

Abstract
Recent research and clinical data have begun to demonstrate the huge potential therapeutic importance of ligands that modulate the activity of the secretin-like, Class II, G-protein coupled receptors (GPCRs). Ligands that can modulate the activity of these Class II GPCRs may have important clinical roles in the treatment of a wide variety of conditions such as osteoporosis, diabetes, amyotrophic lateral sclerosis and autism spectrum disorders. While these receptors present important new therapeutic targets, the large glycoprotein nature of their cognate ligands poses many problems with respect to therapeutic peptidergic drug design. These native peptides often exhibit poor bioavailability, metabolic instability, poor receptor selectivity and resultant low potencies in vivo. Recently, increased attention has been paid to the structural modification of these peptides to enhance their therapeutic efficacy. Successful modification strategies have included D-amino acid substitutions, selective truncation, and fatty acid acylation of the peptide. Through these and other processes, these novel peptide ligand analogs can demonstrate enhanced receptor subtype selectivity, directed signal transduction pathway activation, resistance to proteolytic degradation, and improved systemic bioavailability. In the future, it is likely, through additional modification strategies such as addition of circulation-stabilizing transferrin moieties, that the therapeutic pharmacopeia of drugs targeted towards Class II secretin-like receptors may rival that of the Class I rhodopsin-like receptors that currently provide the majority of clinically used GPCR-based therapeutics. Currently, Class II-based drugs include synthesized analogues of vasoactive intestinal peptide for type 2 diabetes or parathyroid hormone for osteoporosis.
Keywords: G protein-coupled receptor, secretin-like, bioavailability, pharmacotherapeutic, instability, modification
1. INTRODUCTION
Presently, almost 50% of pharmacotherapeutics are targeted towards G protein-coupled receptors (GPCRs). These receptors represent one of the most diverse families of transmembrane sensory proteins in the genome. Their physiological importance is underscored by the finding that nearly 1% of most genomes are given over to code for GPCRs. GPCRs have evolved to facilitate a cell’s or organism’s ability to sense its local environment via interaction with factors such as photons, odorants, tastants, amino acids, saccharides, fatty acids, and complex polypeptides. The global group of GPCRs has recently been re-classified into the GRAFS system of superfamilies (Schioth and Fredriksson, 2005). This system classifies receptors according to their amino acid sequence and structure. The title GRAFS is an acronym for the superfamily exemplar receptors that have typified each phylogenetic branch, i.e. glutamate receptors (G), rhodopsin-like receptors (R), adhesion-family receptors (A), Frizzled/Taste2 receptors (F) and secretin-like receptors (S). Due to their early initial discovery and reinforcement of physiological relevance, the majority of GPCR research has focused upon the rhodopsin-like family (also known as Class I receptors). Through the intense research upon these rhodopsin-like receptors, most of the GPCR-targeted therapeutics clinically in use today are directed towards these receptors. Targeting the rhodopsin-like receptors pharmacologically has proven to be relatively facile due to the chemical nature of compounds to which these receptors are sensitive, e.g. small biogenic amines. Drugs that can mimic or antagonize the actions of endogenous ligands for these receptors (acetylcholine, norepinephrine, histamine) are relatively easy to synthesize and can be readily modified to increase bioavailability and control their metabolism and excretion. Even with rhodopsin-family members that interact with more complex small peptide ligands, the creation of multiple pharmacotherapeutics has been extremely successful, e.g. almost 1000 analogs of the gonadotropin-releasing hormone (GnRH) have been created so far (Millar et al., 2004). Despite the pharmacological pre-eminence of the rhodopsin-like receptors, research over the past decade has demonstrated the therapeutic importance also of the secretin-family (or Class II) of GPCRs (for reviews on this topic, see Martin et al., 2005; 2008). This class of GPCRs typically interacts with large glycoprotein hormones composed of between 20-50 amino acids. Therefore, creating pharmacotherapeutics based on these endogenous ligands poses several problems, not only for synthesis but also for bioavailability and proteolytic degradation. In this review, we will discuss the potential therapeutic importance of Class II receptor targeted-agents and outline the strategies that have been developed to facilitate the creation of viable drug molecules to modulate these receptors.
Peptide ligands for the Class II receptors demonstrate great potential as therapeutic targets for disorders that involve interactions of both neuronal and endocrine systems such as glucose metabolism and energy regulation, cardiac dysfunction, neurodegenerative disorders, obesity, inflammation, and immune dysfunction (see Figure 1 for overview). There is increasing evidence to suggest that for many disorders their etiology/pathology is strongly affected by subtle network interactions between the central neuronal and major somatic endocrine system, in-part mediated by the actions of hormones for Class II GPCRs (Martin et al., 2009, Shin et al., 2008; Wagner et al., 2009; Reglodi et al., 2002; Hanstein et al., 2008; Hill et al., 2007; Macica and Broadus, 2003; Gallai et al., 1995).
Figure 1. Role and targets of Class II GPCR ligands in pathophysiology. A large variety of large peptide hormones are secreted that mediate multiple endocrine functions at various target sites.
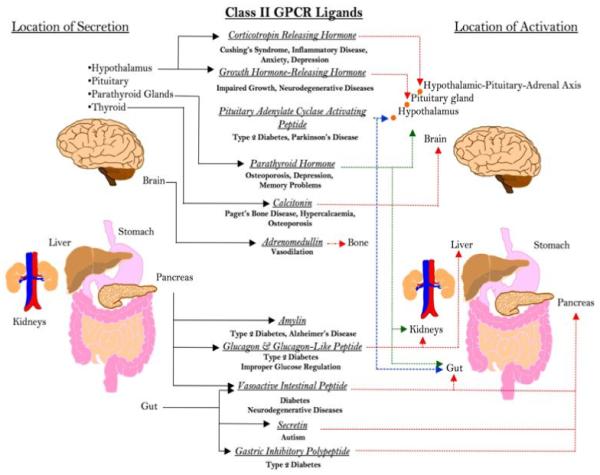
In addition to their physiological role, their pathological secretion or degradation can involve them in multiple acute and chronic disease/pathophysiological processes. The widespread expression and profound activities of these ligands indicate the importance of how a greater understanding of their pharmacological modification may facilitate a new wave of pharmacotherapeutics that may be complementary to or even supersede drugs targeted towards Class I rhodopsin-like receptors.
As many of the hormones that are involved in both autonomic endocrine functions have recently been demonstrated to exert important functional roles in the central nervous system (CNS), their potential exploitation as ‘multi-dimensional’ systemic therapeutics seems enticing. However, it is not always possible to directly use native peptides as therapeutic agents as they often exhibit poor bioavailability, metabolic stability, and potency in vivo. Therefore, it is necessary, in most cases, to modify native peptides to create hormone analogs, which are designed to overcome obstacles such as membrane barriers, rapid enzymatic degradation, and improper ligand-receptor recognition. In recent years, these large hormone derivatives have become more frequently utilized for the treatment of various physiological disorders linked to the direct physiological activity of these hormones at their cognate receptors. In this review, we provide an overview of the physiology and function of several Class II GPCR ligands that are currently the subject of pharmacological investigation. These include secretin, glucagon and glucagon-like peptides (GLPs), growth hormone releasing hormone (GHRH), pituitary adenylate cyclase activating peptide (PACAP), corticotropin-releasing hormone (CRH), vasoactive intestinal peptide (VIP), parathyroid hormone (PTH), calcitonin family ligands, and gastric inhibitory polypeptide (GIP). Additionally, we also discuss several modification strategies that have been used to develop viable peptide analogs and their potential applications in the treatment of various disorders. This review provides a comprehensive overview of Class II GPCR ligand derivatives and their current uses for the treatment of a wide array of disorders. While the classification of G-protein coupled receptors is based on their primary structure, not the function of their endogenous ligands, we have chosen to discuss diverse ligands for a range of Class II GPCRs that are linked via their ability to exert effects at multiple physiological levels, i.e. upon neuronal and endocrine systems. Hence, the peptides the review covers are functionally diverse but are generally related in their functional abilities.
2. SECRETIN
2.1. Physiological secretin activity
Secretin, a 27-residue peptide, was first identified and sequenced from secretory granule-containing endocrine S-cells. Secretin is secreted into the duodenum and proximal jejunum from these endocrine S-cells and acts on epithelial cells lining the pancreatic and biliary ducts to promote the secretion of alkaline bicarbonate-rich fluid. This, in turn, helps to neutralize the acidic chyme emptied from the stomach. Similarly, secretin also slows gastric emptying to further protect the duodenum from excessively acidic chyme (Mutt, 1980).
2.2. Secretin alterations in disease/pathology
Alteration of secretin levels in the gut can have important pathophysiological consequences. Clinical evidence has revealed that patients with disorders such as H. pylori infection and peptic ulcers have increased gastric secretion and motility as well as decreased duodenal bicarbonate response to gastric acid, all of which normalize after eradication of the infection (Love, 2008). These symptoms could be explained by reversible impairment of the secretin mechanism, a theory that is supported by the fact that gastric metaplasia in the duodenum with H. pylori infection is known to reduce secretin secreting S-cells. Secretin may also play an important role in the central nervous system (CNS). In the brain, secretin receptor expression is highest in the cerebellum, intermediate in the cortex, thalamus, striatum, hippocampus, and hypothalamus, and lowest in the brainstem and medulla (Fremeau et al., 1983; Yung et al., 2001). Interestingly, it has been shown that secretin-deficient mice demonstrate impairment in synaptic plasticity in the CA1 area of the hippocampus (Yamagata et al., 2008). Additionally, another study demonstrated that in addition to an alteration in synaptic plasticity, secretin receptor-deficient mice show abnormal social and cognitive behaviors (Nishijima et al., 2006). These findings suggest that the secretin receptor system has important roles in the CNS related to social behavior and learning, and the role of secretin is now being investigated in various CNS disorders, such as autism spectrum disorders. One of the earliest indications that secretin may play a role in the autism phenotype was the report of a single case of the symptomatic improvement of three autistic children after receiving secretin (to improve ductal visualization) as part of a gastro-intestinal/pancreatic evaluation (Horvath et al., 1998).
