetramer. The nuclear localization signal (NLS) and AT hooks (AT) are shown bound to DNA; the PWWP domain is depicted bound to histone proteins. Note that it remains to be established to what extent the PIC is intact at the stage of chromatin tethering.
The demonstrated importance of the association between IN and LEDGF/p75 in HIV-1 integration raises the possibility that this interaction could be exploited as an antiviral target. Indeed, overexpression of the LEDGF/p75 IBD was shown to significantly inhibit HIV-1 replication (De Rijck et al., 2006). Resistance to the IBD fragment arose during virus passaging; this resistance was conferred by mutations in IN that map to the IN/IBD interface. Interestingly, replication of the IBD-resistant virus was even more sensitive to LEDGF/p75 depletion than was that of the WT virus, indicating that the resistant mutant did not replicate in a LEDGF/p75-independent manner (Hombrouck et al., 2007). To identify small molecules that disrupt the interaction between IN and LEDGF/p75, Du and colleagues (Du et al., 2008) performed a small-scale screen based on yeast and mammalian two-hybrid assays in which reductions in IN-LEDGF/p75 binding could be detected. A benzoic acid derivative, 4-[(5-bromo-4-{2,4-dioxo-3-(2-oxo-2-phenylethyl)-1,3-thiazolidin-5-ylidene]methyl}-2-ethoxyphenoxy)methyl]benzoic acid (D77) scored positive in these assays. Surface plasmon resonance (SPR) analysis demonstrated a direct interaction between D77 and the catalytic core domain of IN (with Kd ∼ 6 μM). Molecular docking and mutational analyses suggested that D77 binds at the dimer interface of IN. In cell-based assays, D77 disrupted the nuclear localization of IN and displayed some antiviral activity (EC50 ∼20 μg/ml).
A larger screen for inhibitors of the IN-LEDGF/p75 interaction was performed by using a luminescent proximity (AlphaScreen™) assay with the LEDGF/p75 IBD and the IN catalytic core domain (Hou et al., 2008). A library of 700,000 small molecules was screened and ∼90 compounds were shown to selectively inhibit IN-LEDGF/p75 binding. One representative compound disrupted integration in vitro. The ability of the putative inhibitors to interfere with integration in cell-based assays and to elicit antiviral activity awaits further testing.
Hayouka et al. (Hayouka et al., 2007) used a more directed approach by synthesizing peptides derived from LEDGF/p75. These peptides bound to IN, reportedly shifting the IN multimer from the dimeric form, which is competent to bind DNA, to the tetramer, which is not DNA-binding proficient (Faure et al., 2005). These peptides disrupted in vitro integration and inhibited HIV-1 infectivity and replication in cell culture (Hayouka et al., 2007).
As with any strategy that targets a cellular gene, the toxicity of LEDGF/p75-based inhibitors is an important concern. Although LEDGF/p75 knock-out mice display a high level of embryonic lethality (Sutherland et al., 2006), LEDGF/p75 disruption in cell culture appears to be well tolerated. More importantly, LEDGF/p75-based antiviral strategies currently being developed (see above) are aimed at disrupting the IN-LEDGF/p75 interaction rather than suppressing expression of the host factor itself; such inhibitors could, in principle, target either the host (LEDGF/p75) or viral (IN) protein. The LEDGF/p75 IBD may engage in interactions with vital cellular partners, and indeed, the C-terminus of LEDGF/p75 has been shown to interact with the host factor JP02 (Bartholomeeusen et al., 2007; Maertens, Cherepanov, and Engelman, 2006). However, IN and JP02 appear to utilize non-overlapping interfaces in their interactions with LEDGF/p75 (Bartholomeeusen et al., 2007). It is therefore likely that inhibitors of IN-LEDGF/p75 binding can be developed that do not disrupt the interaction between LEDGF/p75 and its cellular partners.
B3. Assembly and Release
The HIV-1 Gag precursor protein, Pr55Gag, is the sole viral component required to form immature, non-infectious VLPs in Gag-expressing cells. The production of infectious particles requires coexpression of Gag with the Env glycoproteins, and the pol-encoded enzymes PR, RT, and IN. Assembly takes place in a series of discrete steps. The MA domain directs Gag to the host cell plasma membrane where it anchors Gag in the inner leaflet of the lipid bilayer. The MA domain also plays a crucial role in recruiting the viral Env glycoproteins into nascent virus particles. The CA domain, together with SP1 and NC, mediates Gag-Gag interactions that promote particle assembly. NC also interacts with the full-length viral genomic RNA to package two RNA copies into each virus particle. Sequences in p6 known as “late domains” interact with cellular endosomal sorting machinery to promote virus budding and release from the infected cell. Concomitant with virus release, PR cleaves the Gag and GagPol precursor proteins to initiate the maturation process.
There are currently no approved drugs that target Gag or any aspect of the virus assembly pathway. However, as our understanding of assembly has increased, a variety of assembly- and maturation-based targets have emerged as potential targets for novel antiviral therapies.
B3.1 MA
The MA domain of Gag (Fig. 2) serves several important functions in HIV-1 particle production. It provides the signals necessary to target Gag to the site of assembly, which in most instances is the plasma membrane. The N-terminus of MA is modified with a covalently attached myristic acid moiety that directly inserts into the lipid bilayer. Membrane binding is also promoted by a highly basic patch of amino acid residues near the N-terminus of the MA domain (residues ∼17-31); this positively charged region interacts with negatively charged phospholipids on the inner leaflet of the lipid bilayer. Mutations that block myristylation abolish Gag-membrane binding (Bryant and Ratner, 1990; Freed et al., 1994; Gottlinger, Sodroski, and Haseltine, 1989), whereas disruption of the basic residues causes Gag to be relocalized to late endosomes or MVBs (Ono 2004). The localization of a number of cellular proteins that contain bipartite membrane binding domains composed of one or multiple acyl groups and a highly basic patch of amino acids is regulated by a family of lipid molecules known as the phosphoinositides. These lipids form a family of molecules that differ from one another in the position and number of phosphates on the inositol headgroup (De Camilli et al., 1996). Different phosphoinositides are enriched on specific membranes within the cell; for example, phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] is predominantly localized on the inner leaflet of the plasma membrane; PI(3)P is found on early endosomal membranes, and PI(3,5)P2 is concentrated on late endosomes (De Matteis and Godi, 2004). To test whether plasma membrane-localized PI(4,5)P2 plays a role in HIV-1 Gag targeting, PI(4,5)P2 was depleted from the plasma membrane by overexpressing enzymes involved in phospholipid metabolism (Ono et al., 2004). This PI(4,5)P2 disruption induced a relocalization of Gag to late endosomes, recapitulating the phenotype of MA basic residue mutations, and markedly inhibited particle production (Ono et al., 2004). These findings raised the possibility that the basic residues in MA engage in direct interactions with PI(4,5)P2. This hypothesis was validated by nuclear magnetic resonance (NMR) spectroscopy (Saad et al., 2006) and by a mass spectrometric protein footprinting approach (Shkriabai et al., 2006). In the Saad et al. study, the structure of the MA domain bound to a soluble, truncated PI(4,5)P2 derivative was determined (Fig. 6); two particularly interesting observations were made: 1) binding of MA to PI(4,5)P2 induces the exposure of the N-terminal myristate, which in its membrane-unbound state equilibrates between an exposed and a sequestered conformation, and 2) the 2′-acyl chain of PI(4,5)P2 packs into a hydrophobic cleft in MA. Whether such a conformation would be energetically favorable in the context of Gag bound to membrane-embedded PI(4,5)P2 remains to be determined. If so, the hydrophobic cleft into which the 2′-acyl chain packs could be targeted by small molecules that would block PI(4,5)P2 binding. HIV-2 and EIAV MA proteins have also been found to bind PI(4,5)P2 (Chen et al., 2008; Saad et al., 2008) suggesting that interaction with this phosphoinositide may be a general strategy used by retroviruses to target the plasma membrane.
Fig. 6.
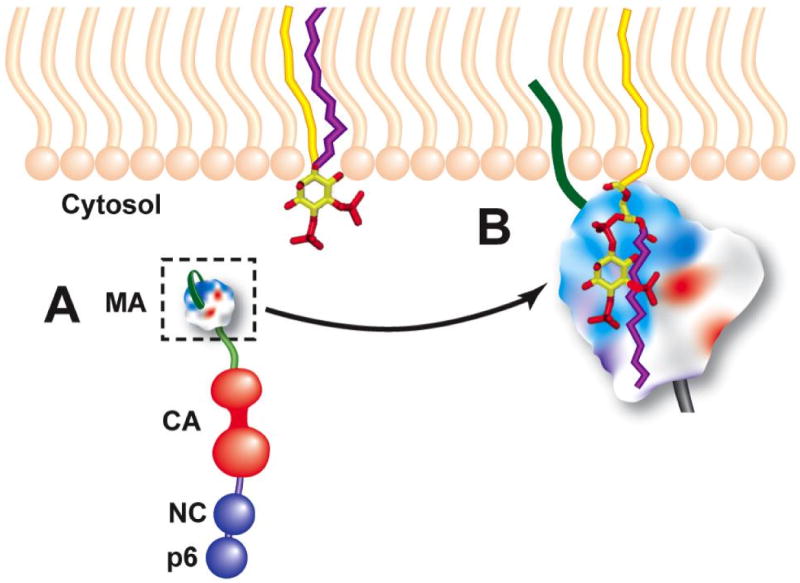
Model for MA binding to PI(4,5)P2. (A) shows the unbound Gag monomer, with the myristic acid moiety (dark green) in the sequestered conformation. Unbound PI(4,5)P2 is shown with both 1′- and 2′-acyl chains (yellow and purple, respectively) embedded in the inner leaflet of lipid bilayer. (B) shows MA bound to PI(4,5)P2, with the myristic acid in the exposed conformation and embedded in the lipid bilayer, basic residues of MA (blue) engaged in electrostatic interactions with negative charges on PI(4,5)P2, and the 2′-acyl chain extruded from the bilayer and packed into a hydrophobic groove in MA. This model is based on the NMR study of Saad et al. (Saad et al., 2006). Reprinted from (Freed, 2006).
As mentioned above, the myristic acid moiety switches between an exposed and a sequestered conformation; in the sequestered conformation it packs into a hydrophobic pocket distinct from the putative PI(4,5)P2-binding cleft (Tang et al., 2004). Successful targeting of this hydrophobic cavity with small molecules would in theory deregulate the myristyl switch, leading to defects in virus assembly. Finally, MA promotes the incorporation of the Env glycoprotein complex into virions, either via a direct gp41-MA interaction or with the assistance of a host factor [e.g., TIP-47 (Diaz and Pfeffer, 1998; Lopez-Verges et al., 2006)] that bridges gp41 and MA. Although we currently have limited structural information about how the gp41 cytoplasmic tail fits in the assembled MA lattice, any molecule that would be capable of disrupting the gp41-MA interaction, and thereby inhibit Env incorporation, would be predicted to display significant antiviral activity.
B3.2 CA
The CA domain (Fig. 2), together with SP1 and NC, mediates Gag-Gag interactions that are required for immature virus particle assembly (Adamson and Freed, 2007). Approximately 5000 Gag molecules multimerize to form a spherical shell (Briggs et al., 2004). Cryo- and high-resolution electron microscopy have allowed visualization of Gag within the immature particle. Gag molecules are rod-shaped and packed side-by-side in a radial arrangement (Fuller et al., 1997; Wilk et al., 2001; Yeager et al., 1998). The N-terminal MA domain is associated with the membrane and the C-terminus of Gag is orientated towards the particle center (Fuller et al., 1997; Wilk et al., 2001; Yeager et al., 1998). In the immature VLP, the Gag molecules form a continuous yet incomplete hexameric lattice with irregular defects that accommodate curvature (Briggs et al., 2009; Wright et al., 2007). The CA domain is the major determinant of the ordered lattice (Briggs et al., 2009; Wright et al., 2007).
CA is composed of two structurally independent and largely helical domains, known as the CA N-terminal (CANTD) and C-terminal (CACTD) domains, which are separated by a short flexible interdomain linker (Fig. 7A) (Adamson, Salzwedel, and Freed, 2009; Ganser-Pornillos, Yeager, and Sundquist, 2008). Mutagenesis of the CANTD suggests that it does not play a major role in driving Gag assembly (Accola, Hoglund, and Gottlinger, 1998; Borsetti, Ohagen, and Gottlinger, 1998; von Schwedler et al., 2003). However, the CACTD plays an important role in Gag multimerization. CACTD forms a dimer (Gamble et al., 1997; Rose et al., 1992; Worthylake et al., 1999), the disruption of which leads to a significant reduction in Gag intermolecular interactions in vitro (Burniston et al., 1999) and particle production in cells (Gamble et al., 1997; von Schwedler et al., 2003). However, assembly is not completely abolished by disruption of the CACTD dimer, demonstrating that the CACTD dimer interface is not absolutely required for VLP assembly. Near the N-terminus of the CACTD is the major homology region (MHR), a stretch of 20 residues highly conserved across retroviral genera (Gamble et al., 1997; Wills and Craven, 1991) that forms an intricate array of hydrogen bonds in the CA crystal structure (Gamble et al., 1997). Amino acid substitutions in the MHR lead to defects in assembly (Mammano et al., 1994; von Schwedler et al., 2003). A high degree of structural homology between the MHR and a mammalian SCAN domain led to the hypothesis that the MHR forms a domain-swapped dimer (Ivanov et al., 2005; Kingston and Vogt, 2005). However, limited experimental evidence has so far corroborated this model. Therefore, the contribution of the MHR to Gag multimerization and assembly remains ill defined.
Fig. 7.
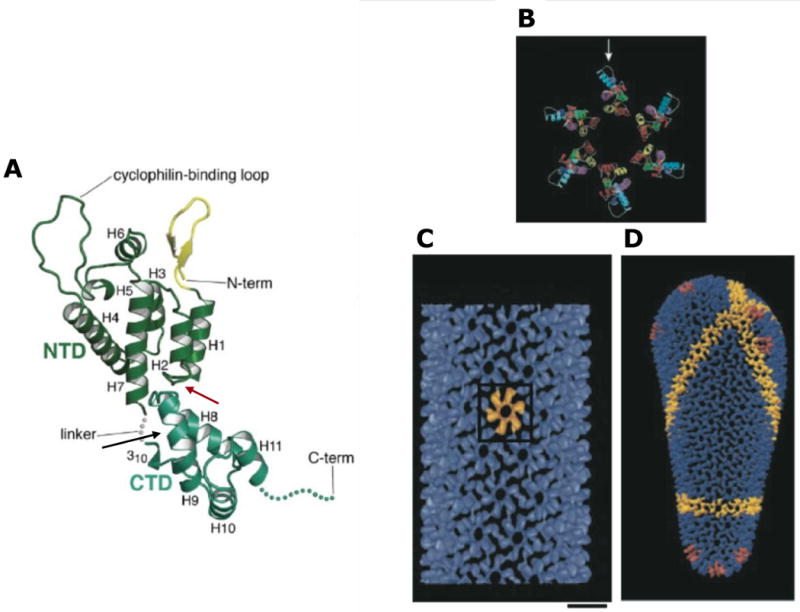
Structure of HIV-1 CA. (A) Structure of monomeric CA, with the CANTD (green) and CACTD (blue/green) indicated. The interdomain linker, N- and C-termini, and cyclophilin A binding loop are shown. Helices 1-11 and the N-terminal β-hairpin (yellow) are labeled. Binding sites for CAP1 and CAI/NYAD-1/NYAD-13 are indicated by red and black arrows, respectively. Reprinted with permission from Elsevier (Ganser-Pornillos, Yeager, and Sundquist, 2008). (B) Molecular model of the CANTD hexameric ring; cyclophilin A binding loop indicated with an arrow. (C) Outside view of an assembled CA tube, showing the CA hexameric lattice. One CA hexamer is shown in yellow. Scale bar = 100 Å. (D) Molecular model of an HIV-1 conical core. A line of hexamers is shown in yellow; pentamers are depicted in red at each end of the conical core. Adapted with permission from Macmillan Publishers Ltd: [Nature], (Li et al., 2000), http://www.nature.com/nature/index.html.