2.3. Secretin tyrosine substitution
Synthetic human and porcine secretin are used frequently in experimental studies, but few highly modified analogs of the compound exist. Banks and colleagues (2002) developed a 131I radioactively-labeled, tyrosine-substituted secretin analog (I-SA). The tyrosine at position 10 within the I-SA peptide was substituted for leucine in standard secretin-27, significantly increasing the bioavailability of the compound. One of the main concerns regarding the synthesized compound was its ability however to cross the blood brain barrier (BBB). Many potentially efficacious compounds are too large to cross this barrier, which prevents molecules such as secretin to act directly on the brain. It was determined that peripherally administered I-SA does indeed cross the BBB at a modest rate by nonsaturable transmembrane diffusion. Additionally, it was also shown to enter the cerebral spinal fluid (CSF) by a saturable transporter. The highest uptake of I-SA in the brain was into the hypothalamus and the hippocampus, whereas the frontal and parietal cortices had the lowest uptake. The extent of I-SA uptake in the brain was similar to that of other proteins that are able to cross the BBB, such as interleukin-1α. Unlike standard secretin, I-SA has not been tested as a pharmacological agent. However, several groups have investigated the use of native secretin for the treatment of Autism Spectrum Disorders in young children (for reviews see Patel et al. 2002; McQueen and Heck 2002; and Kern et al. 2004). Some of these studies have shown successful outcomes with this peptide, whereas others have demonstrated that secretin had no therapeutic effect. Perhaps in the future, I-SA and other novel secretin analogs could be tested for the treatment of Autism Spectrum Disorders (ASDs), as modifications to the bioavailability of these agents could potentially provide a broader efficacy for the treatment of this disorder. However, as demonstrated by the variability of the existing clinical data concerning secretin’s efficacy in ASD, the future usage of secretin or secretin-like analogs should be considered carefully on a case to case basis and with close attention paid to the variability of symptomatic presentations seen in ASDs.
3. GLUCAGON AND GLUCAGON-LIKE PEPTIDES (GLPs)
3.1. Physiological roles of Glucagon and Glucagon-Like Peptides
Glucagon is a 29-amino acid peptide identified as a hyperglycemic factor originating from the pancreas (Young, 2005). Its primary structure is identical in most mammals, including man (Irwin, 2001). It is synthesized mainly in the α-cells of the islets of Langerhans but has also been shown to be expressed in specialized neurons of the CNS (Baum et al., 1962). Isolation of cDNAs encoding glucagon have shown that the peptide is produced from proglucagon, a 160-amino acid precursor that also contains two additional glucagon-like sequences at its carboxyl terminus (Lund et al., 1982). These glucagon-like sequences, known as glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2), have been subsequently shown to display specific biological activities (Drucker, 1998).
Glucagon plays a complex role in digestion and feeding behavior. Production of the hormone is initiated by amino acid nutrients as well as stressful stimuli, such as hypoglycemia and hypovolemia (Young, 2005). Glucagon release results in increased hepatic glucose production, decreased muscle and adipocyte glucose uptake, and increased blood glucose levels (Bensal and Wang, 2008). The glucagon receptor is widely expressed in the body and has been found in the liver and kidneys, and to a lesser extent, in the heart, adipose tissue, spleen, thymus, adrenal glands, pancreas, cerebral cortex, and throughout the gastrointestinal tract of rats (Svoboda et al., 1994; Dunphy et al., 1998).
Another hormone that is known to play a vital role in energy homeostasis is the glucoincretin GLP-1. In humans, GLP-1 is secreted by epithelial intestinal L-cells shortly after ingestion of glucose or lipids and acts on multiple target tissues to enhance energy metabolism (Burcelin et al., 2007). GLP-1 binds with high affinity to glucagon receptors located on pancreatic β-cells and influences insulinotropic actions such as insulin gene transcription, insulin biosynthesis, and insulin secretion (Holz and Chepurny, 2003). Furthermore, GLP-1 inhibits glucagon secretion and decreases gastrointestinal secretions and motility, thus producing a sensation of satiety and reducing food intake (Aaboe, 2008). Shortly after release, the hormone however is quickly degraded (1-2 minutes) by the enzyme dipeptidyl peptidase IV (DPP-IV), rendering it inactive (Burcelin et al., 2007). In addition to its regulatory effects on energy homeostasis, GLP-1 has also been shown to have beneficial effects on neuronal health (Aaboe et al., 2008). When cultured neuronal PC12 cells were exposed to nerve growth factor (NGF) withdrawal, GLP-1 provided the cells with sustained protection from degeneration and death (Biswas et al., 2008). Similarly, it was determined that GLP-1 can completely protect cultured rat hippocampal neurons against glutamate-induced apoptosis. Although GLP-1 receptors are expressed in the brain, particularly in the arcuate nucleus, it is thought that the peptide from the circulating blood, rather than from the CNS, is more likely to act on the receptors in this area. However, it has been demonstrated that a 125I-labeled analog of native GLP-1, [Ser8]GLP-1, readily enters the brain with a transfer rate of nearly 9 × 10-4 mL/g-min. This influx is thought to occur via passive diffusion, as a saturable transport system at the BBB is not established both in vivo and in situ (Kastin et al., 2002). Therefore, by inhibiting the action of DPP-IV or by infusing high doses of GLP-1 to saturate the enzyme, the penetration of the peptide across the BBB should be enhanced. Though the exact mechanism through which GLP-1 exerts its neuroprotective actions presently remains uncertain, it is now becoming apparent that it may play an important role in future neuroprotective therapeutics.
3.2. Glucagon and Glucagon-Like Peptides: fatty acid acylation
Among this peptide family, GLP-1 has by far been the agent that has been the most pharmacotherapeutically derivatized thus far. Several analogs of glucagon-like peptide-1 (GLP-1) have been developed in order to overcome the rapid degradation of the peptide by the enzyme dipeptidyl peptidase IV (DPP-IV), improving its metabolic stability as well as maintaining or enhancing the bioactivity of standard GLP-1. Several modification strategies have been used, e.g. fatty acid acylation of GLP-1 has been shown to extend the efficacy of GLP-1. Based on this approach, analogs of GLP-1 have been created with both short and long fatty acids on positions within the C-terminal part of the peptide. All compounds were full agonists and selective for the GLP-1 receptor. In general, half-lives of the analogs were much improved and reached at least 9 hours when measured in a porcine model (Knudsen et al., 2000). Liraglutide, a specific example of an acylated analog of GLP-1, was shown to have a half-life of 14 hours. Moreover, it demonstrated the ability to lower blood glucose over 24 hours in a porcine model of type 2 diabetes. When tested in diabetic patients over a 12-week period, liraglutide repaired β-cell response, stimulating insulin secretion, and improved glycemic control to a level observed in non-diabetics while maintaining bodyweight. It was determined to be safe and well tolerated, but the exact dosage still remains to be determined (Feinglos et al., 2005). Another example of a fatty acid-modified GLP-1 analog is the DPP IV-resistant mono-poly(ethylene glycol)-conjugated GLP-1 (mono-PEGylated GLP-1) (Lee et al., 2005). It is prepared by combining GLP-1 with excess mPEG2k-SPA and mPEG2k-ALD. The fatty acid is conjugated to either the N-terminal or a lysine residue, making mono-substituted conjugates at Lys26 and Lys34. These analogs were just as potent at stimulating insulin secretion in rats as regular GLP-1 and also have significantly longer half-lives in vitro. The Lys conjugates demonstrated good resistance to proteolytic degradation, as most of the administered compound was still present in plasma after 60 minutes and was still detectable after 2 hours. Therefore, these mono-PEGylated GLP-1 conjugates show significant improvement in plasma exposure and stability compared to native GLP-1, leading to an overall increase in the pharmacokinetic properties of GLP-1. However, the conjugates still do not adequately avoid degradation by DPP-IV, which prevents them from being completely effective anti-diabetic agents.
3.3. Glucagon and Glucagon-Like Peptides: N-terminal modifications
In addition to fatty acid acylation, additional strategies of GLP-1 modification have been adopted in order to develop a pharmacological agent for type 2 diabetes treatment. Firstly, GLP-1 analogs have been synthesized by modifying different positions within the N-terminus of the peptide. D-amino acids were substituted for the naturally occurring amino acids within GLP-1, changing the steric conformation of the peptide. All analogs had at least a ten-fold increase in DPP-IV resistance and retained most of the biological activity of the native peptide. The most successful analogs were derivatives that had D-amino acid substitutions at position 2 of the GLP-1 molecule, which had the lowest rate of degradation and preserved receptor recognition. These analogs were not tested as treatments for type 2 diabetes specifically; however, they exhibited an increased ability to stimulate insulin secretion in response to glucose (Siegel et al., 1999). Similarly, Xiao and colleagues (2001) created analogs of GLP-1 utilizing four modification strategies: N-terminal modification at His1 and at position 2, chimera derivatives that involved replacing the N-terminus of GLP-1 with the corresponding region of several related peptides of the glucagon/secretin family (e.g., glucagon, GIP, PACAP, PHI, secretin, VIP); midchain modification; and alteration of the C-terminus. Overall, the analogs with N-terminal modifications most successfully avoided immediate DPP-IV degradation, and thus increased its bioavailability. These analogs also normalized blood glucose levels for over 2 hrs when tested in diabetic (db/db) mice (Xiao et al., 2001).