The last 12 residues of CA and the adjoining SP1 project from the base of the globular CACTD. This region is highly flexible and thus disordered in crystal structures (Gamble et al., 1997; Newman et al., 2004; Worthylake et al., 1999) but appears to possess helical character (Accola, Hoglund, and Gottlinger, 1998; Morellet et al., 2005; Newman et al., 2004). A recent cryo-electron tomography study of immature particles proposed that this domain forms a six-helix bundle that stabilizes the CA hexamer (Wright et al., 2007). Indeed, genetic studies have demonstrated that this region of Gag forms a critical assembly domain, which mediates strong Gag-Gag interactions that lead to higher-order multimerization (Abdurahman et al., 2004; Accola, Hoglund, and Gottlinger, 1998; Accola, Strack, and Gottlinger, 2000; Guo et al., 2005; Krausslich et al., 1995; Liang et al., 2002; Liang et al., 2003; Melamed et al., 2004; Morikawa et al., 2000; Ono, Demirov, and Freed, 2000; von Schwedler et al., 2003).
In principle, assembly could be disrupted therapeutically by small molecules that bind to critical assembly domains within CA. The mature CA protein also drives a second assembly event to form the viral core following Gag proteolytic cleavage (see section B4.2). Inhibitors that bind CA could therefore disrupt the assembly of both immature VLPs and cores. One such dual inhibitor, CA assembly inhibitor (CAI), has been reported. CAI is a peptide identified in a phage display screen that used both full-length CA and a protein fragment consisting of CACTD/SP1/NC (C-CANC) as bait (Sticht et al., 2005). CAI inhibits in vitro assembly of spherical particles that are structurally analogous to immature VLPs (Sticht et al., 2005). Assembly of tubular structures, in which CA is organized as in the mature viral core, was also disrupted (Sticht et al., 2005). CAI had no effect on virus assembly in cells due to its inherent cell impermeability. This problem was resolved by using hydrocarbon stapling to generate cyclical cell-penetrating derivatives of CAI, termed NYAD-1 and -13 (Bhattacharya et al., 2008; Zhang et al., 2008). In cells, NYAD-1 exhibited an antiviral effect and assembly of both immature and mature-like virus particles was disrupted (Zhang et al., 2008).
CAI and its derivatives target the CACTD (Fig. 7A)(Bhattacharya et al., 2008; Sticht et al., 2005; Ternois et al., 2005). A high-resolution X-ray structure of CAI in complex with CACTD reveals that the peptide binds to a hydrophobic groove in an α-helical conformation (Ternois et al., 2005). Binding of CAI appears to allosterically disrupt the CA dimer interface and interfere with contacts between the CANTD and CACTD (Bartonova et al., 2008; Bhattacharya et al., 2008; Sticht et al., 2005; Ternois et al., 2005). Understanding the consequences of CAI binding for assembly of viral cores has been possible due to detailed structural information on the interactions involved [see section B4.2 and (Adamson, Salzwedel, and Freed, 2009; Ganser-Pornillos, Yeager, and Sundquist, 2008)]. However, the effect of CAI binding on immature particle assembly is less clear given that Gag-Gag interfaces required for VLP assembly are not fully defined. It is tempting to speculate that the effect of CAI binding at the dimer interface may be important, given the role of this region of CACTD in immature particle assembly.
Although the issue of CAI cell permeability has been addressed by the generation of NYAD-1 and 13, these molecules bind with relatively low affinity (Sticht et al., 2005; Zhang et al., 2008) and are therefore not likely to progress to the clinic. They do, however, represent significant leads for the development of future HIV-1 assembly inhibitors. Given that Gag-Gag interactions appear to involve several distinct domains, it might be considered surprising that the screen by Sticht and colleagues identified a group of related peptides that bind to a single reactive site in CA (Sticht et al., 2005). The fact that CAI binds to a hydrophobic groove might explain the favorability of this site, as defined pockets to which an inhibitor can bind are more likely to yield successful drug candidates. The fact that assembly inhibitors will target interacting surfaces that may be relatively flat and cover large areas, and are therefore problematic for drug discovery, represents a significant future challenge for the development of this class of antiretrovirals (Arkin and Wells, 2004).
B3.3 NC
HIV-1 NC (Fig. 2) is a small (7 kDa), highly basic protein characterized by the presence of two zinc-finger motifs. These zinc fingers are critical to several of NC′s functions; indeed, the presence of one or two zinc fingers in NC is one of the most highly conserved features of retroviral Gag proteins. The consensus zinc-coordinating motif found in retroviral NC proteins is Cys-X2-Cys-X4-His-X4-Cys (CCHC); this sequence is somewhat atypical for cellular zinc-finger motifs. Mutations that disrupt the NC zinc fingers abolish genomic RNA encapsidation and virus infectivity (Gorelick et al., 1988; Gorelick et al., 1990). NC performs several important functions during the virus replication cycle (Levin et al., 2005). As a domain of Pr55Gag, it binds and packages the viral RNA genome into nascent virions. This nucleic acid binding property also allows NC to promote Gag-Gag interactions during assembly. After cleavage of NC from the Gag precursor by the viral PR, NC functions as a nucleic acid chaperone to stimulate reverse transcription and integration.
A number of NC-based HIV-1 inhibitors have been reported, most of which target the zinc fingers. Early efforts focused on cell-permeable oxidizing agents that caused zinc to be “ejected” from the zinc fingers (Rice et al., 1993; Rice et al., 1995). One such compound, 2,2-dipyridyl disulfide (Aldrithiol-2 or AT-2), induces extensive cross-linking and has been used in a number of studies as a tool to inactivate HIV-1 virions for experimental purposes [e.g., (Lifson et al., 2004)]. Another compound, N-ethylmaleimide (NEM), inactivates virus infectivity by alkylating the zinc-bound thiols in NC (Chertova et al., 1998). More selective inactivation of NC zinc fingers was observed with the sulfhydryl compound 4-vinylpyridine (4-VP), which, when combined with a membrane-permeable divalent cation chelating agent, potently inactivated HIV-1 infectivity (Morcock et al., 2008). Appella and coworkers have focused on a series of zinc-finger-based inhibitors in the 2-mercaptobezamide thioester class that display low micromolar antiviral activity in culture (Song et al., 2002; Turpin et al., 1999). One of these compounds reduced infectious virus production in an HIV transgenic mouse model system (Schito et al., 2003). Although specificity continues to be an issue with the zinc-ejecting compounds, recent studies have reported specific binding between the 2-mercaptobezamide thioester compounds and NC (Jenkins et al., 2006).
In an alternative strategy that does not involve zinc ejection from the zinc fingers, Shvadchak and colleagues (Shvadchak et al., 2009) screened a chemical library for compounds that block the nucleic acid chaperone activity of NC; specifically, by preventing NC from unfolding a doubly labeled DNA stem-loop structure. This screen identified several compounds that bind the NC zinc fingers and prevent the protein from interacting with the DNA stem-loop. Other studies have focused on compounds in the aminoglycosidic antibiotic class that interact not with NC itself but with the genomic RNA packaging signal, a series of stem-loop structures at the 5′ end of the HIV-1 genome that direct RNA encapsidation into virions (Turner, Hagan, and Fabris, 2006).
B3.4 p6
The budding of HIV-1, like that of other retroviruses, proceeds via the hijacking of cellular endosomal sorting machinery (Fig. 8) (Bieniasz, 2006; Demirov and Freed, 2004; Morita and Sundquist, 2004). This machinery normally promotes the inward budding of vesicles into late endosomes to generate MVBs and also functions during the membrane fission, or “budding-off”, step of cytokinesis (McDonald and Martin-Serrano, 2009). Because virus particle budding from the plasma membrane is topologically equivalent to vesicle budding into late endosomes (oriented away from the cytosol), retroviruses evolved to usurp endosomal sorting machinery to drive the pinching-off of virions from the plasma membrane. At the core of this machinery are three multiprotein complexes known as the endosomal sorting complexes required for transport (ESCRT) I, II, and III. An additional complex, sometimes referred to as ESCRT-0, sorts cargo proteins from early to late endosomes (Hurley and Emr, 2006). Several accessory molecules associate with the ESCRT complexes, including ALG-2-interacting protein X (Alix) and the AAA ATPase Vps4.
Fig. 8.
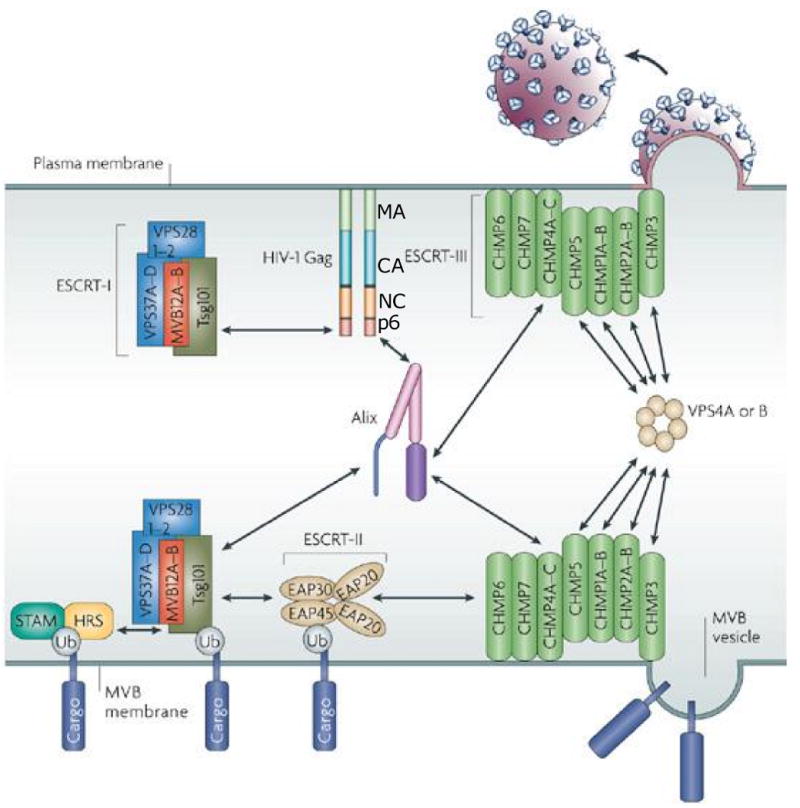
Role of ESCRT and associated machinery in the sorting of cargo proteins to multivesicular bodies (MVBs) and in virus release. At the bottom is depicted the interaction of ubiquitinated (Ub) cargo protein with the STAM/Hrs complex and ESCRT-I, II, and III and the delivery of the cargo protein into a vesicles budding inwardly into the MVB. At the top is depicted the interaction of Gag with ESCRT-I and Alix and the involvement of ESCRT-III and the ATPase Vps4 in HIV-1 budding from the plasma membrane. The major Gag domains – MA, CA, NC, and p6 – are indicated. For additional details, see text, and (Fujii, Hurley, and Freed, 2007). Adapted with permission from Macmillan Publishers Ltd: [Nature Reviews Microbiology], (Fujii, Hurley, and Freed, 2007), http://www.nature.com/nrmicro/index.html.
The interaction between viral Gag proteins and cellular endosomal sorting machinery is mediated by small motifs in Gag known as “late domains” that directly engage their cellular partners. In the case of HIV-1, two late domains are found in the p6 portion of Gag. The major HIV-1 late domain, Pro-Thr/Ser-Ala-Pro (PTAP) binds to the ESCRT-I component Tsg101; a secondary late domain, Tyr-Pro-Xn-Leu (YPXnL, where X is a variable amino acid and n is 1-3 residues) interacts with Alix. Deletion of p6 (Gottlinger et al., 1991) or mutation of the PTAP motif (Huang et al., 1995) is highly detrimental to virus budding and abolishes or attenuates virus replication in most cell types (Demirov, Orenstein, and Freed, 2002). Mutations in the Alix binding site in p6 do not induce a marked defect in virus budding but nevertheless delay virus replication in T-cell lines and in primary T cells and macrophages (Fujii et al., 2009).
The importance of Tsg101 in HIV-1 budding was demonstrated by the findings that i) siRNA-mediated depletion of this ESCRT-I component profoundly reduces virus budding (Garrus et al., 2001), ii) Tsg101 fusion to Gag compensates for p6 mutation (Martin-Serrano, Zang, and Bieniasz, 2001), and iii) overexpression of the N-terminal, Gag-binding domain of Tsg101 exerts a dominant-negative inhibition of virus budding (Demirov et al., 2002). The inhibitory activity of this N-terminal Tsg101 fragment (referred to as TSG-5′) requires a direct TSG-5′-Gag interaction and is PTAP-dependent (Goila-Gaur et al., 2003; Shehu-Xhilaga et al., 2004). Budding and replication of feline immunodeficiency virus (FIV), a feline lentivirus that also encodes a PTAP-type late domain, are strongly inhibited by TSG-5′ (Luttge et al., 2008), demonstrating that this Tsg101 fragment can inhibit a spreading lentiviral infection. Overexpression of the Gag-binding domain of Alix (known as the “V-domain”) also imposes a potent block to HIV-1 budding (Lee et al., 2007; Munshi et al., 2007). It is important to note that disrupting the interaction between HIV-1 Gag late domains and their cellular partners has a more profound effect on virus infectivity than on particle release, since many of the particles that are produced in the absence of a functional late domain are morphologically aberrant and poorly infectious.
While tissue culture experiments demonstrate that TSG-5′ and the Alix V domain are potent inhibitors of virus budding, they are not viable therapeutics. However, high-resolution structural information is available for the p6-Tsg101 and p6-Alix interaction sites (Fig. 9); (Pornillos et al., 2002a; Pornillos et al., 2002b), making the rational design of budding inhibitors possible. As a first step toward this goal, Liu and colleagues engineered PTAP-based peptoid mimetics that display significantly increased affinity for Tsg101 relative to peptides bearing the native PTAP motif (Liu et al., 2006; Liu et al., 2008). These peptoids could, if rendered sufficiently cell permeable, act as competitive inhibitors of the p6-Tsg101 interaction. By using a reverse two-hybrid system designed to detect disruption of a protein-protein interaction, Tavassoli et al. (Tavassoli et al., 2008) screened a large library of cyclic peptides to identify a small number of peptides that interfered with the interaction between p6 and Tsg101. One of these peptides was able to inhibit HIV-1 budding several-fold in cell culture. Clearly, additional work needs to be done to identify small molecules capable of disrupting virus budding. Also to be determined is whether blocking the primary late domain of p6 (PTAP) would be sufficient to confer potent antiviral activity or whether simultaneous disruption of both p6-Tsg101 and p6-Alix interactions would be required for full efficacy.
Fig. 9.
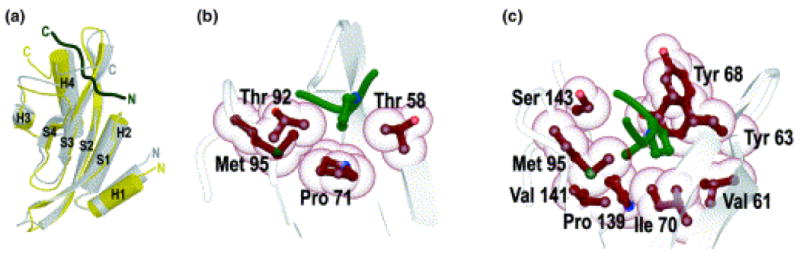
Structure of the ubiquitin enzyme 2 variant (UEV) domain of Tsg101 bound to a PTAP-containing peptide. (a) The structure of the UEV domain is shown in yellow and gray, the PTAP-containing peptide (C, C-terminus; N, N-terminus) is shown in dark green. High-resolution structure of the first Pro (b) and Ala-Pro (c) of PTAP docked in the PTAP-binding groove of Tsg101, viewed from the N-terminus of the peptide. Reprinted with permission form Macmillan Publishers Ltd: [Nature Structural and Molecular Biology], (Pornillos et al., 2002a), http://www.nature.com/nsmb/index.html.