Finally, while fatty acid acylation and D-amino acid substitutions are modifications of the GLP-1 peptide itself, others have developed analogs of GLP-1 receptor agonists, which mimic the actions of the naturally occurring peptide. One example is exendin-4, a naturally occurring GLP-1 receptor agonist that was isolated from the salivary gland venom of the Gila monster (Heloderma suspectum). In this molecule, the second N-terminal amino acid alanine is replaced by serine. This modification substantially increases the half-life and gluco-regulatory effect of native GLP-1 in patients (Pratley and Gilbert, 2008). Recently, Martin and colleagues demonstrated that this long-acting analog improves peripheral glucose regulation and reduces cellular pathology in both the brain and the pancreas in a mouse model of Huntington’s disease (HD). Exendin-4 treated HD mice also exhibited prolonged life spans and improved motor function compared to untreated HD mice (Martin et al., 2009). Exenatide, the synthetically derived version of exendin-4, has been shown to stimulate glucose-dependent insulin secretion, regulate glucagon secretion, delay gastric emptying, and decrease food intake. Additionally, this agonist has a half-life of 2.4 hours and is detectable in patients for up to 10 hours. Exenatide has been approved by the FDA for treatment of type 2 diabetes for patients who prescribe to other therapies, such as metformin and sulfonylurea, but continue to experience insufficient glycemic control (Odegard et al., 2006). With additional studies in animal models and patients, GLP-1 derivatives offer great promise as effective pharmaceutical treatments for type 2 diabetes and other disorders related to inadequate glucose regulation.
4. GROWTH HORMONE-RELEASING HORMONE (GHRH)
4.1. Physiological roles of Growth Hormone-Releasing Hormone
Growth hormone-releasing hormone (GHRH) is a peptide of 42-44 amino acids, depending on the species, that is proteolytically processed from a 103-108 amino acid precursor protein (Mayo et al., 1983). GHRH is released in a pulsatile manner from neurosecretory cells in the arcuate nuclei of the hypothalamus (Merchenthaler et al., 1984; Sawchenko et al., 1985). Upon release, GHRH is transported by the hypothalamo-hypophyseal portal circulation to the somatotrophs of the anterior pituitary, where it stimulates pulsatile growth hormone (GH) secretion into the systemic circulation. GH can then act on a number of peripheral target tissues to alter cellular metabolism, proliferation, and differentiation. Many of the effects of GH are mediated by the generation and action of insulin-like growth factor-I (IGF-I), a strong proliferative growth factor and potent neuroprotective compound (for review, see Mattson et al., 2004). In turn, there is considerable evidence that IGF-I can participate in the physiological regulation of GH and therefore interact with the actions of GHRH. For instance, the IGF-I receptor is expressed both in the hypothalamus and the pituitary (Goodyer et al., 1984; Rosenfeld et al., 1984). In the hypothalamus, IGF-I stimulates the release of somatostatin, a hormone that inhibits GH secretion, into the hypothalamo-hypophysial portal circulation (Shibaski et al., 1986). Consequently, IGF-I may have the ability to reduce the secretion and synthesis of GH from the pituitary (Yamashita and Melmed, 1986).
4.2. Roles of Growth Hormone-Releasing Hormone in disease/pathology
GHRH deficiency can lead to a reduction in GH release, which is caused by a number of factors including GHRH receptor mutation, increased release of somatostatin, or an elevated sensitivity to IGF-I feedback (Thorner et al., 1997). GHRH deficiency can also result from damage to the hypothalamus and can lead to several chronic complications such as short stature, reduced bone mineral density, increased visceral fat, insulin resistance, and increased cardiovascular risk (Doga et al., 2006; Lange et al., 2005). Therefore the early detection of GH deficiency is clinically important. However, as GH secretion is pulsatile and has a half-life of approximately 19 minutes, it is usually undetectable in normal subjects and makes GH deficiency difficult to determine using a single random GH measurement. In the past, GH stimulation, insulin tolerance, and GHRH plus arginine tests have been used to determine GH deficiency with varying degrees of success (Doga et al., 2006). In addition to the impaired GH levels observed with GHRH deficiencies, levels of GH secretion have also been shown to decline with normal aging. Incidentally, the declining activity of the GH-IGF-I axis associated with aging results in changes in body composition, function, and metabolism that show prominent similarities with those of younger adults with pathological GH deficiency (Lanfranco et al., 2003). The age-related changes of the GH-IGF-I axis activity are mainly dependent on variations in the hypothalamic control of somatotroph function. This can also be affected by changes in peripheral hormones and metabolic input. Thus, the condition of “somatopause”, or the dysfunction of GHRH and the age-related decline in GH and IGF-I levels, may be responsible for the changes in body composition, structural functions, and metabolic function, which are characteristic of normal aging.
Dysfunction of the GH-IGF-I axis associated with aging may also play an important role in neurodegenerative disorders. Several studies have reported reduced GH responses to GHRH in patients with Alzheimer’s disease (Nemeroff et al., 1989; Lesch et al., 1990), whereas other studies have been inconclusive (Heuser et al., 1992). Interestingly, in patients with Parkinson’s disease, there appears to be impairment in the stimulation of GH release by serotonin, although GH responses to GHRH may remain intact (Volpi et al., 1997). Studies supporting a role for the GH-IGH-I axis in neurodegenerative diseases claim that IGF-I is a potent neurotrophic and neuroprotective factor in the brain, as it promotes brain development and neuronal survival (Gómez, 2008). Thus, as there are several disorders related to GHRH dysfunction, the development of successful pharmacological treatments targeting this Class II ligand would be potentially beneficial.
4.3 Growth Hormone Releasing Hormone: therapeutic chemical modification for half life extension
GHRH, like many other large neuropeptides is limited by its short circulatory half-life of 7 minutes, and subsequently, several peptide analogs have been developed in order to overcome this trait for use as an efficacious pharmacological agent. One example of a long-acting GHRH analog is CJC-1295. This compound has a core therapeutic moiety of a tetra-substituted GHRH-(1-29)NH2 that allows it to bind to endogenous serum albumin after administration, avoiding rapid proteolytic cleavage. This analog has been tested in several animal species including dogs, pigs, and humans. Studies have shown that CJC-1295 causes sustained, pulsatile GH secretion and increased IGI-I levels for several days after only a single administration. The compound also has a much improved half-life of approximately 6 to 8 days as measured after one dose, which would allow patients to follow a 7 day dosing interval instead of several daily administrations (Teichman et al. 2006; Ionescu and Frohman, 2006). JI-38, a second example of a GHRH analog, is synthesized by incorporating ornithine instead of lysine at two locations within the peptide in addition to substitutions of desaminotyrosine, L-α-aminobutyric acid, agmatine, norleucine, aspartic acid and glutamine (Izdebski et al. 1995). These modifications also increase the compound’s resistance to enzymatic degradation. Additionally, JI-38 has been shown to increase serum GH in mice completely deficient in native GHRH. These animals also exhibited a significant increase in the rate of longitudinal growth, which demonstrates that the analog can partially reverse GH deficiency. However, unlike CJC-1295, JI-38 did not cause a marked increase in serum IGF-I levels (Alba et al. 2005). While CJC-1295 and JI-38 are agonistic derivatives of GHRH, analogs with antagonistic capabilities have also been developed, which have been used for the prevention and treatment of some tumors. Compounds such as MZ-5-156 and JV-1-36 suppress the GH/IGF-1 axis, thereby averting amplified cell proliferation associated with several cancers (Kineman 2000, Kiaris et al. 2000). Some antagonistic GHRH analogs, such as those characterized by Rekasi and colleagues (2000), also interact with VIP receptors (VPAC-R), which unlike GHRH receptors, are present on some tumor cell-types and may provide a more direct inhibition of tumor proliferation. These antagonistic analogs have the common core sequence of human GHRH and contain several substitutions with amino acid residues such as arginine, d-arginine, homoarginine, and norleucine (Varga et al. 1999).
5. PITUITARY ADENYLATE CYCLASE ACTIVATING PEPTIDE (PACAP)
5.1. Physiological actions of Pituitary Adenylate Cyclase Activating Peptide
Pituitary adenylate cyclase activating peptide (PACAP) was first identified as a 38-amino acid peptide (PACAP-38) from ovine hypothalamus that stimulated adenylate cyclase in rat anterior pituitary cells (Miyata et al., 1989). In humans, PACAP has an amino acid sequence homology of 68% with that of vasoactive intestinal polypeptide (VIP) and of 37% with that of secretin, indicating that PACAP is a member of the VIP/glucagon/secretin superfamily (Hashimoto, 2002). PACAP activates several types of Class II receptors; however, this polypeptide exerts the majority of its effects through the PAC1 receptor. The PAC1 receptor exhibits a high affinity for PACAP and a much lower affinity for VIP, whereas the related VPAC1 and VPAC2 receptors have similar affinity for both PACAP and VIP. Distribution analysis and pharmacological studies of PACAP and its receptors have revealed its pleiotropic effects as a hormone, neurotransmitter, neuromodulator, vasodilator, neurotrophic factor and immunomodulator (Fahrenkrug, 2006). The greatest density of PACAP in the brain is found in the magnocellular region of the hypothalamic PVN and SON (Köves et al., 1990; Masuo et al., 1992), where it acts to increase the neuronal firing activity and cause membrane depolarization of magnocellular neurons. Intracerebroventricular and intracisternal injection of PACAP has been shown to cause dose-dependent elevations in plasma vasopressin concentration (Murase et al., 1993; Seki et al., 1995), while introduction of PACAP into the neural lobe of the pituitary stimulates the release of both oxytocin and vasopressin (Lutz-Bucher et al., 1996). PACAP has also been shown to modulate the activity of various other hypothalamic neuronal populations, such as elevating gonadotropin-releasing hormone (GnRH), somatostatin, and corticotropin-releasing hormone (CRH) gene expression (Li et al., 1996; Grinevich et al., 1997). Additionally, PACAP is also known to play an important role in the development of the nervous system and in regeneration following nerve injuries (for review, see Somogyvári-Vigh and Reglodi, 2004). PACAP exerts strong anti-apoptotic effects in several types of cultured neurons and in vivo. It can protect neurons against various toxic insults in vitro, has anti-inflammatory actions, and stimulates the release of neuroprotective substances from astrocytes. In vivo, the protective effects of PACAP have been shown in various models of brain injuries, including cerebral ischemia, Parkinson’s disease, physical trauma, and nerve transections. The upregulation of PACAP following several types of nerve injuries indicates that endogenous PACAP plays a role in the post-traumatic recovery of the nervous system. At the cellular level, PACAP inhibits cerebellar granule neuron apoptosis (Cavallaro et al., 1996; Gonzalez et al., 1997; Villalba et al., 1997; Vaudry et al., 2000) and stimulates neurite outgrowth (Gonzalez et al., 1997). Outside of the brain and CNS, PACAP also affects multiple physiological systems to cause vasorelaxation of smooth muscles, absorption of water and ions, and secretion from the pancreas and intestine (Sherwood et al., 2000).
5.2. Pathophysiological roles of Pituitary Adenylate Cyclase Activating Peptide
Alteration of PACAP and its cognate receptor system have, through epidemiological data and animal models, been linked to multiple pathophysiological conditions such as schizophrenia (Hashimoto et al., 2007), disrupted temperature regulation (Gray et al., 2002), medulloblastoma (Lelievre et al., 2008) and fertility (Shintani et al., 2002). It is interesting to note as well that a small number of diseases and disorders have been linked to PACAP autoimmunity. Autoimmune dysfunction of endogenous vasoactive neuropeptides such as VIP and PACAP has been proposed as a cause for fatigue-related conditions such as multiple sclerosis, amyotrophic lateral sclerosis, and Gulf War Syndrome (Staines, 2005, 2008). These syndromes share common pathogenic features including inflammation, oxidative stress, and mitochondrial dysfunction. PACAP autoimmunity has also been postulated to play a role in Parkinson’s disease pathology due to observed pro-inflammatory responses consistent with apoptotic neurodegeneration (Staines, 2007). As PACAP has neuroprotective effects in Parkinson’s disease models, a defect in vasoactive neuropeptide function may act adversely on neurons to contribute to neurodegeneration. Therefore, it appears that PACAP plays a role in several pathologies, and further research is needed to elucidate the role of the peptide both in the brain and in the periphery.
5.3. Pituitary Adenylate Cyclase-Activating Peptide: therapeutic chemical modification
Native pituitary adenylate cyclase-activating peptide (PACAP) and its receptors are widely distributed within the body, suggesting a role in several key biological activities, e.g. PACAP stimulates insulin secretion upon activation of the PACAP R3 receptor, creating a potential therapeutic use for this peptide in the treatment of type 2 diabetes. However, significant side effects are observed with use, negating any potential improvement in glycemic control. Thus far, many unsuccessful attempts at developing viable derivatives of this compound have been made, for example, R3P66, a leading analog of this peptide, demonstrates increased R3 potency and selectivity; however, this compound has a short half-life and is therefore not an efficacious pharmaceutical agent (Severs and Froland, 2008).
In addition to the treatment of metabolic disorders, PACAP-derived compounds may also serve as promising therapeutic targets for ameliorating pathologies due to various neurodegenerative diseases, such as Parkinson’s disease (Reglodi et al. 2006). These CNS compounds are thought to target the PAC1 receptor, which mediates the neuroprotective effects of the compound, and are designed to avoid rapid enzymatic degradation. It has been shown that modification of the N-terminal domain of the peptide promotes resistance to metabolic breakdown while still preserving biological activity. In particular, acetyl-PACAP derivatives are fully active at both human and rat PAC1 receptors (Bourgault et al., 2008). An additional interesting use of PACAP analogs is in the field of oncology. The native peptide exhibits high affinity for the VIP receptor VPAC1, which is located in high density on the plasma membrane of many human tumor cell-types. Zhang and colleagues developed a radiolabeled version of the PACAP analog TP3805, with the goal of specifically targeting this receptor for in vivo visualization and marking of tumor cells by PET imaging. In this study, the analog was labeled with 64Cu or 99mTc and was found to successfully label VPAC1 receptors on malignant cells of human breast tissue, without compromising the potency and function of the peptide. With additional evaluation, this method may provide patients an alternative route to invasive biopsy surgeries of suspicious masses, particularly those found by mammography (Zhang et al. 2007).
6. CORTICOTROPIN-RELEASING HORMONE (CRH)
6.1. Physiological roles of Corticotropin-Releasing Hormone
Corticotropin-releasing hormone (CRH) is a 41-amino acid peptide that functions as a potent mediator of endocrine, autonomic, and immune responses to stress (Holsboer and Barden, 1996; Owens and Nemeroff, 1991). The native CRH ligand exerts it molecular actions via activation of CRH receptor 1 (CRHR1) or 2 (CRHR2) (Chen et al., 1993; Liaw et al., 1996). It is released from parvocellular neurons of the paraventricular nucleus (PVN) of the hypothalamus, the major site of CRH-containing cell bodies, into portal vessels (Merchenthaler, 1984; Swanson and Simmons, 1989). From there, it activates the hypothalamic-pituitary-adrenal (HPA) axis by triggering the immediate release of adrenocorticotropic hormone (ACTH) from the anterior pituitary (Holsboer and Barden, 1996; Owens and Nemeroff, 1991; Vale et al., 1981). ACTH, in turn, can stimulate the secretion of glucocorticoids from the adrenal gland. Brain regions outside of the PVN with high densities of CRH-containing neurons include the bed nucleus of the stria terminalis and interneurons of prefrontal, cingulate, and insular cortical areas (Owens and Nemeroff, 1991). CRH and CRH-related peptides can also be found peripherally in the immune, cardiovascular, and reproductive systems (Grammatopoulos and Chrousos, 2002). CRH has been implicated in the modulation of a wide range of different types of behavior, including long-term stress response, motor function, arousal, food intake, reproduction, parturition, and anxiety-related behavior (Dunn and Berridge, 1990; McLean and Smith, 2001).
6.2. Pathophysiological roles of Corticotropin-Releasing Hormone
Some studies have suggested that depression may be caused by hypersecretion of CRH from hypothalamic as well as from extrahypothalamic neurons (Arborelius et al., 1999; Grammatopoulos and Chrousos, 2002). This hypersecretion can result in hyperactivity of the HPA axis and mediate some of the behavioral symptoms of depression, including sleep and appetite disturbances, reduced libido, and psychomotor changes. Similar alterations of the HPA axis and of CRH release have been implicated in disorders such as panic attacks, post-traumatic stress disorder, and irritable bowel syndrome (IBS) (Ströhle and Holsboer, 2003). Convincing pre-clinical data has shown that brain CRH administration can mimic acute stress-induced colonic responses and enhance colorectal distension-induced visceral pain in rats. Peripheral CRH has also been shown to reduce the pain threshold to colonic distension and increased colonic motility in humans and rodents. These observations mimic the manifestations of IBS, characterized by abdominal bloating or discomfort and altered bowel habits.
In addition to its potential role in several pathologies, an interesting dichotomy exists with respect to the involvement of CRH in neuronal protection and damage. In some experimental paradigms, CRH appears to be involved in neuronal damage and death, but in other scenarios, it exerts a potent neuroprotective effect. For instance, in rat traumatic injury models, CRH is rapidly upregulated in the PVN and the amygdala. Using a CRH receptor antagonist, a marked neuroprotective effect against a percussion injury was observed, suggesting that CRH is directly involved in the pathogenesis of the brain trauma (Roe et al., 1998). In marked contrast, however, CRH has been shown to exert a PKA-dependent protective action in CRHR1-expressing neurons against oxidative cell death (Lezoualc’h et al., 2000). CRH was also shown to potently prevent glutamate-induced neurotoxicity before or after the actual administration of glutamate to organotypic hippocampal cultures (Elliott-Hunt et al., 2002). Furthermore, CRH protected cultured rat hippocampal neurons from being degraded by glutamate, lipid peroxidation, and amyloid beta-peptide, insults relevant to the pathogenesis of Alzheimer’s disease (Pedersen et al., 2001). Patients with Alzheimer’s disease show morphological abnormalities in the CRH neurons and also dramatic reductions in CRH content (Bissette et al., 1985). Interestingly, cognitive impairment in Alzheimer’s disease patients has been shown to be accompanied by decreased concentrations of CRH in cerebrospinal fluid (Pomara et al., 1989).
6.3. Corticotropin Releasing Hormone: amino acid substitution
Like PACAP, corticotrophin releasing hormone (CRH) and its receptors are widely distributed throughout the body, which implicates that this hormone may play a role in numerous biological processes, particularly those related to the stress response. Several attempts have been made to develop pharmacologically viable peptide derivatives of CRH, but most studies have not produced any fully potent compounds. One such derivative involves amino acid substitutions at two positions within the peptide sequence. Using the Fmoc/tBu solid-phase synthetic protocol, L-phenylalanine is replaced by D-phenylalanine at position 12 and L-leucine is substituted by α-aminoisobutyric acid (AiB) at position 15. This modified peptide did appear to effectively bind to the CRH receptor; however, its biological activity, as measured by the release of catecholamines, was lost. Nevertheless, further development of effective agonistic and antagonistic derivatives may positively impact the treatment of several conditions, such as Cushing’s syndrome, inflammatory diseases, pre-eclampsia, anxiety, and depression (Spyroulias et al. 2002).
Much work has also focused on the development of non-peptidergic CRH receptor antagonists, such as CP-154,526, which has been found to reduce the concentration of norepinephrine and serotonin but not dopamine in the hippocampus after injection (Isogawa et al. 2000). When supplemented with behavioral testing, it was found that the antagonist also significantly reduced the conditioned fear stress induced by freezing behavior, suggesting that this compound had anxiolytic properties (Hikichi et al. 1997). Underlying its importance in clinical therapeutics, in addition to its potent neuroprotective effects, there are many other important beneficial applications for CRH-related ligands (agonists or antagonists) outside the neuroendocrine axis, e.g. for post-operative pain modulation (Likar et al., 2007), sleep quality control (Vgnontzas et al., 2001), prevention of pre-term labor (Chan et al., 1998), drug addiction therapy (Stinus et al., 2005) and even in the treatment of psoriasis (Cao et al., 2005).