B4. Maturation
B4.1 Maturation inhibitors that target Gag processing
HIV-1 particle maturation occurs concomitant with virus release (Fig. 1). PR initiates the maturation process by proteolytically processing the Gag and Gag-Pol polyprotein precursors. Gag is cleaved to generate the MA, CA, NC and p6 proteins and the SP1 and SP2 spacer peptides (Fig. 10A); the Pol portion of Gag-Pol is cleaved into the PR, RT and IN enzymes (Fig. 2) (Adamson and Freed, 2007; Vogt, 1996). Gag cleavage follows a sequential cascade of events that is kinetically controlled by the differential rate of processing at each of the five cleavage sites in Gag (Erickson-Viitanen et al., 1989; Krausslich et al., 1988; Mervis et al., 1988; Pettit et al., 1994; Tritch et al., 1991; Wiegers et al., 1998) (Fig. 10A). Disrupting cleavage at any of the sites in Gag, or altering the order in which the sites are cleaved, results in the formation of particles with an aberrant morphology and significantly reduced infectivity (Accola, Hoglund, and Gottlinger, 1998; Kaplan et al., 1993; Krausslich et al., 1995; Lee, Harris, and Swanstrom, 2009; Li et al., 2003; Pettit et al., 2002; Pettit et al., 1994; Wiegers et al., 1998; Zhou et al., 2004). Disrupting Gag processing thus represents an attractive therapeutic strategy for inhibiting HIV-1 replication.
Fig. 10.
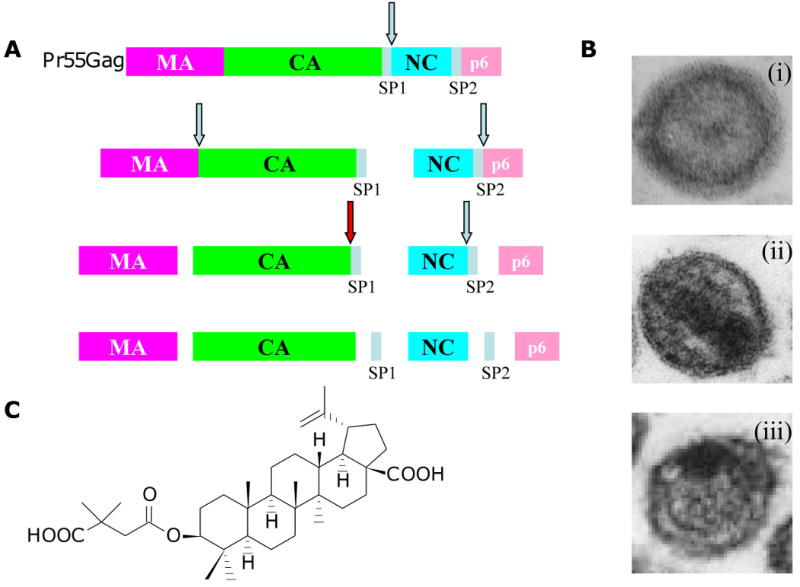

Inhibition of HIV-1 maturation by bevirimat. (A) Gag processing cascade, illustrating the order in which the Gag precursor (Pr55Gag) is cleaved by the viral protease. Red arrow depicts the cleavage event blocked by bevirimat, leading to an accumulation of the CA-SP1 cleavage intermediate. (B) Virion morphology visualized by transmission electron microscopy. Immature (i), mature (ii), and bevirimat-treated (iii) particles are shown. (C) Structure of bevirimat. (D) Amino acid sequence at the CA-SP1 boundary region; the final residue of CA (residue 231) and the first (1) and final (14) residues of SP1 are shown. Amino acids highlighted in red indicate those at which BVM resistance arises in vitro (Adamson et al., 2006); the highly polymorphic SP1 residues 6, 7, and 8 are highlighted in green. Arrows denote the site of CA-SP1 processing. Adapted with permission from Elsevier (Adamson and Freed, 2008).
PIs, which have been in the clinic for more than a decade, block Gag and Gag-Pol processing by directly inhibiting the enzymatic activity of PR [See article by Nijhuis et al. (Nijhuis, 2010) in this issue]. An alternative approach is to target individual Gag cleavage sites. While this strategy could potentially be used to block any of the Gag cleavage sites, thus far only the CA-SP1 cleavage site has been successfully targeted (Adamson, Salzwedel, and Freed, 2009; Aiken and Chen, 2005; Salzwedel, Martin, and Sakalian, 2007). The small molecule 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (DSB), also known PA-457 or bevirimat (BVM), potently inhibits HIV-1 replication by specifically blocking CA-SP1 cleavage (Fig. 10A-C) (Li et al., 2003; Zhou et al., 2004). Processing at the CA-SP1 cleavage site occurs late in the Gag cleavage cascade and when inhibited results in the formation of particles that are non-infectious because they fail to complete maturation (Wiegers et al., 1998). BVM-treated particles exhibit an aberrant morphology typified by an acentric core and an electron-dense Gag crescent inside the viral membrane (Fig. 10B) (Li et al., 2003). This morphology mirrors that induced by mutations at the CA-SP1 cleavage site (Wiegers et al., 1998). Due to its novel mechanism of action, BVM is the first in a new mechanistic class of antiretroviral drug termed maturation inhibitors, which are defined as compounds that target the substrate of the viral PR rather than the enzyme itself (Adamson, Salzwedel, and Freed, 2009; Aiken and Chen, 2005; Salzwedel, Martin, and Sakalian, 2007).
The precise mechanism by which BVM inhibits CA-SP1 cleavage has not been established. However, it is hypothesized that BVM binds to the CA-SP1 junction in Gag and prevents cleavage either by directly inhibiting access of PR to the processing site or by altering the conformation, exposure or flexibility of this region such that it is less efficiently cleaved by PR (Adamson, Salzwedel, and Freed, 2009). Several lines of evidence (discussed below) support this hypothesis, although structural information about BVM bound to its substrate has not yet been obtained. Mapping of residues that confer BVM resistance to the CA-SP1 region (Fig. 10D), and not elsewhere in Gag or in PR, strongly suggests that this region of Gag is the primary molecular target of this compound (Adamson et al., 2006; Fun et al., 2009; Li et al., 2003; Van Baelen et al., 2009; Zhou et al., 2004). Inherent BVM resistance of HIV-2 and simian immunodeficiency virus from rhesus macaques (SIVmac) is due to variability in the amino acid sequence at the CA-SP1 junction (Zhou et al., 2004). Swapping residues between the CA-SP1 junctions of HIV-1 and SIVmac results in exchange of BVM sensitivity between these two viruses (Zhou, Chen, and Aiken, 2004). In addition, some sequence divergence between HIV-1 and SIVmac occurs at residues to which BVM resistance maps in vitro (Adamson et al., 2006; Zhou et al., 2004).
BVM has been shown to bind immature but not mature HIV-1 particles, suggesting that Gag processing leads to disruption of the BVM binding site (Zhou et al., 2005). Further, BVM binds to a pocket formed during Gag oligomerization, as BVM does not inhibit CA-SP1 processing in the context of monomeric Gag in solution (Li et al., 2003), but rather requires Gag assembly for its activity (Li et al., 2003; Sakalian et al., 2006; Zhou et al., 2005). A reduction in Gag binding to immature particles has been observed in the presence of several mutations that confer resistance to BVM, suggesting that resistance is acquired by prevention of BVM-Gag binding (Zhou, Chen, and Aiken, 2006; Zhou et al., 2005). However, blocking BVM binding may not be the only mechanism by which resistance is acquired as some binding capacity is retained by two of the mutants tested (Zhou, Chen, and Aiken, 2006), and a degree of drug dependence is associated with other mutations that confer resistance to BVM (Adamson et al., 2006).
Characterization of the putative BVM binding pocket has been hindered by the lack of high-resolution structural information for the CA-SP1 junction. This region of Gag, while disordered in CA crystals, has been proposed to be α-helical (Accola, Hoglund, and Gottlinger, 1998; Gamble et al., 1997; Morellet et al., 2005; Newman et al., 2004; Worthylake et al., 1999; Wright et al., 2007). The oligomeric state of CA-SP1 region of Gag in immature particles remains unresolved, although a recent cryo-electron tomography study proposed that it forms as six-helix bundle (Wright et al., 2007). Further structure-function analysis of the CA-SP1 region of Gag will be required to more fully understand the mechanism of action of BVM. Such information may also provide a rational basis for the design of additional compounds that target this cleavage site.
The potent in vitro activity of BVM and its novel mechanism of action have encouraged its clinical development. Testing in HIV-1-infected patients was initiated following promising pharmacological and safety studies in animal models and in Phase I clinical trials (Martin, Salzwedel, and Allaway, 2008). Statistically significant, dose-dependent viral load reductions were demonstrated in initial Phase II clinical trials (Smith et al., 2007). However, further Phase II studies showed that, despite optimal BVM plasma concentrations, not all BVM-treated patients exhibited significant viral load reductions. In a Phase IIb functional monotherapy trial, ∼50% of patients receiving BVM were defined as non-responders with vial load reductions of < 0.5 log (McCallister et al., 2008). Virological parameters were hypothesized to be responsible for the observed variable clinical outcome; the presence of base-line polymorphisms at SP1 residues 6, 7 and 8 (Fig. 10D) appeared to correlate with a patient's failure to respond (Margot, Gibbs, and Miller, 2009; McCallister et al., 2008; Salzwedel et al., 2009; Salzwedel et al., 2008; Van Baelen et al., 2009). Residues 6-8 are located in the relatively non-conserved C-terminal half of SP1 and the occurrence of polymorphisms at these positions appears to be independent of prior treatment with antiretrovirals (Knapp, Huang, and Harrington, 2009; Yebra and Holgun, 2008). In vitro testing demonstrated reduced BVM-susceptibility associated with key polymorphisms located at residues 7 and 8 but not residue 6 (Van Baelen et al., 2009)(Adamson et al., unpublished data). Further studies are required to understand the relationship between these polymorphisms and HIV-1 susceptibility to BVM. However, a genotypic assay is now readily available to identify those patients that are most likely to respond to BVM treatment.
The type of resistance acquired by those patients who respond to BVM remains an outstanding question. Virus isolates with key polymorphisms at SP1 residues 6-8 are known to replicate in patients and a recent study has shown the acquisition of resistance mutations at SP1 residues 7 and 8 in in vitro selection studies with BVM (Fun et al., 2009). Therefore, it is likely that mutations at these positions will contribute to BVM resistance in patients. However, it is noteworthy that in vitro selection studies have identified a panel of six other BVM-resistance mutations that arise at positions different from SP1 residues 6-8 (Adamson et al., 2006; Li et al., 2003; Zhou et al., 2004) (Fig. 10D). Specifically, these mutations mapped to highly conserved residues at or near the C-terminus of CA (CA-H226Y, CA-L231M, CA-L231F) and the first and third residues of SP1 (SP1-A1V, SP1-A3V, SP1-A3T). It has been predicted that, of this panel, the SP1-A1V substitution is most likely to arise in vivo because it was selected most frequently and replicated robustly even at a high BVM concentration (Adamson et al., 2006; Adamson et al., 2009). The highly conserved nature of SP1 residue 1 amongst HIV-1 isolates suggests that a fitness cost would be associated with mutations at this position in vivo. However, the SP1-A1V mutant replicates efficiently in primary macrophages (Adamson, Ablan and Freed, unpublished data) and in SCID-hu Thy/Liv mice (Stoddart et al., 2007). Furthermore, SP1-A1V has been observed in isolates from two of the 46 patients participating in BVM Phase II clinical trials (Adamson, Salzwedel, and Freed, 2009). It is also noteworthy that the CA-L231M substitution has been reported in the context of one PI-experienced patient sample and the SP1-A3T mutation is present in one viral isolate listed in the Los Alamos sequence database (Malet et al., 2007; Salzwedel, Martin, and Sakalian, 2007).
Two studies have also investigated the in vitro acquisition of BVM resistance in the context of viral isolates with preexisting mutations in PR that confer resistance to PIs (Adamson et al., 2009; Fun et al., 2009). This is a significant question as BVM is likely to be used as salvage therapy for patients failing first-line drug regimens due to multi-drug resistance and are therefore likely to be PI-experienced. The impact of the PR mutations on the temporal acquisition of BVM resistance compared to wild-type (WT) virus differed between the two studies and may be dependent on the type of PR mutations or the study systems used. In both studies, however, the SP1-A1V substitution was acquired frequently. A spectrum of other mutations were also frequently acquired in the study by Fun et al. Interestingly, these mutations mapped to SP1 residues 5, 7 and 8 and to CA residue 230 (Fun et al., 2009). Ongoing clinical trials will ultimately reveal the types of resistance mutations that arise in patients who respond to BVM. The emergence of A1V, as well as the preexistence of polymorphisms at SP1 residues 6-8, will present a challenge for the successful development of BVM as a clinically effective antiretroviral drug.
BVM is the first Gag-targeted compound that has undergone clinical development. Thus, BVM opens the way for the discovery and development of other maturation inhibitors. These may include second- and third-generation inhibitors that target the CA-SP1 cleavage site. Other cleavage sites in Gag may also be viable targets. A recent study explores this possibility by introducing mutations that independently block each of the five Gag processing sites and analyzing their dominant-negative effect on virus maturation and infectivity when mixed at different ratios with the WT counterpart (Lee, Harris, and Swanstrom, 2009). Blocking cleavage at the MA-CA cleavage site resulted in the most potent inhibition, with very small amounts of the uncleaved MA-CA protein poisoning correct virus maturation and thus significantly suppressing viral infectivity (Lee, Harris, and Swanstrom, 2009). The MA-CA cleavage site therefore represents a promising future drug development target.
B4.2 Maturation inhibitors that target core assembly
HIV-1 maturation generates a condensed conical core composed of a CA lattice (Fig. 7B-D) surrounding the viral RNA genome in complex with NC, RT, and IN. The function of the core is to facilitate the delivery and reverse transcription of the viral RNA genome following infection of the target cell. The core is formed by a CA reassembly event triggered by the liberation of the CA domain from Gag upon proteolytic processing (Adamson, Salzwedel, and Freed, 2009; Ganser-Pornillos, Yeager, and Sundquist, 2008). Correct core formation and stability are essential for virus infectivity (Fitzon et al., 2000; Forshey et al., 2002; Ganser-Pornillos et al., 2004; Lee, Harris, and Swanstrom, 2009; Li et al., 2003; Reicin et al., 1996; Tang et al., 2003; von Schwedler et al., 1998; von Schwedler et al., 2003; Wiegers et al., 1998). Disrupting the CA-CA interactions required for core formation thus represents a potential therapeutic strategy for inhibiting HIV-1.
A detailed understanding of HIV-1 core morphology, organization of the CA lattice, and the CA-CA interactions required for core formation (Fig. 7B-D) has laid a solid foundation for the identification and development of maturation inhibitors that disrupt core formation (Adamson, Salzwedel, and Freed, 2009; Ganser-Pornillos, Yeager, and Sundquist, 2008). This understanding originates from the important observation that purified CA assembles in vitro to form long tubes organized in a manner similar to that of authentic cores (Ganser et al., 1999; Li et al., 2000). Furthermore, the in vitro assembly systems offer a tractable model to screen for CA-based inhibitors of core formation (Sticht et al., 2005; Zhang et al., 2008).
HIV-1 cores are usually cone-shaped, although a range of related structures have been observed (Benjamin et al., 2005; Briggs et al., 2006; Briggs et al., 2003; Welker et al., 2000). The cone is formed by a curved hexagonal CA lattice, which is closed by the strategic positioning of 12 pentamers at the ends of the cone (Fig. 7D). This model is based on the geometrical principles of a fullerene-cone (Ganser et al., 1999) and has been validated in vitro by elegant crytallographic structural and modeling studies of the CA lattice (Ganser et al., 1999; Ganser-Pornillos, Cheng, and Yeager, 2007; Ganser-Pornillos et al., 2004; Li et al., 2000; Pornillos et al., 2009) and by imaging of authentic, mature retroviral particles (Benjamin et al., 2005; Briggs et al., 2006; Briggs et al., 2003; Butan et al., 2008). The in vitro structure-based studies have provided significant insights into the CA-CA interactions that are required for core formation.
Both the CANTD and CACTD play a role in CA hexameric lattice assembly. The lattice is constructed from rigid rings of six CANTD's connected to the neighboring hexameric rings via more flexible CACTD dimer interactions (Ganser-Pornillos, Cheng, and Yeager, 2007; Li et al., 2000; Pornillos et al., 2009). This organization requires three types of interfaces: (i) a CANTD-CANTD six-fold symmetric interface that creates the hexameric rings, (ii) a CANTD-CACTD intermolecular interface between the two domains that reinforces the hexamer and (iii) a CACTD-CACTD dimeric interface that links the hexameric rings to form the lattice (Ganser-Pornillos, Cheng, and Yeager, 2007; Pornillos et al., 2009). Various genetic, biochemical and biophysical studies support the existence of these interacting interfaces (Dorfman et al., 1994; Fitzon et al., 2000; Reicin et al., 1996; Scholz et al., 2005; Tang et al., 2001; von Schwedler et al., 1998; von Schwedler et al., 2003). Each of the three major CA-CA interfaces represents a possible target for disrupting core formation.