7. VASOACTIVE INTESTINAL PEPTIDE (VIP)
7.1. Physiological roles of Vasoactive Intestinal Peptide
Vasoactive intestinal peptide (VIP) is a 28-amino acid peptide produced in many areas of the body, including the gut, pancreas, and suprachiasmatic nucleus (SCN) of the hypothalamus. VIP has many functions, including stimulation of pepsinogen secretion by the chief cells of the gut, smooth muscle relaxation and vasodilation, water secretion into pancreatic juice and bile, and secretion of water and electrolytes into the intestine (Felley et al., 1992; Onoue et al., 2008; Riepl and Lehnert, 1993; Wapnir and Teichberg, 2002). Together, these reactions facilitate overall gut motility and digestion. In addition to its role in the gastrointestinal tract, VIP has been shown to be involved in a wide range of biological functions, e.g. VIP is an important immunomodulator that influences numerous processes responsible for controlling the homeostasis of the immune system (Gonzalez-Rey et al., 2007). It is released within lymphoid organs from nerve terminals and/or immune cells and plays a significant, anti-inflammatory role by inhibiting macrophage-induced inflammatory reactions and promoting T-helper cell type 2 responses (Delgado et al., 2004). VIP also induces prolactin secretion from the pituitary and catecholamine release from the adrenal medulla (Reichlin, 1988; Malhotra et al., 1988). In a bronchial airway system where VIP-immunoreactive nerve fibers are present, VIP can also act as a neurotransmitter or neuromodulator of the inhibitory non-adrenergic and non-cholinergic airway nervous system and thus influence many aspects of pulmonary biology (Onoue et al., 2007).
With specific respect to VIP’s effects in the CNS, recent evidence indicates that VIP is critical in the normal functioning of the SCN, the master internal clock located in the hypothalamus of mammals that is responsible for synchronizing daily rhythms (Reed et al., 2001; Harmar et al. 2002; Aton et al., 2005; Brown et al., 2007). It was also shown that VIP plays an important role in driving circadian rhythms in the hypothalamic-pituitary-adrenal axis. VIP-deficient mice lost circadian rhythms in ACTH and corticosterone and had an impaired ability to produce a corticosterone response in the presence of light (Loh et al., 2008). VIP also possesses a crucial role in neuroprotective mechanisms as it can potently promote neuronal survival and regulate glycogen metabolism in the cerebral cortex (Brenneman and Eiden, 1986; Sorg and Magistretti, 1992). VIP has also been implicated as a neuroprotective factor for a number of neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease. Alzheimer’s disease is described by several pathologies including extracellular deposits of fibrillar beta-amyloid (Aβ) in the brain, increased microglial-mediated inflammatory reactions in senile plaques, selective neuronal death, and cognitive deficits. VIP inhibits Aβ-induced neurodegeneration by indirectly preventing the production of a large group of inflammatory and neurotoxic agents by activated microglia cells (Delgado et al., 2008). It has also been shown that VIP can ameliorate the toxicity of dopamine, 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenylpyridinium ion (MPP) in rat PC12 cells. This neuroprotection may be mediated by the mechanism of raising cellular resistance against oxidative stress (Offen et al., 2000).
7.2. Pharmacotherapeutic potential Vasoactive Intestinal Peptide-based peptides
VIP has been suggested as a potential treatment for numerous clinical disorders, such as diabetes, neurodegenerative disorders, asthma, lung injury, and multiple types of tumors. However, treatment of these disorders with VIP is limited due to the extremely rapid degradation of the peptide in vivo, leading to the low potency and short duration of any therapeutic effects. In that light, the development of VIP analogs has focused mainly on maintaining high stability against destructive digestive enzymes (Dangoor et al., 2008). Following chemical modification of the native peptide, VIP analogs have been proposed as promising treatments for treating acute and chronic inflammatory and autoimmune diseases, such as septic shock, multiple sclerosis, Crohn’s disease, and autoimmune diabetes (Gonzalez-Rey et al., 2007).
One common pharmacotherapeutic modification of VIP includes alteration of the N-terminus in order to affect binding to the cognate receptors for VIP (VPAC1, VPAC2). Compared to the native peptide, a more potent and selective agonist for the VPAC2 receptor was discovered by synthesizing a VIP analog through N-terminal hexanoylation. The manipulation resulted in higher selectivity and potency by increasing the affinity for the VPAC2 receptor and decreasing the affinity for the VPAC1 receptor (Langer et al., 2004). Similarly to the selectivity accomplished for the VPAC2 receptor, a rapid dose-dependent internalization of an agonist for the VPAC2 receptor occurred with the addition of a monoclonal antibody on the N-terminus segment of VIP (Langlet et al., 2004). More specifically, structural modifications, such as linear tandem extension and parallel branching, have been investigated with respect to the effects of adenylate cyclase coupling to the VPAC1 and VPAC2 receptors. The two branched N-terminal VIP sequences exhibited higher binding and activation of the receptor in respect to native VIP and N-terminal multiplication analogs. The higher affinity of these structural analogs could have potential therapeutic value for multiple disorders, even though no correlation has been proven to the increase of cAMP production (Dangoor et al., 2006).
Similar to the multiplication modifications performed with the N-terminus of the VIP ligand, tandem extension and additional branching methodologies have been carried out on the C-terminal domain, in an attempt to amplify VPAC1 binding. However, these alterations at the C-terminus showed no significant difference in the activation of the VPAC1 receptor or the cAMP production when compared to unmodified VIP (Dangoor et al., 2007). Nonetheless, manipulations at the C-terminal region of VIP have been shown to affect discrimination between VPAC1 and VPAC2 receptors. While modifications of the N-terminus have shown increased affinity for the VPAC2 receptor, the transglutaminase catalyzed analog VIP-Diaminopropane (VIP-DAP) has exhibited maximal effect and affinity for the VPAC1 receptor in comparison to VIP (Caraglia et al., 2008).
8. PARATHYROID HORMONE (PTH)
8.1. Physiological roles of Parathyroid Hormone
Parathyroid hormone (PTH) is a large, 84-residue glycoprotein hormone secreted from the parathyroid glands. Despite its large size, it has been shown that the biological activity of PTH primarily resides within its first 35 residues. Two other peptides related to PTH have also been identified in humans, parathyroid hormone-related peptide (PTHrP) and the 39-residue tuberoinfundibular peptide (TIP39). PTHrP was first isolated from tumors associated with hypercalcaemia (Mangin et al., 1990), whereas TIP39 was isolated from bovine hypothalamus (Usdin et al., 1999). PTH secretion from the parathyroid glands is controlled by feedback via calcium levels in the blood (Poole and Reeve, 2005). PTH functions to increase the concentration of calcium in the blood by modulating absorption or resorption of calcium from the bones, kidneys, and intestines. At the same time, PTH reduces resorption of phosphate from the kidney and enhances the uptake of phosphate from the intestine and bones, resulting in a small net drop in phosphate serum concentration (Parkinson and Thakker, 1992). Additionally, PTHrP affects a wide variety of organs and has been shown to regulate skeletal, pancreatic, epidermal, and mammary gland differentiation as well as bladder and vascular smooth muscle relaxation. Both PTH and PTHrP primarily interact with PTH1 receptors, while TIP39 interacts with PTH2 receptors.
PTH and related peptides, as well as their receptors, are widely expressed in the CNS (Clemens et al., 2001). PTH is expressed in hypothalamic nuclei with projections to the portal system, implicating a role in the regulation of prolactin secretion (Clemens et al., 2001; Harvey and Hayer, 1993). PTH2 receptor expression, however, is concentrated in limbic, hypothalamic, and sensory areas, particularly hypothalamic paraventricular neurons, nerve terminals in the median eminence, superficial layers of the spinal cord dorsal horn, and the caudal part of the sensory trigeminal nucleus (Weaver et al., 1995). Based upon the specific locations of PTH2 receptors and TIP39 in the CNS as well as recent neurobehavioral studies, it appears likely that one of the important actions of TIP39 is to facilitate the response to painful stimuli (LaBuda and Usdin, 2004). In addition, it has been noted that several neuronal populations in which the PTHrP gene is highly expressed share the features of abundant L-type voltage-sensitive calcium channels (L-VSCCs) as well as excitatory amino acid content and a known sensitivity to excitotoxicity (Weir et al., 1990). It has also been reported that when high levels of PTH are sustained in hippocampal organotypic cultures, a toxic increase in intracellular calcium results. Therefore, in the brain, PTH may induce degeneration due to calcium overload via activation of dihydropyridine-sensitive L-VSCCs (Hirasawa et al., 2000).
8.2. Roles of Parathyroid Hormone in disease/pathophysiology
A variety of disorders, including damage to the parathyroid gland, cancer, chronic renal failure, autoimmune disorders, and inborn errors of metabolism can cause a systematic alteration of PTH levels. These altered levels pose potentially fatal consequences, as low levels of PTH can cause hypocalcemia, muscle spasms known as tetany, and seizures, whereas high levels of PTH can result in lack of energy, memory problems, depression, kidney stones and osteoporosis (Okazaki, 2005).
8.3. Parathyroid Hormone: therapeutic chemical modification
An important clinical application of PTH-related therapy relates to its ability to increase vertebral trabecular bone mineral density in older women with osteoporosis (Chunxiao et al., 2007). Numerous attempts have been made to modify the structure of PTH in order to increase the affinity of this ligand for its receptor, thereby enhancing its therapeutic potential. In terms of hormone modification, various groups have identified segments of the ligand that are essential to successful receptor binding. The binding of PTH to the PTH-1 receptor has been suggested to occur at residue 19 of the hormone (Gensure et al., 2003). In a later study, the assumption developed that subsequent activation of the PTH-1 receptor relied heavily on the N-terminal portion of the parathyroid hormone. Manipulation of the N-terminus via constraint of the di-alkyl and alpha amino-isobutyric acid located at positions 1 and 3 showed an increase in receptor binding. Nevertheless, these modifications could impact side chain effects in an inhibitory manner (Shimizu et al. 2004).
Another method of PTH modification involves fragmentation of the human hormone for altered receptor activation. PTH proteolysis occurs in the liver in vivo and produces carboxyl fragments from the parathyroid hormone. Even though these fragments exist in much higher concentrations compared to intact PTH, these segments do not contain the parts necessary to successfully bind and activate the PTH receptor. However, specific receptors exist for these CPTH segments in OC59 bone cells, suggesting a potential application in the treatment of osteoporosis (Selim et al., 2006). On the other hand, engineered fragmentation has been used to produce a human parathyroid fragment, Pro-Pro-hPTH(1-34), which has been shown to significantly induce calcium increment compared to native PTH. The Pro-Pro-hPTH analog has even been approved by the FDA as a viable treatment for osteoporosis, which acts to increase bone mineral density and prevent fractures (Chunxiao et al. 2007).
While some analogs of PTH bind to the receptor in the native GTP-gamma-S-resistant fashion, the analog PTH(1-34) does not bind through this mechanism. Rather, the analog showed higher affinity for the receptor in its R* (active) confirmation, which does not depend on G-protein coupling, and demonstrated sustained cAMP responses and prolonged calcemic and phosphate responses. Prolonged exposure to the M-PTH analog, a derivative of PTH with several amino acid substitutions, produced increased bone volume and turnover, providing further evidence that a unique receptor conformation establishes stable complexes with PTH analogs for extended signaling and responses in target cells (Okazaki et al., 2008).
9. CALCITONIN FAMILY LIGANDS
9.1. Physiological roles of Calcitonin Family Ligands
The calcitonin family of peptides is comprised of four main members: calcitonin (CT), calcitonin gene-related peptide (CGRP), adrenomedullin, and amylin. These hormones differ in their primary sequences but have analogous secondary structures (i.e., amidated carboxyl termini and a six to seven amino acid ring structure formed by an intramolecular disulphide bond). These peptides are widely distributed in various peripheral tissues as well as in the peripheral and central nervous systems.
Calcitonin is a 32-amino acid peptide that is released from thyroid parafollicular C-cells (Huang et al., 2006). It is an endogenous inhibitor of osteoclast development and function and consequently of bone resorption as well (Qin and Yang, 2008). Specific calcitonin receptors are expressed on osteoclasts, and by activating these receptors, osteoclast development and function is inhibited. Alternatively, splicing of the calcitonin gene leads to the production of α-CGRP, a 37-amino acid peptide, especially in nervous tissues (Amara et al., 1982). α-CGRP has been implicated in neuromodulation and in the physiological regulation of blood flow (Zaidi et al, 1990). Adrenomedullin (AM), is a vasoactive peptide of 52 residues that was first isolated from human adrenal pheochromocytomas (Kitamura et al., 1993). AM is synthesized by several mammalian tissues, including endothelial and vascular smooth muscle cells, myocardium, and within the CNS (Beltowski and Jamroz, 2004). AM shares 24% homology with CGRP and carries out similar biological functions (Wimalawansa, 1997). Amylin, another calcitonin gene peptide, shares 46% amino acid sequence homology with CGRP and 20% with human CT. It is a 37-amino acid peptide released from the beta cells of the islets of the pancreas along with insulin. It plays an important role in glucose homeostasis by inhibiting gastric emptying and postprandial hepatic glucose production (Otto-Buczkowska et al., 2008).
Low levels of circulating calcitonin are associated with bone loss and osteoporosis (Zaidi et al., 1990), and therapeutic treatment with calcitonin has proven effective in increasing bone mass and preventing fractures (Siminoski and Josse, 1996). Because of its potent anti-resorptive effects, calcitonin has been strongly implicated as a treatment for disorders such as Paget’s bone disease, osteoporosis, and hypercalcaemia (Huang et al., 2006). Interestingly, emerging evidence suggests that in addition to reducing the occurrence of fractures, calcitonin may also have a direct effect on bone pain. One study examining back pain caused by vertebral collapse found that calcitonin significantly reduced the severity of pain at rest as early as one week into treatment. This effect endured for up to four weeks and was noted in pain scores associated with sitting, standing, and walking (Knopp et al., 2005).
Other members of the calcitonin superfamily also play important roles in a number of diseases and disorders. For instance, CGRP is the most potent vasodilator known in isolated cerebral blood vessels and can induce migraine attacks (Lassen et al., 2008). Similarly, AM is involved in circulatory homeostasis, and studies have found that AM can cause vasodilation and increases in cardiac contractility, cardiac output, diuresis, and natriuresis (Nishikimi and Matsuoka, 2005). Studies also report an increase in AM gene expression and/or immunoreactivity in the ventricles of cardiac hypertrophy and heart failure, as well as increased plasma AM levels in arterial hypertension and acute coronary syndromes (Beltowski and Jamroz, 2004; Brain and Grant, 2004; Kato et al., 2003).
Amylin also appears to play an important pathophysiological role. Normally, amylin synergizes with insulin to regulate plasma glucose concentrations in the bloodstream, suppresses the postprandial secretion of glucagon, and limits the rate of gastric emptying. It has been observed that people with type 2 diabetes, however, have a deficiency in the secretion of amylin that parallels the deficiency in insulin secretion. This causes an excessive inflow of glucose into the bloodstream after eating a meal (Kruger et al., 1999). The cause of this deficiency in amylin secretion is unclear but could be due to abnormal production of the peptide that causes it to form amyloid fibrils. Deposits of amylin have been found in pancreatic islets of 90% of diabetic subjects at postmortem (Clark et al., 1996). Additionally, amylin and the amylin receptor may be directly involved in Alzheimer’s disease. Researchers have demonstrated that antagonists of the human amylin receptor can effectively block the deleterious neuronal effects of amyloid β-peptide, introducing an important and novel therapeutic target for the treatment of AD (Jhamanadas and MacTavish, 2004).
9.2. Calcitonin Family Ligands: therapeutic chemical modification
Initially considerable interest was shown in calcitonin and CGRP as therapeutic possibilities, however in recent years the less appreciated potential of amylin and adrenomedullin has risen in prominence. The hypotensive peptide, adrenomedullin, exists in multiple organ systems throughout the body and exhibits vasodilator activity in a number of species. The replacement in the peptide of the cysteine-14 residue with mercaptoproprionic acid produces a ring-structure without a free N-terminus known as Mpr14-rADM. This analog shows more potent activity at the receptor compared to the native adrenomedullin peptide. In addition, Mpr14-rADM exhibits dose-related decreases in the hind limb perfusion pressure of the cat with controlled blood flow and greater recovery half-times for all doses. Unlike adrenomedullin, the effects of the ring-structure analog were not altered by the antagonist CGRP, thus indicating that Mpr14-rADM acts independently from CGRP, and contributes to a longer-lasting and more effective activation of vasodilatation (Champion et al., 1996).
In a similar manner to insulin, amylin is attenuated or absent in diabetic patients (Schmitz et al., 2004). Recent treatments for diabetes have focused on regulating glucose homeostasis within the body rather than using exogenous agents to enhance insulin secretion. Unfortunately, dosing with exogenous amylin results in aggregation of the peptide into unusable amyloid fibers within the body. However, a stable analog of the peptide has been developed, known as pramlintide, which contains most of the same properties as native amylin but without being susceptible to aggregation (Schmitz et al., 2004). Pramlintide works to regulate glucose in the bloodstream by slowing gastric emptying, inducing proper secretion of glucagon after food intake, and increasing satiety. Pramlintide has been able to improve postprandial glucose levels and glycosylated hemoglobin levels while reducing the amount of insulin needed in the body. Thus, this synthetic analog of amylin could potentially be a beneficial treatment for patients suffering from diabetes (Edelman, 2008).
9.3. Pharmacotherapeutic modifications of calcitonin
Calcitonin’s ability to regulate calcium homeostasis has heightened the interest in it as a potential treatment for bone disorders. The amino acid sequence of calcitonin differs across species, and it has been shown that salmon calcitonin interacts more strongly with phospholipids than human calcitonin due to its amphipathic helix. Thus, combining the ideal helix of salmon calcitonin with the remainder of the human peptide has the potential to produce an analog with more biological potency than the native human hormone. Several analogs have been manufactured using the amphipathic helical region of the salmon calcitonin and the carboxyl and amino termini of the human calcitonin (Epand et al., 2004). It has been shown that by replacing leucine with glycine at position 19 within the peptide, which creates a carboxyl terminus more similar to salmon calcitonin, there is significantly reduced hypocalcemic activity. Among all the manufactured analogs, the change in the thermodynamic properties, as well as secondary structure, proves to enhance biological potency when compared to the calcitonin human hormone. Additionally, further research has been performed to produce analogs of salmon calcitonin alone (Cheng and Lim, 2009). One example, Mal-sCT, demonstrates comparable hypocalcemic activity to native sCT in vivo. Analogs of this peptide derivative, known as PEGylated Mal-sCT, showed robust helical conformation and self-aggregating properties in aqueous solutions. In addition, the analogs 1 PEGylated Mal-sCT and 2 PEGylated Mal-sCT showed more stability in intestinal fluids than sCT or Mal-sCT. Even though these lipidized analogs showed greater stability against enzyme degradation, the hypocalcemic activity remained consistent with sCT and Mal-sCT in both oral and injection administration trials.
9.4. Pharmacotherapeutic modifications of CGRP
It has been demonstrated that perhaps multiple stable isoforms of GPCRs can exist that mediate specific signaling cascade events (Maudsley et al., 2005). For example in MG63 osteoblastic cells the CGRP agonist analog [Cys(Acm)2,7]CGRPR induced ERK dephosphorylation at a similar maximal level to native CGRP but showed antagonistic actions with respect to CGRP-induced cAMP production and p38 modulation. On the other hand, CGRP8-37 had antagonistic properties in regards to cAMP production and CREB and p38 MAPK phosphorylation with a more minor effect on ERK dephosphorylation. The agonist and antagonist properties support the theory that these MG63 osteoblastic cells possess two distinct receptor subtypes that have the same affinity for native CGRP but with greatly differing affinities for xenobiotic CGRP analogs (Kawase et al., 2005). Similar to calcitonin, CGRP comparative physiology has also yielded benefits with respect to altering pharmacological activity. In a recent study, comparisons were made between CGRP present in the skin of the frog species Phyllomedusa bicolor (pbCGRP) and humans using multiple cell types. In terms of the binding affinity to C-terminus, fragments of the frog CGRP (8-37 and 27-37) were much more potent than the human fragments (8-37 and 27-37). In addition, the antagonistic properties of frog CGRPs were much greater than human CGRPs at the CCRP-1 receptor. Thus, pbCGRP8-37 of Phyllomedusa bicolor currently exists as the most potent CGRP-1 competitive antagonist of all reported species in nature (Ladram et al., 2008).