Two key studies used different approaches to identify molecules that target CA. One study used in silico modeling to identify compounds that potentially bind clefts on the CA surface, followed by a binding screen using NMR titration spectroscopy (Tang et al., 2003). The other study, already discussed in section B3.2, used in vitro assembly assays to conduct high-throughput screening of a random peptide phage display library (Sticht et al., 2005). These studies identified two lead candidates, CAP-1 (N-(3-chloro-4-methylphenyl)-N′-{2-[({5-[(dimethylamino)-methyl]-2-furyl}-methyl)sulfanyl]ethyl}urea) and CAI, respectively. The primary mode of action of CAP-1 is to disrupt core formation (Tang et al., 2003) whereas CAI and its cell-permeable derivatives NYAD-1 and NYAD-13 inhibit both core formation and assembly of immature particles (Barklis et al., 2009; Sticht et al., 2005; Zhang et al., 2008).
High-resolution structural studies of CAP1, CAI, and NYAD-13 in complex with CA have provided valuable insights into their mechanisms of action (Bhattacharya et al., 2008; Sticht et al., 2005). CAP-1 interacts with the base of the CANTD (Fig. 7A). Binding induces a conformational change that creates the CAP-1 binding site by displacement of CANTD residue F32 from its buried position in the protein core (Kelly et al., 2007). The aromatic ring of CAP-1 is sequestered within a hydrophobic pocket vacated by F32 (Kelly et al., 2007). The position of CAP-1 and F32 suggests that CAP-1 binding inhibits the formation of the CANTD-CACTD interface of the CA lattice (Ganser-Pornillos, Cheng, and Yeager, 2007; Ganser-Pornillos, Yeager, and Sundquist, 2008; Kelly et al., 2007; Pornillos et al., 2009; Tang et al., 2003). Mutagenesis of key CA residues that interact with CAI suggests that this peptide inhibits core formation by two mechanisms (Bartonova et al., 2008). First, CAI blocks the CANTD-CACTD interaction by competing for the natural binding region in the CANTD and, second, CAI alters the CACTD-CACTD dimer interface that is important for connecting the hexameric rings in the CA lattice (Bartonova et al., 2008; Ganser-Pornillos, Cheng, and Yeager, 2007; Ganser-Pornillos, Yeager, and Sundquist, 2008; Pornillos et al., 2009; Ternois et al., 2005). As CAP-1 and CAI (and derivatives NYAD-1 and NYAD-13) interfere with the CANTD-CACTD interface, this region of CA may act as a ‘hot-spot’ for inhibitors targeting CA-CA interactions. Despite the fact that neither CAP1 nor CAI and related peptides are drug candidates, the extensive knowledge gained in studying these inhibitors and their molecular targets will likely be exploited for future rational design of more effective inhibitors that target CA-CA interactions and core formation.
B5. Accessory Proteins as Targets
In addition to the structural proteins and pol-encoded enzymes, HIV-1 encodes several accessory proteins – Vif, Vpu, Nef, and Vpr (Fig. 2). Under some circumstances, these proteins are dispensable for replication in culture (hence their designation as accessory proteins); however, in vivo these proteins appear to be crucial for virus propagation and disease induction. Recent progress has greatly increased our understanding of the mechanism by which the HIV-1 accessory proteins function, providing new opportunities for the development of antiviral agents. As mentioned in the Introduction, HIV-1 also encodes two regulatory proteins, Rev and Tat. Efforts to develop inhibitors against these proteins will not be discussed here, as this topic is covered elsewhere (Baba, 2004; Bannwarth and Gatignol, 2005; Richter and Palu, 2006).
B5.1 Vif and APOBEC-family proteins
As mentioned in the Introduction, HIV-1 and a number of other lentiviruses encode an accessory protein known as Vif that markedly enhances virus infectivity. Early studies demonstrated that this infection-stimulating effect was producer-cell-type dependent; virus derived from Vif-permissive cells was fully infectious regardless of whether or not it encoded Vif, whereas the infectivity of virus produced from Vif-nonpermissive cells displayed a strong requirement for Vif expression (Gabuzda et al., 1992). Importantly, relevant primary cell types (PBMCs and macrophages) are Vif-nonpermissive. Heterokaryons formed between Vif-permissive and Vif-nonpermissive cells exhibited the Vif-nonpermissive phenotype, suggesting that Vif enhances infectivity by counteracting a dominant restriction factor expressed in the virus-producing cell (Madani and Kabat, 1998; Simon et al., 1998). This restriction factor was ultimately identified as APOBEC3G (A3G), a member of a large family of cytosine deaminases that functions in mRNA editing and immunoglobulin gene diversification (Harris and Liddament, 2004; Sheehy et al., 2002). The related deaminase, APOBEC3F (A3F), also displays antiviral activity that is counteracted by Vif. In the absence of Vif expression, A3G is incorporated into virus particles in the producer cell, and, during reverse transcription in the next round of infection, converts cytosines to uracils (Fig. 11). This cytosine deamination leads to G-to-A hypermutation in the newly synthesized viral DNA, potentially leading to its instability (Malim, 2009). A3G also induces defects in reverse transcription and DNA integration (Bishop et al., 2008; Mbisa et al., 2007). Vif counteracts this cellular defense mechanism by recruiting components of the proteasomal pathway, e.g., cullin 5 and elongins B and C, to induce the polyubiquitination and degradation of A3G (Yu et al., 2003). The net effect of Vif-induced A3G degradation is to prevent the incorporation of A3G into virions.
Fig. 11.
Schematic representation of the counteraction of APOBEC3G by Vif. In the Vif+ setting (top), Vif (yellow) induces the proteasomal degradation of APOBEC3G (red star) in the virus-producing cell (left), enabling productive infection to occur in the target cell (right). In the absence of Vif expression (bottom), APOBEC3G is packaged into virus particles, and in the next round of infection induces the deamination of cytosines to uracils, resulting in G-to-A hypermutation. The presence of APOBEC3G also impairs reverse transcription and integration. Reprinted with permission from Elsevier (Freed, 2004).
The potent antiviral activity of A3G raises the possibility that inhibitors could be developed that prevent Vif from counteracting this cellular factor. To this end, Nathans and colleagues (Nathans et al., 2008) devised a cell-based screen in which A3G fused to yellow fluorescent protein (YFP) was expressed in cells in the presence of Vif. A library of 30,000 compounds was tested for their ability to counteract Vif-mediated degradation of A3G and thereby increase the A3G-YFP fluorescent signal. After performing secondary screens designed to exclude compounds that simply increase fluorescence or elevate gene expression, several dozen compounds were obtained. A set of these molecules was then tested for antiviral activity in permissive (A3G-deficient) or nonpermissive (A3G-expressing) cells. Two compounds were identified that displayed antiviral activity only in nonpermissive cells, suggesting that they act by preventing Vif from counteracting A3G. The more potent compound, termed RN-18, was selected for further study. Analysis of Vif and APOBEC levels demonstrated that RN-18 decreased Vif expression in the presence but not the absence of a Vif-APOBEC interaction. This APOBEC-specific loss of Vif expression led to increased levels of A3G in virions and inhibition of virus infectivity. Additional studies will be required to define in more detail the mechanism by which RN-18 leads to reduced Vif expression. In an earlier, independent report, it was observed that the membrane-permeable zinc chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN) inhibited cullen 5 recruitment by Vif and Vif-mediated A3G degradation (Xiao et al., 2007). Gabuzda and colleagues have developed a scalable fluorescence resonance energy transfer (FRET)-based Vif-A3G binding assay and have used it to identify peptides and monoclonal antibodies that block this protein-protein interaction (Mehle et al., 2007; Pery et al., 2009). This assay is useful not only for mapping the determinants of Vif-A3G binding but also, potentially, for developing a high throughput screen for inhibitors that block this binding event.
APOBEC-mediated restriction is not confined to lentiviruses. Mice encode four APOBEC family members (as compared to 11 in humans) one of which, APOBEC3, blocks infection by mouse mammary tumor virus (MMTV). APOBEC3 knock-out mice support increased MMTV replication relative to their WT litter mates (Okeoma et al., 2007) demonstrating the importance of this host antiviral defense mechanism in vivo. Treatment of mice with lipopolysaccharide (LPS) or interferon increases APOBEC3 expression, leading to suppression of MMTV replication (Okeoma et al., 2009). Interestingly, the murine leukemia virus resistance factor encoded by the recovery from Friend virus 3 (Rfv3) allele (Chesebro and Wehrly, 1979) was recently identified as APOBEC3 (Miyazawa, Tsuji-Kawahara, and Kanari, 2008; Santiago et al., 2008). Together, these studies indicate that modulation of APOBEC expression can elicit an antiviral effect in vivo. However, it remains to be determined whether such an approach would be practical in humans, and there is concern that increased expression of APOBEC family proteins could be accompanied by host cell DNA damage (Harris, Petersen-Mahrt, and Neuberger, 2002). Nevertheless, the Vif-APOBEC axis holds much promise for the development of novel antiretroviral agents and will likely continue to be the focus of active investigation.
B5.2 Vpu and tetherin
In addition to A3G and TRIM5α, a third component of the innate immune response functions at the level of virus particle release from the cell surface. Early studies on the HIV-1 accessory protein Vpu demonstrated that Vpu-deficient mutants exhibit an accumulation of mature virus particles apparently stuck to the plasma membrane of the virus-producer cell and in internal vesicles. As observed for Vif, the requirement for Vpu is producer cell-dependent, and fusions between Vpu-permissive and Vpu-nonpermissive cells exhibit the nonpermissive phenotype (Varthakavi et al., 2003). These results suggested that Vpu counteracts a dominant factor that in some way retains virus particles on the cell surface after they have budded from the plasma membrane. It was also noted that treatment of Vpu-permissive cells with interferon recapitulates the Vpu-nonpermissive phenotype, suggesting that the putative restriction factor is interferon-inducible. Two groups independently identified a protein known as CD317 or bone marrow stromal cell antigen 2 (BST-2) as the host factor counteracted by Vpu (Neil, Zang, and Bieniasz, 2008; Van Damme et al., 2008). Depletion of CD317/BST-2 from Vpu-nonpermissive cells greatly stimulated the release of Vpu(-) HIV-1; conversely, in Vpu-permissive cells that do not express CD317/BST-2, overexpression of this factor inhibited the release of Vpu(-) HIV-1 (Neil, Zang, and Bieniasz, 2008; Van Damme et al., 2008). In accordance with its apparent function as a virus-tethering factor, CD317/BST-2 was given the name tetherin (Neil, Zang, and Bieniasz, 2008). A second major function for Vpu is to induce the proteasomal degradation of CD4 (Bour and Strebel, 2003).
Shortly after the identification of tetherin as the host factor counteracted by Vpu, several interesting observations were made that increased our understanding of the tetherin-mediated restriction: 1) Tetherin proteins encoded by a variety of non-human primates and from rodents inhibit virus release but are not counteracted by HIV-1 Vpu (Goffinet et al., 2009; Gupta et al., 2009; Jia et al., 2009; McNatt et al., 2009; Rong et al., 2009; Sato et al., 2009). 2) Tetherin restricts the release of not only HIV-1 but also of a wide range of retroviruses (Jouvenet et al., 2009) and non-retroviral enveloped viruses [e.g., Ebola; (Kaletsky et al., 2009)]. Because Vpu expression is limited to HIV-1 and a small number of closely related SIVs, other viruses have likely evolved distinct approaches to counteracting this host restriction factor. For example, the Env glycoprotein of some strains of HIV-2 possesses Vpu-like activity (Bour et al., 1996; Ritter et al., 1996) and the Nef proteins of several SIVs antagonize tetherin (Jia et al., 2009).
The precise mechanism by which tetherin retains virus particles on the cell surface remains to be defined. Tetherin displays a somewhat unusual topology (Kupzig et al., 2003) (Fig. 12); it bears an N-terminal cytosolic domain, a transmembrane anchor sequence, and an extracellular coiled-coil domain. The transmembrane domain reportedly determines whether or not a particular tetherin protein can be antagonized by Vpu (Jia et al., 2009; McNatt et al., 2009; Rong et al., 2009). At its C-terminus, tetherin possesses another membrane anchor, a glycosylphosphatidylinositol (GPI) moiety, which serves to recruit the protein to lipid rafts (Kupzig et al., 2003). If tetherin does in fact function as a protein tether, the presence of two membrane anchors allows one to envision several distinct topologies that the protein could adopt to retain virions on the cell surface (Fig. 12). Additional studies will be required to determine whether tetherin does act as a tethering molecule and, if so, to define the orientation of the protein in both cellular and viral membranes.
Fig. 12.

Hypothetical models for the tethering of HIV-1 virions to the cell surface by CD317/BST-2/tetherin. The virion is shown in green, with core in blue and viral RNA in red. BST-2/CD317/tetherin is shown on the left anchored in the lipid bilayer of the plasma membrane, with the cytoplasmic tail (CT), transmembrane ™, and coiled-coil (CC) domains and the GPI anchor indicated. In model (i), two molecules of BST-2/CD317/tetherin are aligned in parallel, with the TM domains in the producer cell plasma membrane and the GPI anchors in the viral membrane. In model (ii), one molecule is embedded in the plasma membrane, the other in the viral membrane. The two molecules associate via their coiled-coil domains.
Also unclear is how Vpu counteracts tetherin to stimulate virus release. Initial reports either did not detect significant reductions in tetherin levels upon Vpu expression (Neil, Zang, and Bieniasz, 2008), or observed a Vpu-induced downregulation of tetherin from the cell surface (Van Damme et al., 2008). Subsequent studies, in contrast, were able to measure a significant reduction in total tetherin levels resulting from Vpu expression (Mitchell et al., 2009; Rong et al., 2009). Vpu reportedly removes tetherin from the cell surface and induces its degradation in the lysosome by serving as an adaptor between tetherin and the β-TrCP/SCF E3 ubiquitin ligase complex (Mitchell et al., 2009) or induces its degradation in the proteasome (Goffinet et al., 2009). Despite its ability to induce tetherin internalization and degradation, it remains to be established how critical this downregulation is in Vpu's ability to counteract tetherin and promote virus release; some evidence has been presented suggesting that Vpu can antagonize tetherin even in the absence of clear downregulation of either surface or total expression of the host factor (Miyagi et al., 2009).
Several approaches could be attempted in developing Vpu as a therapeutic target. Vpu has been reported to form ion channels, an activity that has been implicated in its virus-release activity (Bour and Strebel, 2003). Compounds that block Vpu ion channel activity have been reported to display anti-Vpu activity (Ewart et al., 2002; Park and Opella, 2007). Stephens and colleagues demonstrated that the presence of the vpu gene in an SIV/HIV-1 (SHIV) chimera contributes to SHIV pathogenesis in macaques (Stephens et al., 2002). Interestingly, substitution of the transmembrane domain of Vpu with that of the influenza A viroporin M2, or mutation of a key residue in the Vpu transmembrane domain, rendered virus replication sensitive to M2 ion channel blockers (Hout et al., 2006a; Hout et al., 2006b). These findings illustrate the potential for developing anti-HIV agents that act by blocking Vpu ion channel activity. An alternative approach to disrupting Vpu function is to target the Vpu-tetherin interface with small molecules or transmembrane peptide decoys that bind the Vpu membrane-spanning domain and block its interactions with tetherin (Montal, 2009). The interface between Vpu and host factors that help mediate the lysosomal or proteasomal degradation of tetherin (e.g., β-TrCP) could also be targeted. The cholesterol-binding compound AME, discussed above (section B1), was shown to inhibit virus release in a Vpu-dependent fashion, suggesting that it might disrupt the ability of Vpu to antagonize tetherin (Waheed et al., 2008). The relative importance of Vpu's two major functions - tetherin antagonism and CD4 downregulation – in HIV-1 pathogenesis remains to be defined. This question will be important in determining which function and domain of Vpu to target in developing inhibitors.