10. GASTRIC INHIBITORY POLYPEPTIDE (GIP)
10.1. Physiological roles of Gastric Inhibitory Polypeptide
The incretin gastric inhibitory polypeptide (GIP), also known as glucose-dependent insulinotropic polypeptide, is a physiological gut peptide that mediates insulin-releasing and extra-pancreatic glucoregulatory actions (Green and Flatt, 2007). Secreted from the K cells of the upper small intestine (Gautier et al., 2008), GIP stimulates glucose-dependent insulin secretion and enhances pancreatic beta-cell mass through regulation of beta-cell proliferation, neogenesis, and apoptosis (Hansotia and Drucker, 2005). The effect is similar to that seen with GLP-1 secretion; however, unlike GLP-1, GIP does not inhibit gastric emptying, glucagon secretion, or food intake and is much less potent than GLP-1. Despite the difference in their potencies, GIP and GLP-1 seem to contribute nearly equally to the incretin effect measured in healthy humans. This is accounted for by differences in secretion concentration of the two peptides (Vilsbøll et al., 2003a). GIP may also play a role in bone formation, as GIP receptors have been found in both osteoblasts and osteoclasts (Xie et al., 2007). This peptide has been shown to increase collagen type I synthesis and osteoblast activity, as well as inhibit osteoclastic activity and differentiation. Moreover, GIP receptor knockout mice exhibit a low bone mass phenotype, whereas GIP transgenic mice have significant increases in bone mass (Xie et al., 2005). Together, these data suggest that GIP may be able to inhibit bone resorption and stimulate bone formation.
Recently, evidence has emerged to suggest that GIP may also have important effects in the CNS. The GIP receptor is widely expressed in the rat brain stem, telencephalon, diencephalon, olfactory bulb, pituitary, and cerebellum (Nyberg et al., 2005). Using immunohistochemical techniques, GIP-producing cells were localized to the olfactory bulb, hippocampus, and Purkinje cells in the cerebellum, while a moderate but distinct GIP immunoreactivity was observed in the cerebral cortex, amygdala, substantia nigra, and lateral septal nucleus (Nyberg et al., 2007). Moreover, a study examining a GIP overexpressing mouse model found that, compared to age-matched controls, GIP transgenic mice displayed enhanced exploratory behavior as well as increased motor performance (Ding et al., 2006).
10.2. Pathophysiological actions of Gastric Inhibitory Polypeptide
In addition to its roles in bone formation and within the CNS, alterations in GIP secretion have been associated primarily with type 2 diabetes. Studies have shown that although the effect of GLP-1 is preserved in the typical middle-aged, obese, insulin-resistant type 2 diabetic patient, there is defective amplification of the late-phase plasma insulin response to glucose stimulated by GIP (Vilsbøll el al., 2003b). Interestingly, there has been some evidence suggesting the use of GIP as a diagnostic tool in the identification of patients in a pre-diabetic state. For instance, polycystic ovary syndrome (PCOS) has been linked to a high risk of type 2 diabetes mellitus. One study found that increased total GIP concentrations during oral glucose tolerance test characterized PCOS women with higher C-peptide secretion, a peptide associated with insulin release, in comparison with healthy controls, and may be an early marker of a pre-diabetic state (Vrbikova et al., 2008). Thus, due to the wide distribution of GIP and its receptor in the body, this peptide could serve as a potential target in the treatment of several metabolic disorders.
10.3. Gastric Inhibitory Polypeptide: therapeutic chemical modification for degradative protection
Due to the widespread effects that GIP has on muscle, adipose tissue, liver tissue, and the minimalistic risk of hypoglycemia, GIP has become a prime candidate for the treatment of diabetes. However, GIP is rapidly degraded in circulation, which proves to be a significant hindrance towards its use as a therapeutic agent (Irwin et al., 2005). DPP IV metabolizes GIP into an N-terminal dipeptide and a degradation fragment GIP (3-42). Not only does the N-terminal fragment have no biological activity, but it may even serve as an antagonist to the GIP receptor. Thus, developing a DPP IV resistant analog of GIP could potentially prolong the half-life of the peptide and prevent antagonism of the GIP receptor in order to produce an effective therapeutic drug (Irwin et al., 2005). In addition to the prevention of degradation, manipulation of the peptide through NH2 and fatty acid modifications creates analogs with the ability to act as antagonists that decrease plasma glucose levels, increase glucose tolerance and insulin sensitivity, and decrease pancreatic insulin release. With these effects, the modified analogs could be used to prevent the onset of diabetes stemming from obesity (O’Hartem et al., 2007).
More recently, it has been shown that manipulation of GIP through C-terminal mini-PEGylation (GIP [mPEG]) increases the biological potency of this peptide. In addition, this analog had increased circulation time due to its improved resistance to degradation by DPP-IV. Similar to the native GIP peptide, the C-terminally altered analog still stimulated cAMP production and insulin secretion in pancreatic cells. When this analog was injected in conjunction with glucose, the plasma glucose level decreased, while the insulin response increased. However, the sensitivity to insulin, pancreatic insulin content, and adiponectin levels remained the same. Modification of the C-terminus of GIP appears to produce a peptide derivative with therapeutic potential for patients with type 2 diabetes due to its prolonged metabolic stability and slightly improved biological action, compared to the native ligand (Gault et al., 2008).
11. CONCLUSION
While extensive pharmacological attention has been paid over the last thirty years to the targeting of rhodopsin-like GPCRs, Class II secretin-like receptor research has been relatively overlooked. Initial concerns over the physiological importance and drugability of these receptors, connected to a lesser understanding of their molecular activity (compared to rhodopsin-like receptors), have in recent times been superseded. Multiple lines of research have shown that Class II receptor systems are equally as important as rhodopsin-like systems and through careful ligand modification, present themselves as viable pharmacological targets.
The modifications applied to these Class II G-protein coupled receptor ligands have proven to promote potential therapeutic effects for numerous neurological and physiological diseases. The majority of these peptide analogs are created by substitutions within the amino acid sequence of the native ligands. Additionally, the metabolic stability of native ligands, such as GLP-1, is improved upon fatty acid acylation and can overcome degradation by the enzyme DPP IV, whereas native PACAP avoids enzyme degradation by modifying its N-terminus. Nevertheless, Class II GPCR ligand analogs that are created by substitution and acylation modifications appear to have enhanced bioavailability and stability (see Table 1 and Figure 2 for summaries).
Table 1. Modification strategies used to develop efficacious analogs of G-protein coupled receptor (GPCR) Class II ligands.
| Modification strategy | Result of modification | Analog examples |
|---|---|---|
| Fatty acid acylation | Increased metabolic stability; enhanced biological activity | Liraglutidea; PEGylated GLP-1b; GIP [mPEG]c |
| Amino acid substitution | Increased metabolic stability; improvement in bioavailability; enhanced biological activity (agonistic analogs) | I-SAd; Mpr14-rADMe |
| N-terminal modifications | Increased metabolic stability; enhanced biological activity | [dAla2]GLP-1f |
| Hybridization (chimera compounds) | Maintained or slightly enhanced biological activity | Glucagon(1-5)GLP-1f; sCT(9-23)-hCT(1-8,24-31)g |
| Fragmentation | Enhanced biological activity | Pro-Pro-hPTH(1-34)h |
Several modification strategies have been employed in order to develop analogs of ligands that bind to Class II GPCRs. These derivatives are designed with the main goals of increasing metabolic stability, improving bioavailability, and enhancing biological activity of their corresponding native peptides. Some examples of these analogs are listed above. Abbreviations used: GLP-1, glucagon-like peptide-1; mPEG, mono-poly(ethylene glycol); GIP, glucose-dependent insulinotropic polypeptide; I-SA, 131I radioactively-labeled, tyrosine-substituted secretin analog; Mpr14-rADM, mercaptoproprionic acid(14)-substituted analog of rat adrenomedullin; dAla2, D-alanine(2); sCT, salmon calcitonin; hCT, human calcitonin; Pro-pro-hPTH(1-34), proline-proline-human parathyroid hormone(1-34). Subscripted numbers in parentheses indicate the positions of amino acid residues of interest within the native peptide.
Figure 2. Chemical modifications of Class II GPCR ligands.

The primary chemical modifications applied to Class II GPCR ligands and subsequent analogs are illustrated. A) Amino acid substitution of GHRH-(1-29)-NH2 within the peptide produces the tetrasubstituted analog CJC-1295 (Teichman et al., 2006). Two amino acid substitutions within the primary sequence of CRH produce the derivative [D-Phe12, Aib15]CRH (Spyroulias et al., 2002). B) Fatty acid acylation of GLP-1 at Lys26 yields the analog liraglutide (Feinglos et al., 2005). C) N-terminal modification of GLP-1-(1-18) at Ala2 produces the analog [D-Ala2]GLP-1-(1-18) (Xiao et al., 2001). D) Chimera formation of the CT derivative sCT-(9-23)-hCT-(1-8, 24-31) occurs by joining segments of hCT and sCT (Epand et al., 2004).