B5.3 Nef
The nef gene encodes a 27-kDa membrane-associated protein that is expressed at high levels in infected cells and is incorporated into virus particles. Nef was initially considered to be a negative regulatory factor (hence the name negative factor) but was ultimately shown to be an important positive determinant of lentiviral pathogenesis. A role for Nef in disease induction was demonstrated by two key observations: 1) Deletion of the nef gene in SIVmac profoundly impairs progression to AIDS in infected rhesus macaques (Kestler et al., 1991), and 2) individuals infected with nef-defective strains of HIV-1 in some cases exhibit very low viral loads and a long-term nonprogressor phenotype in which they remain healthy for extended periods of time postinfection (Deacon et al., 1995; Kirchhoff et al., 1995). The apparent importance of Nef in disease induction implies that disruption of Nef function would provide antiretroviral activity.
A complicating factor in developing Nef-based inhibitors is the complexity and diversity of the reported functions of this protein. A number of Nef activities have been reported, including: 1) downregulation of surface expression of the major histocompatibility complex I (MCH-I), 2) downmodulation of CD4 expression, 3) alteration of cellular activation pathways, including stimulation of p21-activated protein kinase 2 (Pak2), and 4) enhancement of virus infectivity [for reviews, see (Foster and Garcia, 2007; Foster and Garcia, 2008)]. These four major activities appear to be genetically separable (Foster and Garcia, 2008), suggesting the presence of multiple potential drug targets. It remains to be determined which of these functions is critical for the ability of Nef to increase viral loads and stimulate disease induction in vivo, although there is some evidence that CD4 downregulation is important for HIV-1 pathogenesis (Carl et al., 2000). Nef has been reported to interact with a large number of cellular partners, including CD3, CD4, the clathrin adapter protein complex 2 (AP-2), the vacuolar ATPase, COP1 coatomers, ADP ribosylation factor 1 (Arf1), p53, actin, p56lck, Pak2, and Hck (for reviews see (Foster and Garcia, 2007; Foster and Garcia, 2008; Freed, 2007).
The structure of the Nef core has been determined in its unliganded state and in complex with Src homology 3 (SH3) domains or the cytoplasmic tail of CD4 (Arold et al., 1997; Grzesiek et al., 1996a; Grzesiek et al., 1997; Grzesiek et al., 1996b; Lee et al., 1996). Nef contains an unstructured N-terminus, followed by a poly-Pro type II helix that bears the Pro-X-X-Pro motif responsible for binding SH3 domains, several helical regions, and a five-stranded antiparallel β-sheet. Dileucine and diacidic motifs in a 30-amino acid flexible loop in Nef contribute to AP-2 binding. In addition to the Pro-X-X-Pro motif, SH3 binding also requires a hydrophobic groove referred to as the RT loop-binding region (RTLBR); this region interacts with the RT loop of the SH3 domain (Arold et al., 1997; Lee et al., 1996). Betzi and colleagues performed an in silico screen to identify compounds that bind Nef and prevent its interaction with the SH3 domain of Hck (Betzi et al., 2007). This virtual screen was followed up with binding studies and a mammalian-cell-based two-hybrid assay to identify compounds that block Nef-SH3 binding. This study identified two drug-like compounds that bind Nef in the micromolar range and effectively compete for SH3 binding. It remains to be determined whether these compounds will move forward to clinical development; however, the importance of Nef in HIV disease progression will continue to make this protein an interesting target for drug discovery.
C. Conclusions
The ultimate objectives of HIV research are a safe and effective vaccine to prevent viral infection, and the ability to eradicate HIV from infected patients. However, achieving these goals, if they are even possible, may take many years if not decades. The most feasible approach to managing HIV infection will therefore continue to center on combination antiretroviral therapy aimed at suppressing viral loads in infected individuals. The remarkable ability of HIV-1 to evade antiretroviral agents, even when administered in combination, together with problems of drug toxicity, will likely necessitate the continued development of novel therapeutics. Success will require a sustained and multidisciplinary effort involving virology and chemical, structural, molecular, and cell biology. As drug discovery expands from its focus on purely viral targets (e.g., RT, PR, IN, gp41) to include viral/host protein interactions or cellular targets, a more in-depth understanding of the cell biology of HIV-1 replication will be required. High-throughput screens will have to be devised to identify small-molecule inhibitors of still-uncharacterized viral/host and host/host interactions and medicinal chemistry efforts will be required to translate hits in these screens to effective and non-toxic drugs. In this article, we provide an overview of recent progress in understanding fundamental aspects of HIV-1 replication and discuss how this information is being used, and could, in theory, be extended in the future, to develop to new drugs. These efforts will enhance our ability to treat HIV-1 infection and will also reap enormous benefits in terms of fundamental new insights into the molecular and cell biology of retroviral replication and pathogenesis.
Acknowledgments
We thank Jason Rausch and members of the Freed laboratory for helpful discussions and critical review of the manuscript, and Kalyan Das for providing the RT structure. Research in our lab is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by the Intramural AIDS Targeted Antiviral Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdurahman S, Hoglund S, Goobar-Larsson L, Vahlne A. Selected amino acid substitutions in the C-terminal region of human immunodeficiency virus type 1 capsid protein affect virus assembly and release. J Gen Virol. 2004;85(Pt 10):2903–13. doi: 10.1099/vir.0.80137-0. [DOI] [PubMed] [Google Scholar]
- Accola MA, Hoglund S, Gottlinger HG. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J Virol. 1998;72(3):2072–8. doi: 10.1128/jvi.72.3.2072-2078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accola MA, Strack B, Gottlinger HG. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol. 2000;74(12):5395–402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Nagashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat) J Virol. 2006;80(22):10957–71. doi: 10.1128/JVI.01369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Freed EO. HIV-1 Assembly, Release and Maturation. In: Jeang KT, editor. Advances in Pharmacolgy, HIV-1: Molecular Biology and Pathogenesis: Viral Mechansims. Vol. 55. Elsevier; 2007. [Google Scholar]
- Adamson CS, Freed EO. Recent progress in antiretrovirals—lessons from resistance. Drug Discov Today. 2008;13(910):424–32. doi: 10.1016/j.drudis.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Salzwedel K, Freed EO. Virus maturation as a new HIV-1 therapeutic target. Expert Opin Ther Targets. 2009;18:895–908. doi: 10.1517/14728220903039714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Waki K, Ablan SD, Salzwedel K, Freed EO. Impact of human immunodeficiency virus type 1 resistance to protease inhibitors on evolution of resistance to the maturation inhibitor bevirimat (PA-457) J Virol. 2009;83(10):4884–94. doi: 10.1128/JVI.02659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C, Chen CH. Betulinic acid derivatives as HIV-1 antivirals. Trends Mol Med. 2005;11(1):31–6. doi: 10.1016/j.molmed.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Aloia RC, Curtain CC, Jensen FC. Membrane Cholesterol and Human Immunodeficiency Virus Infectivity. In: Aloia RC, Curtain CC, editors. Advances in Membrane Fluidity. Vol. 6. Wiley-Liss, Inc.; New York: 1992. pp. 283–304. [Google Scholar]
- Aloia RC, Tian H, Jensen FC. Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc Natl Acad Sci U S A. 1993;90(11):5181–5185. doi: 10.1073/pnas.90.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Akkina R. TRIM5alpharh expression restricts HIV-1 infection in lentiviral vector-transduced CD34+-cell-derived macrophages. Mol Ther. 2005;12(4):687–96. doi: 10.1016/j.ymthe.2005.07.291. [DOI] [PubMed] [Google Scholar]
- Anderson J, Akkina R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5alpha (TRIM 5alpha) in CD34(+) cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum Gene Ther. 2008;19(3):217–28. doi: 10.1089/hum.2007.108. [DOI] [PubMed] [Google Scholar]
- Anderson JL, Campbell EM, Wu X, Vandegraaff N, Engelman A, Hope TJ. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J Virol. 2006;80(19):9754–60. doi: 10.1128/JVI.01052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3(4):301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Arold S, Franken P, Strub MP, Hoh F, Benichou S, Benarous R, Dumas C. The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure. 1997;5(10):1361–72. doi: 10.1016/s0969-2126(97)00286-4. [DOI] [PubMed] [Google Scholar]
- Asaoka K, Ikeda K, Hishinuma T, Horie-Inoue K, Takeda S, Inoue S. A retrovirus restriction factor TRIM5alpha is transcriptionally regulated by interferons. Biochem Biophys Res Commun. 2005;338(4):1950–6. doi: 10.1016/j.bbrc.2005.10.173. [DOI] [PubMed] [Google Scholar]
- Baba M. Inhibitors of HIV-1 gene expression and transcription. Curr Top Med Chem. 2004;4(9):871–82. doi: 10.2174/1568026043388466. [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3(1):61–71. doi: 10.2174/1570162052772924. [DOI] [PubMed] [Google Scholar]
- Barklis E, Alfadhli A, McQuaw C, Yalamuri S, Still A, Barklis RL, Kukull B, Lopez CS. Characterization of the in vitro HIV-1 capsid assembly pathway. Journal of Molecular Biology. 2009;387:376–389. doi: 10.1016/j.jmb.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K, De Rijck J, Busschots K, Desender L, Gijsbers R, Emiliani S, Benarous R, Debyser Z, Christ F. Differential interaction of HIV-1 integrase and JPO2 with the C terminus of LEDGF/p75. J Mol Biol. 2007;372(2):407–21. doi: 10.1016/j.jmb.2007.06.090. [DOI] [PubMed] [Google Scholar]
- Bartonova V, Igonet S, Sticht J, Glass B, Habermann A, Vaney MC, Sehr P, Lewis J, Rey FA, Krausslich HG. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J Biol Chem. 2008;283(46):32024–33. doi: 10.1074/jbc.M804230200. [DOI] [PubMed] [Google Scholar]
- Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol. 2009;388(3):462–74. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J Mol Biol. 2005;346(2):577–88. doi: 10.1016/j.jmb.2004.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Besnier C, Takeuchi Y, Towers G. Restriction of lentivirus in monkeys. Proc Natl Acad Sci U S A. 2002;99(18):11920–5. doi: 10.1073/pnas.172384599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzi S, Restouin A, Opi S, Arold ST, Parrot I, Guerlesquin F, Morelli X, Collette Y. Protein protein interaction inhibition (2P2I) combining high throughput and virtual screening: Application to the HIV-1 Nef protein. Proc Natl Acad Sci U S A. 2007;104(49):19256–61. doi: 10.1073/pnas.0707130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Zhang H, Debnath AK, Cowburn D. Solution structure of a hydrocarbon stapled peptide inhibitor in complex with monomeric C-terminal domain of HIV-1 capsid. J Biol Chem. 2008;283(24):16274–8. doi: 10.1074/jbc.C800048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol. 2004;5(11):1109–15. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344(1):55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD. The cell biology of HIV-1 virion genesis. Cell Host Microbe. 2009;5(6):550–8. doi: 10.1016/j.chom.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4(12):e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolinger C, Boris-Lawrie K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology. 2009;6:8. doi: 10.1186/1742-4690-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkow G, Fletcher RS, Barnard J, Arion D, Motakis D, Dmitrienko GI, Parniak MA. Inhibition of the ribonuclease H and DNA polymerase activities of HIV-1 reverse transcriptase by N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone. Biochemistry. 1997;36(11):3179–85. doi: 10.1021/bi9624696. [DOI] [PubMed] [Google Scholar]
- Borsetti A, Ohagen A, Gottlinger HG. The C-terminal half of the human immunodeficiency virus type 1 Gag precursor is sufficient for efficient particle assembly. J Virol. 1998;72(11):9313–7. doi: 10.1128/jvi.72.11.9313-9317.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour S, Schubert U, Peden K, Strebel K. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: a Vpu-like factor? J Virol. 1996;70(2):820–9. doi: 10.1128/jvi.70.2.820-829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour S, Strebel K. The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect. 2003;5(11):1029–39. doi: 10.1016/s1286-4579(03)00191-6. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Grunewald K, Glass B, Forster F, Krausslich HG, Fuller SD. The mechanism of HIV-1 core assembly: insights from three-dimensional reconstructions of authentic virions. Structure. 2006;14(1):15–20. doi: 10.1016/j.str.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. Structure and assembly of immature HIV. Proc Natl Acad Sci U S A. 2009;106(27):11090–5. doi: 10.1073/pnas.0903535106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11(7):672–5. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD. Structural organization of authentic, mature HIV-1 virions and cores. Embo J. 2003;22(7):1707–15. doi: 10.1093/emboj/cdg143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–36. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68(3):533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A. 2006;103(8):2641–6. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun S, Solignat M, Gay B, Bernard E, Chaloin L, Fenard D, Devaux C, Chazal N, Briant L. VSV-G pseudotyping rescues HIV-1 CA mutations that impair core assembly or stability. Retrovirology. 2008;5:57. doi: 10.1186/1742-4690-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant M, Ratner L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A. 1990;87(2):523–7. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33(4):1249–56. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J Virol. 1999;73(10):8527–40. doi: 10.1128/jvi.73.10.8527-8540.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butan C, Winkler DC, Heymann JB, Craven RC, Steven AC. RSV capsid polymorphism correlates with polymerization efficiency and envelope glycoprotein content: implications that nucleation controls morphogenesis. J Mol Biol. 2008;376(4):1168–81. doi: 10.1016/j.jmb.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Perez O, Anderson JL, Hope TJ. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J Cell Biol. 2008;180(3):549–61. doi: 10.1083/jcb.200706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Gaus K, Bittman R, Jessup W, Crowe S, Mak J. The raft-promoting property of virion-associated cholesterol, but not the presence of virion-associated Brij 98 rafts, is a determinant of human immunodeficiency virus type 1 infectivity. J Virol. 2004;78(19):10556–65. doi: 10.1128/JVI.78.19.10556-10565.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SM, Crowe SM, Mak J. Virion-associated cholesterol is critical for the maintenance of HIV-1 structure and infectivity. Aids. 2002;16(17):2253–2261. doi: 10.1097/00002030-200211220-00004. [DOI] [PubMed] [Google Scholar]
- Carl S, Daniels R, Iafrate AJ, Easterbrook P, Greenough TC, Skowronski J, Kirchhoff F. Partial “repair” of defective NEF genes in a long-term nonprogressor with human immunodeficiency virus type 1 infection. J Infect Dis. 2000;181(1):132–40. doi: 10.1086/315187. [DOI] [PubMed] [Google Scholar]
- Champoux JJ, Schultz SJ. Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009;276(6):1506–16. doi: 10.1111/j.1742-4658.2009.06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol. 2008;82(22):11228–38. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Bachtiar I, Piszczek G, Bouamr F, Carter C, Tjandra N. Solution NMR characterizations of oligomerization and dynamics of equine infectious anemia virus matrix protein and its interaction with PIP2. Biochemistry. 2008;47(7):1928–37. doi: 10.1021/bi701984h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Hoy J, Lewin SR. Ten years of highly active antiretroviral therapy for HIV infection. Med J Aust. 2007;186(3):146–51. doi: 10.5694/j.1326-5377.2007.tb00839.x. [DOI] [PubMed] [Google Scholar]
- Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem. 2003;278(1):372–81. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol. 2005;12(6):526–32. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- Chertova EN, Kane BP, McGrath C, Johnson DG, Sowder RC, 2nd, Arthur LO, Henderson LE. Probing the topography of HIV-1 nucleocapsid protein with the alkylating agent N-ethylmaleimide. Biochemistry. 1998;37(51):17890–7. doi: 10.1021/bi980907y. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K. Identification of a non-H-2 gene (Rfv-3) influencing recovery from viremia and leukemia induced by Friend virus complex. Proc Natl Acad Sci U S A. 1979;76(1):425–9. doi: 10.1073/pnas.76.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihlar T. NRTIs. Antiviral Res 2010 [Google Scholar]
- Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med. 2005;11(12):1287–9. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci U S A. 2002;99(18):11914–9. doi: 10.1073/pnas.162299499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofaro JV, Rausch JW, Le Grice SF, DeStefano JJ. Mutations in the ribonuclease H active site of HIV-RT reveal a role for this site in stabilizing enzyme-primer-template binding. Biochemistry. 2002;41(36):10968–75. doi: 10.1021/bi025871v. [DOI] [PubMed] [Google Scholar]
- Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A. 2008;105(5):1466–71. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JF, 2nd, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science. 1991;252(5002):88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- de bethune MP. NNRTIs. Antiviral Res. 2010 doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271(5255):1533–9. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Godi A. PI-loting membrane traffic. Nat Cell Biol. 2004;6(6):487–92. doi: 10.1038/ncb0604-487. [DOI] [PubMed] [Google Scholar]
- De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F, Debyser Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol. 2006;80(23):11498–509. doi: 10.1128/JVI.00801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270(5238):988–91. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- del Real G, Jimenez-Baranda S, Mira E, Lacalle RA, Lucas P, Gomez-Mouton C, Alegret M, Pena JM, Rodriguez-Zapata M, Alvarez-Mon M, Martinez AC, Manes S. Statins inhibit HIV-1 infection by down-regulating Rho activity. J Exp Med. 2004;200(4):541–7. doi: 10.1084/jem.20040061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delelis O, Carayon K, Saib A, Deprez E, Mouscadet JF. Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology. 2008;5:114. doi: 10.1186/1742-4690-5-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106(2):87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A. 2002;99(2):955–60. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirov DG, Orenstein JM, Freed EO. The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol. 2002;76(1):105–117. doi: 10.1128/JVI.76.1.105-117.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz E, Pfeffer SR. TIP47: a cargo selection device for mannose 6-phosphate receptor trafficking. Cell. 1998;93(3):433–43. doi: 10.1016/s0092-8674(00)81171-x. [DOI] [PubMed] [Google Scholar]
- Didierjean J, Isel C, Querre F, Mouscadet JF, Aubertin AM, Valnot JY, Piettre SR, Marquet R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob Agents Chemother. 2005;49(12):4884–94. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW. Beyond receptor expression: the influence of receptor conformation, density, and affinity in HIV-1 infection. Virology. 2000;276(2):229–37. doi: 10.1006/viro.2000.0612. [DOI] [PubMed] [Google Scholar]
- Doms RW. Entry Inhibitors. Antiviral Res. 2010 doi: 10.1016/j.antiviral.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Dorfman T, Bukovsky A, Ohagen A, Hoglund S, Gottlinger HG. Functional domains of the capsid protein of human immunodeficiency virus type 1. J Virol. 1994;68(12):8180–7. doi: 10.1128/jvi.68.12.8180-8187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Zhao Y, Chen J, Yang L, Zheng Y, Tang Y, Shen X, Jiang H. D77, one benzoic acid derivative, functions as a novel anti-HIV-1 inhibitor targeting the interaction between integrase and cellular LEDGF/p75. Biochem Biophys Res Commun. 2008;375(1):139–44. doi: 10.1016/j.bbrc.2008.07.139. [DOI] [PubMed] [Google Scholar]
- Emiliani S, Mousnier A, Busschots K, Maroun M, Van Maele B, Tempe D, Vandekerckhove L, Moisant F, Ben-Slama L, Witvrouw M, Christ F, Rain JC, Dargemont C, Debyser Z, Benarous R. Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV-1 replication. J Biol Chem. 2005;280(27):25517–23. doi: 10.1074/jbc.M501378200. [DOI] [PubMed] [Google Scholar]
- Emini EA, Fan HY. Immunological and pharmacological approaches to the control of retroviral infections. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- Engelman A, Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008;4(3):e1000046. doi: 10.1371/journal.ppat.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson-Viitanen S, Manfredi J, Viitanen P, Tribe DE, Tritch R, Hutchison CA, 3rd, Loeb DD, Swanstrom R. Cleavage of HIV-1 gag polyprotein synthesized in vitro: sequential cleavage by the viral protease. AIDS Res Hum Retroviruses. 1989;5(6):577–91. doi: 10.1089/aid.1989.5.577. [DOI] [PubMed] [Google Scholar]
- Ewart GD, Mills K, Cox GB, Gage PW. Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur Biophys J. 2002;31(1):26–35. doi: 10.1007/s002490100177. [DOI] [PubMed] [Google Scholar]
- Faure A, Calmels C, Desjobert C, Castroviejo M, Caumont-Sarcos A, Tarrago-Litvak L, Litvak S, Parissi V. HIV-1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Res. 2005;33(3):977–86. doi: 10.1093/nar/gki241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzon T, Leschonsky B, Bieler K, Paulus C, Schroder J, Wolf H, Wagner R. Proline residues in the HIV-1 NH2-terminal capsid domain: structure determinants for proper core assembly and subsequent steps of early replication. Virology. 2000;268(2):294–307. doi: 10.1006/viro.1999.0178. [DOI] [PubMed] [Google Scholar]
- Forshey BM, Shi J, Aiken C. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J Virol. 2005;79(2):869–75. doi: 10.1128/JVI.79.2.869-875.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002;76(11):5667–77. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JL, Garcia JV. Role of Nef in HIV-1 replication and pathogenesis. Adv Pharmacol. 2007;55:389–409. doi: 10.1016/S1054-3589(07)55011-8. [DOI] [PubMed] [Google Scholar]
- Foster JL, Garcia JV. HIV-1 Nef: at the crossroads. Retrovirology. 2008;5:84. doi: 10.1186/1742-4690-5-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 and the host cell: an intimate association. Trends Microbiol. 2004;12(4):170–7. doi: 10.1016/j.tim.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Freed EO. HIV-1 Gag: flipped out for PI(4,5)P(2) Proc Natl Acad Sci U S A. 2006;103(30):11101–2. doi: 10.1073/pnas.0604715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol. 1994;68(8):5311–20. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EOM, M A. HIVs and their replication. In: Knipe DMH, M P, editors. Fields Virology. Lippincott, Williams and Wilkins; 2007. pp. 2107–2185. [Google Scholar]
- Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol. 2007;5(12):912–6. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Functional role of Alix in HIV-1 replication. Virology. 2009 doi: 10.1016/j.virol.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller SD, Wilk T, Gowen BE, Krausslich HG, Vogt VM. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr Biol. 1997;7(10):729–38. doi: 10.1016/s0960-9822(06)00331-9. [DOI] [PubMed] [Google Scholar]
- Fun A, van Maarseveen NM, Maas REM, Nijhuis M. Resistance mutations in the viral protease alter bevirimat resistance patterns in vitro. 18th HIV Drug Resisatnce Workshop; Ft Myeres, Florida, USA. 2009. [Google Scholar]
- Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J Virol. 1992;66(11):6489–95. doi: 10.1128/jvi.66.11.6489-6495.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87(7):1285–94. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science. 1997;278(5339):849–53. doi: 10.1126/science.278.5339.849. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Daniels T, Casiano CA. LEDGF/p75: a novel nuclear autoantigen at the crossroads of cell survival and apoptosis. Autoimmun Rev. 2003;2(5):290–7. doi: 10.1016/s1568-9972(03)00063-6. [DOI] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283(5398):80–3. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131(1):70–9. doi: 10.1016/j.cell.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol. 2004;78(5):2545–52. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18(2):203–17. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Goffinet C, Allespach I, Homann S, Tervo HM, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-Locker J, Banting G, Krausslich HG, Fackler OT, Keppler OT. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5(3):285–97. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77(11):6507–19. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick RJ, Henderson LE, Hanser JP, Rein A. Point mutants of Moloney murine leukemia virus that fail to package viral RNA: evidence for specific RNA recognition by a “zinc finger-like” protein sequence. Proc Natl Acad Sci U S A. 1988;85(22):8420–4. doi: 10.1073/pnas.85.22.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick RJ, Nigida SM, Jr, Bess JW, Jr, Arthur LO, Henderson LE, Rein A. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J Virol. 1990;64(7):3207–11. doi: 10.1128/jvi.64.7.3207-3211.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A. 1991;88(8):3195–9. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1989;86(15):5781–5. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DR, Chertova E, Hilburn JM, Arthur LO, Hildreth JE. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J Virol. 2003;77(15):8237–48. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene WC, Debyser Z, Ikeda Y, Freed EO, Stephens E, Yonemoto W, Buckheit RW, Este JA, Cihlar T. Novel targets for HIV therapy. Antiviral Res. 2008;80(3):251–65. doi: 10.1016/j.antiviral.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996a;3(4):340–5. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Bax A, Hu JS, Kaufman J, Palmer I, Stahl SJ, Tjandra N, Wingfield PT. Refined solution structure and backbone dynamics of HIV-1 Nef. Protein Sci. 1997;6(6):1248–63. doi: 10.1002/pro.5560060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996b;35(32):10256–61. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- Guo X, Roldan A, Hu J, Wainberg MA, Liang C. Mutation of the SP1 sequence impairs both multimerization and membrane-binding activities of human immunodeficiency virus type 1 Gag. J Virol. 2005;79(3):1803–12. doi: 10.1128/JVI.79.3.1803-1812.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Hue S, Schaller T, Verschoor E, Pillay D, Towers GJ. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009;5(5):e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyader M, Kiyokawa E, Abrami L, Turelli P, Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J Virol. 2002;76(20):10356–10364. doi: 10.1128/JVI.76.20.10356-10364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang JQ, Rajendran S, Yang Y, Li Y, In PW, Overton H, Parkes KE, Cammack N, Martin JA, Klumpp K. Activity of the isolated HIV RNase H domain and specific inhibition by N-hydroxyimides. Biochem Biophys Res Commun. 2004;317(2):321–9. doi: 10.1016/j.bbrc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Hannoush RN, Carriero S, Min KL, Damha MJ. Selective inhibition of HIV-1 reverse transcriptase (HIV-1 RT) RNase H by small RNA hairpins and dumbbells. Chembiochem. 2004;5(4):527–33. doi: 10.1002/cbic.200300831. [DOI] [PubMed] [Google Scholar]
- Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, Cherepanov P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009;5(1):e1000259. doi: 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–77. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10(5):1247–53. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD. Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J Virol. 2004a;78(11):6005–12. doi: 10.1128/JVI.78.11.6005-6012.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Perez-Caballero D, Yang A, Cowan S, Bieniasz PD. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci U S A. 2004b;101(29):10774–9. doi: 10.1073/pnas.0402361101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayouka Z, Rosenbluh J, Levin A, Loya S, Lebendiker M, Veprintsev D, Kotler M, Hizi A, Loyter A, Friedler A. Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc Natl Acad Sci U S A. 2007;104(20):8316–21. doi: 10.1073/pnas.0700781104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol. 2006;1(11):702–12. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, Witvrouw M, Engelborghs Y, Christ F, Gijsbers R, Debyser Z. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 2007;3(3):e47. doi: 10.1371/journal.ppat.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, McGuinness DE, Prongay AJ, Feld B, Ingravallo P, Ogert RA, Lunn CA, Howe JA. Screening for antiviral inhibitors of the HIV integrase-LEDGF/p75 interaction using the AlphaScreen luminescent proximity assay. J Biomol Screen. 2008;13(5):406–14. doi: 10.1177/1087057108317060. [DOI] [PubMed] [Google Scholar]
- Hout DR, Gomez LM, Pacyniak E, Miller JM, Hill MS, Stephens EB. A single amino acid substitution within the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein renders simian-human immunodeficiency virus (SHIV(KU-1bMC33)) susceptible to rimantadine. Virology. 2006a;348(2):449–61. doi: 10.1016/j.virol.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Hout DR, Gomez ML, Pacyniak E, Gomez LM, Fegley B, Mulcahy ER, Hill MS, Culley N, Pinson DM, Nothnick W, Powers MF, Wong SW, Stephens EB. Substitution of the transmembrane domain of Vpu in simian-human immunodeficiency virus (SHIVKU1bMC33) with that of M2 of influenza A results in a virus that is sensitive to inhibitors of the M2 ion channel and is pathogenic for pig-tailed macaques. Virology. 2006b;344(2):541–59. doi: 10.1016/j.virol.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Hu WS, Rhodes T, Dang Q, Pathak V. Retroviral recombination: review of genetic analyses. Front Biosci. 2003;8:d143–55. doi: 10.2741/940. [DOI] [PubMed] [Google Scholar]
- Huang M, Orenstein JM, Martin MA, Freed EO. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69(11):6810–8. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35:277–98. doi: 10.1146/annurev.biophys.35.040405.102126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov D, Stone JR, Maki JL, Collins T, Wagner G. Mammalian SCAN domain dimer is a domain-swapped homolog of the HIV capsid C-terminal domain. Mol Cell. 2005;17(1):137–43. doi: 10.1016/j.molcel.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Javanbakht H, Yuan W, Yeung DF, Song B, Diaz-Griffero F, Li Y, Li X, Stremlau M, Sodroski J. Characterization of TRIM5alpha trimerization and its contribution to human immunodeficiency virus capsid binding. Virology. 2006;353(1):234–46. doi: 10.1016/j.virol.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Jenkins LM, Durell SR, Maynard AT, Stahl SJ, Inman JK, Appella E, Legault P, Omichinski JG. Comparison of the specificity of interaction of cellular and viral zinc-binding domains with 2-mercaptobenzamide thioesters. J Am Chem Soc. 2006;128(36):11964–76. doi: 10.1021/ja063329e. [DOI] [PubMed] [Google Scholar]
- Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5(5):e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmans D. Novel HIV-1 reverse transcriptase inhibitors. Virus Res. 2008;134(12):171–85. doi: 10.1016/j.virusres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J Virol. 2005;79(18):12088–94. doi: 10.1128/JVI.79.18.12088-12094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83(4):1837–44. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106(8):2886–91. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AH, Zack JA, Knigge M, Paul DA, Kempf DJ, Norbeck DW, Swanstrom R. Partial inhibition of the Human Immunodeficiency Virus Type 1 protease results in abbreant virus assembly and the formation of non-infectious particles. Journal of Virology. 1993;67:4050–4055. doi: 10.1128/jvi.67.7.4050-4055.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J Virol. 2008;82(23):11669–81. doi: 10.1128/JVI.01559-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keckesova Z, Ylinen LM, Towers GJ. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl Acad Sci U S A. 2004;101(29):10780–5. doi: 10.1073/pnas.0402474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol. 2007;373(2):355–66. doi: 10.1016/j.jmb.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestler HW, 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65(4):651–62. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- Khanna KV, Whaley KJ, Zeitlin L, Moench TR, Mehrazar K, Cone RA, Liao Z, Hildreth JE, Hoen TE, Shultz L, Markham RB. Vaginal transmission of cell-associated HIV-1 in the mouse is blocked by a topical, membrane-modifying agent. J Clin Invest. 2002;109(2):205–11. doi: 10.1172/JCI13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston RL, Vogt VM. Domain swapping and retroviral assembly. Mol Cell. 2005;17(2):166–7. doi: 10.1016/j.molcel.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332(4):228–32. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong Kai In P, Overton H, Parkes KE, Cammack N, Martin JA. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003;31(23):6852–9. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K, Mirzadegan T. Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr Pharm Des. 2006;12(15):1909–22. doi: 10.2174/138161206776873653. [DOI] [PubMed] [Google Scholar]
- Knapp DJHF, Huang S, Harrington R. Stable prevelance of bevirimat-related Gag polymorphisms both before and after HAART exposure. 16th conference on retroviruses and opportunistic infections; Montreal, Canada. 2009. [Google Scholar]
- Krausslich HG, Facke M, Heuser AM, Konvalinka J, Zentgraf H. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J Virol. 1995;69(6):3407–19. doi: 10.1128/jvi.69.6.3407-3419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausslich HG, Schneider H, Zybarth G, Carter CA, Wimmer E. Processing of in vitro-synthesized gag precursor proteins of human immunodeficiency virus (HIV) type 1 by HIV proteinase generated in Escherichia coli. J Virol. 1988;62(11):4393–7. doi: 10.1128/jvi.62.11.4393-4397.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4(10):694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI. Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J Virol. 2008;82(23):11682–94. doi: 10.1128/JVI.01562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. 1996;85(6):931–42. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH. Structural basis for viral late-domain binding to Alix. Nat Struct Mol Biol. 2007;14(3):194–9. doi: 10.1038/nsmb1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Harris J, Swanstrom R. A Strongly Transdominant Mutation in the Human Immunodeficiency Virus Type 1 gag Gene Defines An Achilles Heel in the Virus Life Cycle. J Virol. 2009 doi: 10.1128/JVI.00317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucleic Acid Res Mol Biol. 2005;80:217–86. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A. 2003;100(23):13555–60. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hill CP, Sundquist WI, Finch JT. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407(6802):409–13. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- Li X, Sodroski J. The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol. 2008;82(23):11495–502. doi: 10.1128/JVI.01548-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li X, Stremlau M, Lee M, Sodroski J. Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J Virol. 2006;80(14):6738–44. doi: 10.1128/JVI.00270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J Virol. 2002;76(22):11729–37. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Hu J, Whitney JB, Kleiman L, Wainberg MA. A structurally disordered region at the C terminus of capsid plays essential roles in multimerization and membrane binding of the gag protein of human immunodeficiency virus type 1. J Virol. 2003;77(3):1772–83. doi: 10.1128/JVI.77.3.1772-1783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses. 2001;17(11):1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- Lifson JD, Rossio JL, Piatak M, Jr, Bess J, Jr, Chertova E, Schneider DK, Coalter VJ, Poore B, Kiser RF, Imming RJ, Scarzello AJ, Henderson LE, Alvord WG, Hirsch VM, Benveniste RE, Arthur LO. Evaluation of the safety, immunogenicity, and protective efficacy of whole inactivated simian immunodeficiency virus (SIV) vaccines with conformationally and functionally intact envelope glycoproteins. AIDS Res Hum Retroviruses. 2004;20(7):772–87. doi: 10.1089/0889222041524661. [DOI] [PubMed] [Google Scholar]
- Liu F, Stephen AG, Adamson CS, Gousset K, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr Hydrazone- and hydrazide-containing N-substituted glycines as peptoid surrogates for expedited library synthesis: application to the preparation of Tsg101-directed HIV-1 budding antagonists. Org Lett. 2006;8(22):5165–8. doi: 10.1021/ol0622211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Stephen AG, Waheed AA, Aman MJ, Freed EO, Fisher RJ, Burke TR., Jr SAR by oxime-containing peptide libraries: application to Tsg101 ligand optimization. Chembiochem. 2008;9(12):2000–4. doi: 10.1002/cbic.200800281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314(5798):461–4. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- Lopez-Verges S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C. Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci U S A. 2006;103(40):14947–52. doi: 10.1073/pnas.0602941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J Virol. 2007;81(3):1054–61. doi: 10.1128/JVI.01519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73(6):1067–78. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- Luttge BG, Shehu-Xhilaga M, Demirov DG, Adamson CS, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Molecular characterization of feline immunodeficiency virus budding. J Virol. 2008;82(5):2106–19. doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N, Kabat D. An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J Virol. 1998;72(12):10251–5. doi: 10.1128/jvi.72.12.10251-10255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens GN, Cherepanov P, Engelman A. Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J Cell Sci. 2006;119(Pt 12):2563–71. doi: 10.1242/jcs.02995. [DOI] [PubMed] [Google Scholar]
- Malet I, Wirden M, Derache A, Simon A, Katlama C, Calvez V, Marcelin AG. Primary genotypic resistance of HIV-1 to the maturation inhibitor PA-457 in protease inhibitor-experienced patients. Aids. 2007;21(7):871–3. doi: 10.1097/QAD.0b013e3280b079d9. [DOI] [PubMed] [Google Scholar]
- Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364(1517):675–87. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammano F, Ohagen A, Hoglund S, Gottlinger HG. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J Virol. 1994;68(8):4927–36. doi: 10.1128/jvi.68.8.4927-4936.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes S, del Real G, Lacalle RA, Lucas P, Gomez-Mouton C, Sanchez-Palomino S, Delgado R, Alcami J, Mira E, Martinez AC. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000;1(2):190–196. doi: 10.1093/embo-reports/kvd025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margot NA, Gibbs CS, Miller MD. Phenotyphic Susceptibility to Bevirimat among HIV-Infected Patient Isolates without Prior Exposure to Bevirimat. 16th Conference on Retroviruses and Opportunistic Infections; Montreal, Canada. 2009. [Google Scholar]
- Marsden MD, Zack JA. Eradication of HIV: current challenges and new directions. J Antimicrob Chemother. 2009;63(1):7–10. doi: 10.1093/jac/dkn455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, Bickmore W, Poeschla E, Bushman FD. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2(12):e1340. doi: 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Salzwedel K, Allaway GP. Bevirimat: a novel maturation inhibitor for the treatment of HIV-1 infection. Antivir Chem Chemother. 2008;19(3):107–13. doi: 10.1177/095632020801900301. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7(12):1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81(13):7099–110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallister S, Lalezari J, Richmond G, Thompson M, Harrigan R, Martin D, Salzwedel K, Allaway G. HIV-1 Gag Polymorphisms Determine Treatment Response to Bevirimat (PA-457). XVII International HIV Drug Resistance Workshop; Sitges, Spain. 2008. [Google Scholar]
- McDonald B, Martin-Serrano J. No strings attached: the ESCRT machinery in viral budding and cytokinesis. J Cell Sci. 2009;122(Pt 13):2167–77. doi: 10.1242/jcs.028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJ, Bieniasz PD. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5(2):e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Wilson H, Zhang C, Brazier AJ, McPike M, Pery E, Gabuzda D. Identification of an APOBEC3G binding site in human immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J Virol. 2007;81(23):13235–41. doi: 10.1128/JVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed D, Mark-Danieli M, Kenan-Eichler M, Kraus O, Castiel A, Laham N, Pupko T, Glaser F, Ben-Tal N, Bacharach E. The conserved carboxy terminus of the capsid domain of human immunodeficiency virus type 1 gag protein is important for virion assembly and release. J Virol. 2004;78(18):9675–88. doi: 10.1128/JVI.78.18.9675-9688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervis RJ, Ahmad N, Lillehoj EP, Raum MG, Salazar FH, Chan HW, Venkatesan S. The gag gene products of human immunodeficiency virus type 1: alignment within the gag open reading frame, identification of posttranslational modifications, and evidence for alternative gag precursors. J Virol. 1988;62(11):3993–4002. doi: 10.1128/jvi.62.11.3993-4002.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min BS, Miyashiro H, Hattori M. Inhibitory effects of quinones on RNase H activity associated with HIV-1 reverse transcriptase. Phytother Res. 2002;16 1:S57–62. doi: 10.1002/ptr.808. [DOI] [PubMed] [Google Scholar]
- Mische CC, Javanbakht H, Song B, Diaz-Griffero F, Stremlau M, Strack B, Si Z, Sodroski J. Retroviral restriction factor TRIM5alpha is a trimer. J Virol. 2005;79(22):14446–50. doi: 10.1128/JVI.79.22.14446-14450.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RS, Katsura C, Skasko MA, Fitzpatrick K, Lau D, Ruiz A, Stephens EB, Margottin-Goguet F, Benarous R, Guatelli JC. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009;5(5):e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi E, Andrew AJ, Kao S, Strebel K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A. 2009;106(8):2868–73. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa M, Tsuji-Kawahara S, Kanari Y. Host genetic factors that control immune responses to retrovirus infections. Vaccine. 2008;26(24):2981–96. doi: 10.1016/j.vaccine.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Moncunill G, Negredo E, Bosch L, Vilarrasa J, Witvrouw M, Llano A, Clotet B, Este JA. Evaluation of the anti-HIV activity of statins. Aids. 2005;19(15):1697–700. doi: 10.1097/01.aids.0000183517.60384.db. [DOI] [PubMed] [Google Scholar]
- Montal M. Vpu matchmakers as a therapeutic strategy for HIV infection. PLoS Pathog. 2009;5(5):e1000246. doi: 10.1371/journal.ppat.1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcock DR, Thomas JA, Sowder RC, 2nd, Henderson LE, Crise BJ, Gorelick RJ. HIV-1 inactivation by 4-vinylpyridine is enhanced by dissociating Zn(2+) from nucleocapsid protein. Virology. 2008;375(1):148–58. doi: 10.1016/j.virol.2008.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. Helical structure determined by NMR of the HIV-1 (345-392)Gag sequence, surrounding p2: implications for particle assembly and RNA packaging. Protein Sci. 2005;14(2):375–86. doi: 10.1110/ps.041087605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Hockley DJ, Nermut MV, Jones IM. Roles of matrix, p2, and N-terminal myristoylation in human immunodeficiency virus type 1 Gag assembly. J Virol. 2000;74(1):16–23. doi: 10.1128/jvi.74.1.16-23.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Munk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci U S A. 2002;99(21):13843–8. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115(4):377–88. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- Munshi UM, Kim J, Nagashima K, Hurley JH, Freed EO. An Alix fragment potently inhibits HIV-1 budding: characterization of binding to retroviral YPXL late domains. J Biol Chem. 2007;282(6):3847–55. doi: 10.1074/jbc.M607489200. [DOI] [PubMed] [Google Scholar]
- Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM. Small-molecule inhibition of HIV-1 Vif. Nat Biotechnol. 2008;26(10):1187–92. doi: 10.1038/nbt.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425–30. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Nekhai S, Jeang KT. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: role of cellular factors for Tat and Rev. Future Microbiol. 2006;1:417–26. doi: 10.2217/17460913.1.4.417. [DOI] [PubMed] [Google Scholar]
- Newman JL, Butcher EW, Patel DT, Mikhaylenko Y, Summers MF. Flexibility in the P2 domain of the HIV-1 Gag polyprotein. Protein Sci. 2004;13(8):2101–7. doi: 10.1110/ps.04614804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74(7):3264–3272. doi: 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Taub D. CXCR4 function requires membrane cholesterol: implications for HIV infection. J Immunol. 2002;168(8):4121–4126. doi: 10.4049/jimmunol.168.8.4121. [DOI] [PubMed] [Google Scholar]
- Nijhuis M. Protease inhibitors. Antiviral Res. 2010 doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Nisole S, Lynch C, Stoye JP, Yap MW. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A. 2004;101(36):13324–8. doi: 10.1073/pnas.0404640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121(7):1005–16. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol Cell. 2007;28(2):264–76. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25(9):1924–33. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445(7130):927–30. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- Okeoma CM, Low A, Bailis W, Fan HY, Peterlin BM, Ross SR. Induction of APOBEC3 in vivo causes increased restriction of retrovirus infection. J Virol. 2009;83(8):3486–95. doi: 10.1128/JVI.02347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101(41):14889–94. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Demirov D, Freed EO. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J Virol. 2000;74(11):5142–50. doi: 10.1128/jvi.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98(24):13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO. Role of lipid rafts in virus replication. Adv Virus Res. 2005;64:311–58. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]
- Ono A, Waheed AA, Freed EO. Depletion of cellular cholesterol inhibits membrane binding and higher-order multimerization of human immunodeficiency virus type 1 Gag. Virology. 2007;360(1):27–35. doi: 10.1016/j.virol.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol. 2004;78(10):5423–37. doi: 10.1128/JVI.78.10.5423-5437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8(11):849–60. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Opella SJ. Conformational changes induced by a single amino acid substitution in the trans-membrane domain of Vpu: implications for HIV-1 susceptibility to channel blocking drugs. Protein Sci. 2007;16(10):2205–15. doi: 10.1110/ps.073041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmegiani RM, Loebenberg D, Antonacci B, Yarosh-Tomaine T, Scupp R, Wright JJ, Chiu PJ, Miller GH. Comparative in vitro and in vivo evaluation of N-D-ornithyl amphotericin B methyl ester, amphotericin B methyl ester, and amphotericin B. Antimicrob Agents Chemother. 1987;31(11):1756–60. doi: 10.1128/aac.31.11.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD. Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J Virol. 2005;79(14):8969–78. doi: 10.1128/JVI.79.14.8969-8978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perno CF, Moyle G, Tsoukas C, Ratanasuwan W, Gatell J, Schechter M. Overcoming resistance to existing therapies in HIV-infected patients: the role of new antiretroviral drugs. J Med Virol. 2008;80(4):565–76. doi: 10.1002/jmv.21034. [DOI] [PubMed] [Google Scholar]
- Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J Virol. 2007;81(5):2138–48. doi: 10.1128/JVI.02318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron MJ, Stremlau M, Song B, Ulm W, Mulligan RC, Sodroski J. TRIM5alpha mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc Natl Acad Sci U S A. 2004;101(32):11827–32. doi: 10.1073/pnas.0403364101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pery E, Rajendran KS, Brazier AJ, Gabuzda D. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J Virol. 2009;83(5):2374–81. doi: 10.1128/JVI.01898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Henderson GJ, Schiffer CA, Swanstrom R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J Virol. 2002;76(20):10226–33. doi: 10.1128/JVI.76.20.10226-10233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J Virol. 1994;68(12):8017–27. doi: 10.1128/jvi.68.12.8017-8027.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickl WF, Pimentel-Muinos FX, Seed B. Lipid rafts and pseudotyping. J Virol. 2001;75(15):7175–7183. doi: 10.1128/JVI.75.15.7175-7183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006;47(7):1597–8. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- Poeschla EM. Integrase, LEDGF/p75 and HIV replication. Cell Mol Life Sci. 2008;65(9):1403–24. doi: 10.1007/s00018-008-7540-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol. 2002;76(10):4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002a;9(11):812–7. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Alam SL, Rich RL, Myszka DG, Davis DR, Sundquist WI. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 2002b;21(10):2397–406. doi: 10.1093/emboj/21.10.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137(7):1282–92. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probasco JC, Spudich SS, Critchfield J, Lee E, Lollo N, Deeks SG, Price RW. Failure of atorvastatin to modulate CSF HIV-1 infection: results of a pilot study. Neurology. 2008;71(7):521–4. doi: 10.1212/01.wnl.0000325006.84658.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri A, Blumenthal R. HIV type 1 entry correlates with plasma membrane microdomain lateral assemblies of receptors: implications in HIV/AIDS treatment. J Viral Entry. 2008;3:10–20. [Google Scholar]
- Rabson AB, Graves BJ. Synthesis and processing of viral RNA. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- Reicin AS, Ohagen A, Yin L, Hoglund S, Goff SP. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J Virol. 1996;70(12):8645–52. doi: 10.1128/jvi.70.12.8645-8652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. The tripartite motif family identifies cell compartments. EMBO J. 2001;20(9):2140–51. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WG, Schaeffer CA, Harten B, Villinger F, South TL, Summers MF, Henderson LE, Bess JW, Jr, Arthur LO, McDougal JS, et al. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature. 1993;361(6411):473–5. doi: 10.1038/361473a0. [DOI] [PubMed] [Google Scholar]
- Rice WG, Supko JG, Malspeis L, Buckheit RW, Jr, Clanton D, Bu M, Graham L, Schaeffer CA, Turpin JA, Domagala J, Gogliotti R, Bader JP, Halliday SM, Coren L, Sowder RC, 2nd, Arthur LO, Henderson LE. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science. 1995;270(5239):1194–7. doi: 10.1126/science.270.5239.1194. [DOI] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323(5919):1304–7. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- Richter SN, Palu G. Inhibitors of HIV-1 Tat-mediated transactivation. Curr Med Chem. 2006;13(11):1305–15. doi: 10.2174/092986706776872989. [DOI] [PubMed] [Google Scholar]
- Ritter GD, Jr, Yamshchikov G, Cohen SJ, Mulligan MJ. Human immunodeficiency virus type 2 glycoprotein enhancement of particle budding: role of the cytoplasmic domain. J Virol. 1996;70(4):2669–73. doi: 10.1128/jvi.70.4.2669-2673.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rold CJ, Aiken C. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 2008;4(5):e1000074. doi: 10.1371/journal.ppat.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, Zhang J, Lu J, Pan Q, Lorgeoux RP, Aloysius C, Guo F, Liu SL, Wainberg MA, Liang C. The transmembrane domain of BST-2 determines its sensitivity to down-modulation by human immunodeficiency virus type 1 Vpu. J Virol. 2009;83(15):7536–46. doi: 10.1128/JVI.00620-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose S, Hensley DJ, O'Shannessy J, Culp C, Debouck C, Chaiken I. Characterisation of HIV-1 p24 self-association using affinity chromatography. Proteins. 1992;13:112–119. doi: 10.1002/prot.340130204. [DOI] [PubMed] [Google Scholar]
- Roux KH, Taylor KA. AIDS virus envelope spike structure. Curr Opin Struct Biol. 2007;17(2):244–52. doi: 10.1016/j.sbi.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, Soheilian F, Freed EO, Summers MF. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J Mol Biol. 2008;382(2):434–47. doi: 10.1016/j.jmb.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103(30):11364–9. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakalian M, McMurtrey CP, Deeg FJ, Maloy CW, Li F, Wild CT, Salzwedel K. 3-o-(3′,3′-dimethysuccinyl) betulinic Acid inhibits maturation of the human immunodeficiency virus type 1 gag precursor assembled in vitro. J Virol. 2006;80(12):5716–22. doi: 10.1128/JVI.02743-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma R, Mael AA, Ikeda Y. Alpha interferon enhances TRIM5alpha-mediated antiviral activities in human and rhesus monkey cells. J Virol. 2007;81(18):10201–6. doi: 10.1128/JVI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma R, Noser JA, Ohmine S, Ikeda Y. Inhibition of HIV-1 replication by simian restriction factors, TRIM5alpha and APOBEC3G. Gene Ther. 2007;14(2):185–9. doi: 10.1038/sj.gt.3302852. [DOI] [PubMed] [Google Scholar]
- Salzwedel K, Hamy F, Louvel S, Sakalian M, Reddick M, Finnegan C, Martin D, McCallister S, Klimkait T, Allaway G. Suceptibility of diverse HIV-1 Patient Isolates to the Maturation Inhibitor Bevirimat (MPC-4326) is Determined by Clade-Specific Polymorphisms in Gag CA-SP1. 16th Conference on Retroviruses and Opportunistic Infections; Montreal, Canada. 2009. [Google Scholar]
- Salzwedel K, Martin DE, Sakalian M. Maturation inhibitors: a new therapeutic class targets the virus structure. AIDS Rev. 2007;9(3):162–72. [PubMed] [Google Scholar]
- Salzwedel K, Reddick M, Matallana C, Finnegan C, Adamson C, Sakalian M, Stanley D, Martin D, McCallister S, Freed E, Allaway G. Role of Gag Polymorphisms in HIV-1 Sensitivity to the Maturation Inhibitor Bevirimat. XVII International HIV Drug Resistance Workshop; Sitges, Spain. 2008. [Google Scholar]
- Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science. 2008;321(5894):1343–6. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Das K, Tantillo C, Clark AD, Jr, Ding J, Whitcomb JM, Boyer PL, Hughes SH, Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20(6):1449–61. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385(3):693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Yamamoto SP, Misawa N, Yoshida T, Miyazawa T, Koyanagi Y. Comparative study on the effect of human BST-2/Tetherin on HIV-1 release in cells of various species. Retrovirology. 2009;6:53. doi: 10.1186/1742-4690-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 2005;102(8):2832–7. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430(6999):569–73. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- Schito ML, Goel A, Song Y, Inman JK, Fattah RJ, Rice WG, Turpin JA, Sher A, Appella E. In vivo antiviral activity of novel human immunodeficiency virus type 1 nucleocapsid p7 zinc finger inhibitors in a transgenic murine model. AIDS Res Hum Retroviruses. 2003;19(2):91–101. doi: 10.1089/088922203762688595. [DOI] [PubMed] [Google Scholar]
- Scholz I, Arvidson B, Huseby D, Barklis E. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J Virol. 2005;79(3):1470–9. doi: 10.1128/JVI.79.3.1470-1479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110(4):521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Schultz SJ, Champoux JJ. RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 2008;134(12):86–103. doi: 10.1016/j.virusres.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian S, Luban J. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology. 2005;2:40. doi: 10.1186/1742-4690-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J Biol Chem. 2003;278(5):2777–80. doi: 10.1074/jbc.C200621200. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO. Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol. 2004;78(2):724–32. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Aiken C. Saturation of TRIM5 alpha-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virology. 2006;350(2):493–500. doi: 10.1016/j.virol.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Shibata R, Sakai H, Kawamura M, Tokunaga K, Adachi A. Early replication block of human immunodeficiency virus type 1 in monkey cells. J Gen Virol. 1995;76(Pt 11):2723–30. doi: 10.1099/0022-1317-76-11-2723. [DOI] [PubMed] [Google Scholar]
- Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry. 2006;45(13):4077–83. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21(14):1767–78. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvadchak V, Sanglier S, Rocle S, Villa P, Haiech J, Hibert M, Van Dorsselaer A, Mely Y, de Rocquigny H. Identification by high throughput screening of small compounds inhibiting the nucleic acid destabilization activity of the HIV-1 nucleocapsid protein. Biochimie. 2009;91(7):916–23. doi: 10.1016/j.biochi.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Simon JH, Miller DL, Fouchier RA, Soares MA, Peden KW, Malim MH. The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J. 1998;17(5):1259–67. doi: 10.1093/emboj/17.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon V, Ho DD, Abdool Karim Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet. 2006;368(9534):489–504. doi: 10.1016/S0140-6736(06)69157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar PA, Masur H, Grubb JR, Voell J, Witek J, Ono A, Freed EO, Maldarelli F. Pravastatin does not have a consistent antiviral effect in chronically HIV-infected individuals on antiretroviral therapy. Aids. 2005;19(10):1109–11. doi: 10.1097/01.aids.0000174461.31794.50. [DOI] [PubMed] [Google Scholar]
- Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother. 2007;51(10):3574–81. doi: 10.1128/AAC.00152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasunderam A, Ferguson MR, Rojo DR, Thiviyanathan V, Li X, O'Brien WA, Gorenstein DG. Combinatorial selection, inhibition, and antiviral activity of DNA thioaptamers targeting the RNase H domain of HIV-1 reverse transcriptase. Biochemistry. 2005;44(30):10388–95. doi: 10.1021/bi0507074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Goel A, Basrur V, Roberts PE, Mikovits JA, Inman JK, Turpin JA, Rice WG, Appella E. Synthesis and biological properties of amino acid amide ligand-based pyridinioalkanoyl thioesters as anti-HIV agents. Bioorg Med Chem. 2002;10(5):1263–73. doi: 10.1016/s0968-0896(01)00392-3. [DOI] [PubMed] [Google Scholar]
- Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, Nothnick W, Wong SW, Gunderson R, Berman NE, Singh DK. Deletion of the vpu sequences prior to the env in a simian-human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology. 2002;293(2):252–61. doi: 10.1006/viro.2001.1244. [DOI] [PubMed] [Google Scholar]
- Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich HG. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12(8):671–7. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- Stoddart CA, Joshi P, Sloan B, Bare JC, Smith PC, Allaway GP, Wild CT, Martin DE. Potent activity of the HIV-1 maturation inhibitor bevirimat in SCID-hu Thy/Liv mice. PLoS ONE. 2007;2(11):e1251. doi: 10.1371/journal.pone.0001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP, Yap MW. Chance favors a prepared genome. Proc Natl Acad Sci U S A. 2008;105(9):3177–8. doi: 10.1073/pnas.0800667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427(6977):848–53. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci U S A. 2006;103(14):5514–9. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Welikala S, Sodroski J. Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol. 2005;79(5):3139–45. doi: 10.1128/JVI.79.5.3139-3145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland HG, Newton K, Brownstein DG, Holmes MC, Kress C, Semple CA, Bickmore WA. Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol Cell Biol. 2006;26(19):7201–10. doi: 10.1128/MCB.00459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Craigie R. The road to chromatin - nuclear entry of retroviruses. Nat Rev Microbiol. 2007;5(3):187–96. doi: 10.1038/nrmicro1579. [DOI] [PubMed] [Google Scholar]
- Svarovskaia ES, Cheslock SR, Zhang WH, Hu WS, Pathak VK. Retroviral mutation rates and reverse transcriptase fidelity. Front Biosci. 2003;8:d117–34. doi: 10.2741/957. [DOI] [PubMed] [Google Scholar]
- Swanstrom R, Wills JW. Synthesis, assembly and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003;327(5):1013–20. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101(2):517–22. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J Virol. 2001;75(19):9357–66. doi: 10.1128/JVI.75.19.9357-9366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen SN. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag-TSG101 interaction. ACS Chem Biol. 2008;3(12):757–64. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- Temesgen Z, Cainelli F, Poeschla EM, Vlahakis SA, Vento S. Approach to salvage antiretroviral therapy in heavily antiretroviral-experienced HIV-positive adults. Lancet Infect Dis. 2006;6(8):496–507. doi: 10.1016/S1473-3099(06)70550-3. [DOI] [PubMed] [Google Scholar]
- Ternois F, Sticht J, Duquerroy S, Krausslich HG, Rey FA. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat Struct Mol Biol. 2005;12(8):678–82. doi: 10.1038/nsmb967. [DOI] [PubMed] [Google Scholar]
- Tisdale M, Schulze T, Larder BA, Moelling K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J Gen Virol. 1991;72(Pt 1):59–66. doi: 10.1099/0022-1317-72-1-59. [DOI] [PubMed] [Google Scholar]
- Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O. A conserved mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci U S A. 2000;97(22):12295–9. doi: 10.1073/pnas.200286297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers GJ. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007;4:40. doi: 10.1186/1742-4690-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med. 2003;9(9):1138–43. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- Tramontano E, Esposito F, Badas R, Di Santo R, Costi R, La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005;65(2):117–24. doi: 10.1016/j.antiviral.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Tritch RJ, Cheng YE, Yin FH, Erickson-Viitanen S. Mutagenesis of protease cleavage sites in the human immunodeficiency virus type 1 gag polyprotein. J Virol. 1991;65(2):922–30. doi: 10.1128/jvi.65.2.922-930.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KB, Hagan NA, Fabris D. Inhibitory effects of archetypical nucleic acid ligands on the interactions of HIV-1 nucleocapsid protein with elements of Psi-RNA. Nucleic Acids Res. 2006;34(5):1305–16. doi: 10.1093/nar/gkl004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin JA, Song Y, Inman JK, Huang M, Wallqvist A, Maynard A, Covell DG, Rice WG, Appella E. Synthesis and biological properties of novel pyridinioalkanoyl thiolesters (PATE) as anti-HIV-1 agents that target the viral nucleocapsid protein zinc fingers. J Med Chem. 1999;42(1):67–86. doi: 10.1021/jm9802517. [DOI] [PubMed] [Google Scholar]
- Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ. HIV-1 Susceptibility to the Maturation Inhibitor Bevirimat Is Modulated by Baseline Polymorphisms in Gag SP1. Antimicrob Agents Chemother. 2009 doi: 10.1128/AAC.01650-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3(4):245–52. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandegraaff N, Engelman A. Molecular mechanisms of HIV integration and therapeutic intervention. Expert Rev Mol Med. 2007;9(6):1–19. doi: 10.1017/S1462399407000257. [DOI] [PubMed] [Google Scholar]
- Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci U S A. 2003;100(25):15154–9. doi: 10.1073/pnas.2433165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt VM. Proteolytic processing and particle maturation. Curr Top Microbiol Immunol. 1996;214:95–131. doi: 10.1007/978-3-642-80145-7_4. [DOI] [PubMed] [Google Scholar]
- von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. Embo J. 1998;17(6):1555–68. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol. 2003;77(9):5439–50. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Ablan SD, Mankowski MK, Cummins JE, Ptak RG, Schaffner CP, Freed EO. Inhibition of HIV-1 replication by amphotericin B methyl ester: selection for resistant variants. J Biol Chem. 2006;281(39):28699–711. doi: 10.1074/jbc.M603609200. [DOI] [PubMed] [Google Scholar]
- Waheed AA, Ablan SD, Roser JD, Sowder RC, Schaffner CP, Chertova E, Freed EO. HIV-1 escape from the entry-inhibiting effects of a cholesterol-binding compound via cleavage of gp41 by the viral protease. Proc Natl Acad Sci U S A. 2007;104(20):8467–71. doi: 10.1073/pnas.0701443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Ablan SD, Soheilian F, Nagashima K, Ono A, Schaffner CP, Freed EO. Inhibition of human immunodeficiency virus type 1 assembly and release by the cholesterol-binding compound amphotericin B methyl ester: evidence for Vpu dependence. J Virol. 2008;82(19):9776–81. doi: 10.1128/JVI.00917-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Freed EO. Lipids and membrane microdomains in HIV-1 replication. Virus Res. 2009 doi: 10.1016/j.virusres.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrilow D, Harrich D. HIV-1 replication from after cell entry to the nuclear periphery. Curr HIV Res. 2007;5(3):293–9. doi: 10.2174/157016207780636579. [DOI] [PubMed] [Google Scholar]
- Welker R, Hohenberg H, Tessmer U, Huckhagel C, Krausslich HG. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J Virol. 2000;74(3):1168–77. doi: 10.1128/jvi.74.3.1168-1177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeler M, Lee HF, Bermingham A, Miller JT, Chertov O, Bona MK, Baichoo NS, Ehteshami M, Beutler J, O'Keefe BR, Gotte M, Kvaratskhelia M, Le Grice S. Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem Biol. 2008;3(10):635–44. doi: 10.1021/cb8001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegers K, Rutter G, Kottler H, Tessmer U, Hohenberg H, Krausslich HG. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J Virol. 1998;72(4):2846–2854. doi: 10.1128/jvi.72.4.2846-2854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, Krausslich HG, Boulanger P, Fuller SD. Organization of immature human immunodeficiency virus type 1. J Virol. 2001;75(2):759–71. doi: 10.1128/JVI.75.2.759-771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills JW, Craven RC. Form, function, and use of retroviral gag proteins. AIDS. 1991;5:639–654. doi: 10.1097/00002030-199106000-00002. [DOI] [PubMed] [Google Scholar]
- Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. Structures of the HIV-1 capsid protein dimerization domain at 2.6 A resolution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 1):85–92. doi: 10.1107/S0907444998007689. [DOI] [PubMed] [Google Scholar]
- Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. Embo J. 2007;26(8):2218–26. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci U S A. 2006;103(19):7465–70. doi: 10.1073/pnas.0510483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu XF. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J. 2007;21(1):217–22. doi: 10.1096/fj.06-6773com. [DOI] [PubMed] [Google Scholar]
- Xu L, Yang L, Moitra PK, Hashimoto K, Rallabhandi P, Kaul S, Meroni G, Jensen JP, Weissman AM, D'Arpa P. BTBD1 and BTBD2 colocalize to cytoplasmic bodies with the RBCC/tripartite motif protein, TRIM5delta. Exp Cell Res. 2003;288(1):84–93. doi: 10.1016/s0014-4827(03)00187-3. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T. Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 2008;275(7):1540–55. doi: 10.1111/j.1742-4658.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol Cell. 2006;22(1):5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Yap MW, Dodding MP, Stoye JP. Trim-cyclophilin A fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J Virol. 2006;80(8):4061–7. doi: 10.1128/JVI.80.8.4061-4067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MW, Mortuza GB, Taylor IA, Stoye JP. The design of artificial retroviral restriction factors. Virology. 2007;365(2):302–14. doi: 10.1016/j.virol.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Yap MW, Nisole S, Lynch C, Stoye JP. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci U S A. 2004;101(29):10786–91. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MW, Nisole S, Stoye JP. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol. 2005;15(1):73–8. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular organisation of immature and mature murine leukemia virus revealed by electron cryo-microscopy: implications for retroviral assembly mechansims. Proceedings in the National Academy of Science (USA) 1998;95:7299–7304. doi: 10.1073/pnas.95.13.7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yebra G, Holgun A. The maturation inhibitor bevirimat (PA-457) can be active in patients carrying HIV type-1 non-B subtypes and recombinants. Antiviral therapy. 2008;13(8):1083–5. [PubMed] [Google Scholar]
- Yu F, Liu X, Zhan P, De Clercq E. Recent advances in the research of HIV-1 RNase H inhibitors. Mini Rev Med Chem. 2008;8(12):1243–51. doi: 10.2174/138955708786141052. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol. 2008;378(3):565–80. doi: 10.1016/j.jmb.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chen CH, Aiken C. The sequence of the CA-SP1 junction accounts for the differential sensitivity of HIV-1 and SIV to the small molecule maturation inhibitor 3-O-{3′,3′-dimethylsuccinyl}-betulinic acid. Retrovirology. 2004;1(1):15. doi: 10.1186/1742-4690-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chen CH, Aiken C. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J Virol. 2006;80(24):12095–101. doi: 10.1128/JVI.01626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Huang L, Hachey DL, Chen CH, Aiken C. Inhibition of HIV-1 maturation via drug association with the viral Gag protein in immature HIV-1 particles. J Biol Chem. 2005;280(51):42149–55. doi: 10.1074/jbc.M508951200. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee KH, Aiken C, Chen CH. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol. 2004;78(2):922–9. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]