Many of these peptide derivatives have been used as therapeutic agents in attempts to treat disorders related to neurodegeneration, bone metabolism, and energy homeostasis of the body. Analogs of ligands such as secretin, CRH, GIP, GHRH, and PACAP are all created primarily by modifying the native ligand. Due to its ability to cross the blood-brain barrier (BBB), the 131I radioactively-labeled secretin analog (I-SA) has significantly improved bioavailability compared to the native peptide. Furthermore, secretin has been implicated as a potential treatment for autistic children. However, studies are inconclusive with regards to the peptide’s actual therapeutic effect. Perhaps in the future, I-SA and other prospective secretin analogs will deem more successful in treating autism in children. On the same token, the ability of CRH analogs to effectively bind to receptors allows for possible treatment of certain inflammatory diseases, anxiety, Cushing’s disease, and even depression. Similarly, GIP analogs exhibit a prolonged half-life in vivo compared to the unmodified peptide and also resist enzyme degradation to a greater degree. These characteristics make the peptide an excellent treatment choice for patients suffering from type 2 diabetes. The substitution modifications made to GHRH and PACAP both focus on avoiding rapid enzyme degradation, and subsequently, GHRH analogs provide aid in the prevention and treatment of tumors (in terms of cancer) while PACAP analogs show promise as treatments for type 2 diabetes and Parkinson’s disease.
While we have focused on the chemical modification of the native peptide ligands for Class II GPCRs, it is also worthwhile to acknowledge the recent developments in ‘small molecule’ therapeutic agents targeted to these receptors. From a clinical and therapeutic aspect, one of the first receptors to be targeted has been the GLP-1 receptor. For example T-0632, a non-peptide modulator of the GLP-1 receptor, binds with micromolar affinity to the human GLP-1R and blocks GLP-1-induced cAMP production (Tibaduiza et al., 2001). Knudsen and colleagues (2007) have also described several small, ‘ago-allosteric’ modulators of the GLP-1 receptor which behave as both allosteric activators of the receptor as well as independent agonists. In addition to GLP-1, small molecule agents have been targeted to the CRH receptor (Chen et al., 1996) and also to CGRP receptors (Aiyar et al., 2001). These small molecule agents are likely to possess an impact upon Class II receptor clinical biology both by themselves and also potentially in conjunction with modified peptide analogs. As with the peptide ligand analogs, a better understanding of how these small, non-peptide molecules interact with receptors may lead to the development of future Class II receptor-modulating agents with increased efficacy and minimized side-effects.
Although there has been great progress made in the development of potential pharmaceutical agents by modifying these large peptide ligands, further work is needed to elucidate the eventual clinical effects of these peptide analogs in terms of disease progression and treatment. Furthermore, a cautious approach towards the use of such pharmacological compounds is warranted as it is important to acknowledge the potential for adverse side effects of these pluripotent agents. For example, recent clinical use of exenatide (Byetta®) the exendin-4 analog of GLP-1 prescribed as a therapy for type 2 diabetes has been tenuously associated with the development of acute pancreatitis (Denker and Dimarco, 2006). Due to the relative dearth of epidemiological data in this matter a strong proven correlation between exenatide and pancreatitis may take several years to realize (Wajcberg and Tavaria, 2009). Nevertheless, with further careful research into more biologically stable and perhaps receptor-isoform selective analogs, a Class II ligand pharmacopeia may one day rival that generated for the Class I rhodopsin-like receptors.
Acknowledgements
This work was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging.
Abbreviations used
- GPCR
G protein-coupled receptor
- GHRH
growth hormone-releasing hormone
- CRH
corticotropin-releasing hormone
- D-Phe12
D-phenylalanine 12
- Aib15
α-aminoisobutyric acid 15
- GLP-1
glucagon-like peptide-1
- Lys26
lysine 26
- Ala2
alanine 2
- CT
calcitonin
- sCT
salmon calcitonin
- hCT
human calcitonin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaboe K, Krarup T, Madsbad S, Holst J. GLP-1: physiological effects and potential therapeutic applications. Diabetes Obes Metab. 2008;10:994–1003. doi: 10.1111/j.1463-1326.2008.00853.x. [DOI] [PubMed] [Google Scholar]
- Aiyar N, Daines RA, Disa J, Chambers PA, Sauermelch CF, Quiniou M, et al. Pharmacology of SB-273779, a nonpeptide calcitonin gene-related peptide 1 receptor antagonist. J Pharmacol Exp Ther. 2001;296:768–775. [PubMed] [Google Scholar]
- Alba M, Schally A, Salvatori R. Partial reversibility of growth hormone (GH) deficiency in the GH-releasing hormone (GHRH) knockout mouse by postnatal treatment with a GHRH analog. Endocrinology. 2005;146:1506–1513. doi: 10.1210/en.2004-1044. [DOI] [PubMed] [Google Scholar]
- Amara S, Jonas V, Rosenfeld M. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 1982;298:240–244. doi: 10.1038/298240a0. [DOI] [PubMed] [Google Scholar]
- Arborelius L, Owens M, Plotsky P, Nemeroff C. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Aton S, Colwell C, Harmar A, Waschek J, Herzog E. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci. 2005;8:476–483. doi: 10.1038/nn1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks W, Goulet M, Rusche J, Niehoff M, Boismenu R. Differential transport of a secretin analog across the blood-brain and blood-cerebrospinal fluid barriers of the mouse. J Pharmacol Exp Ther. 2002;302:1062–1069. doi: 10.1124/jpet.102.036129. [DOI] [PubMed] [Google Scholar]
- Baum J, Simons B, Jr., Unger R, Madison L. Localization of glucagon in the alpha cells in the pancreatic islet by immunofluorescent technics. Diabetes. 1962;11:371–374. [PubMed] [Google Scholar]
- Beltowski J, Jamroz A. Adrenomedullin—what do we know 10 years since its discovery? Pol J Pharmacol. 2004;56:5–27. [PubMed] [Google Scholar]
- Bensal P, Wang Q. Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab. 2008;295:E751–E761. doi: 10.1152/ajpendo.90295.2008. [DOI] [PubMed] [Google Scholar]
- Bissette G, Reynolds G, Kilts C, Widerlöv E, Nemeroff C. Corticotropin-releasing factor-like immunoreactivity in senile dementia of the Alzheimer type. Reduced cortical and striatal concentrations. JAMA. 1985;254:3067–3069. [PubMed] [Google Scholar]
- Biswas S, Buteau J, Greene L. Glucagon-like peptide-1 (GLP-1) diminishes neuronal degeneration and death caused by NGF deprivation by suppressing Bim induction. Neurochem Res. 2008;33:1845–1851. doi: 10.1007/s11064-008-9646-4. [DOI] [PubMed] [Google Scholar]
- Bourgault S, Vaudry D, Botia B, Couvineau A, Laburthe M, Vaudry H, et al. Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides. 2008;29:919–932. doi: 10.1016/j.peptides.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Brain S, Grant A. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- Brenneman D, Eiden L. Vasoactive intestinal peptide and electrical activity influence neuronal survival. Proc Natl Acad Sci U S A. 1986;83:1159–1162. doi: 10.1073/pnas.83.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T, Colwell C, Waschek J, Piggins H. Disrupted neuronal activity rhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficient mice. J Neurophysiol. 2007;97:2553–2558. doi: 10.1152/jn.01206.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcelin R, Cani P, Knauf C. Glucagon-like peptide-1 and energy homeostasis. J Nutr. 2007;137:2534S–2538S. doi: 10.1093/jn/137.11.2534S. [DOI] [PubMed] [Google Scholar]
- Cao J, Papadopoulou N, Kempuraj D, Boucher WS, Sugimoto K, Cetrulo CL, et al. Human mast cells express corticotropin-releasing hormone (CRH) receptors and CRH leads to selective secretion of vascular endothelial growth factor. J Immunol. 2005;174:7665–7675. doi: 10.4049/jimmunol.174.12.7665. [DOI] [PubMed] [Google Scholar]
- Caraglia M, Carteni M, Dicitore A, Cassesse D, De Maria S, Ferranti P, et al. Experimental study on vasoactive intestinal peptide (VIP) and its diaminopropane bound (VIP-DAP) analog in solution. Amino Acids. 2008;35:275–281. doi: 10.1007/s00726-007-0567-3. [DOI] [PubMed] [Google Scholar]
- Cavallaro S, Copani A, D’Agata V, Musco S, Petralia S, Ventra C, et al. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol Pharmacol. 1996;50:60–66. [PubMed] [Google Scholar]
- Champion H, Duperier C, Fitzgerald W, Lambert D, Murphy W, Coy D, et al. [MPR14]-rADM (14-50), A novel analog of adrenomedullin possesses potent vasodilator activity in the hindlimb vascular bed of the cat. Life Sciences. 1996;59:1–7. doi: 10.1016/0024-3205(96)00258-5. PL. [DOI] [PubMed] [Google Scholar]
- Chan E-C, Falconer J, Madsen G, Rice KC, Webster EL, Chrousos GP, et al. A corticotrophin-releasing hormone Type I receptor antagonist delays parturition in sheep. Endocrinology. 1998;139:3357–3360. doi: 10.1210/endo.139.7.6189. [DOI] [PubMed] [Google Scholar]
- Chen C, Dagnino R, Desouza EB, Grigoriadis DE, Huang CQ, Kim KI, et al. Design and synthesis of a series of non-peptide high-affinity human corticotropin-releasing factor(1) receptor antagonists. J Med Chem. 1996;39:4358–4360. doi: 10.1021/jm960149e. [DOI] [PubMed] [Google Scholar]
- Chen R, Lewis K, Perrin M, Vale W. Expression and cloning of a human corticotropin-releasing factor receptor. Proc Natl Acad Sci U S A. 1993;90:8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Lim L. Synthesis, characterization and in vivo activity of salmon calcitonin coconjugated with lipid and polyethylene glycol. J of Pharm Sci. 2009;98:1438–1451. doi: 10.1002/jps.21524. [DOI] [PubMed] [Google Scholar]
- Chunxiao W, Jingjing L, Yire X, Min D, Zhaohui W, Gaofu Q, Xiangchun S, et al. Study on preparation and activity of a novel recombinant human parathyroid hormone(1-34) analog with N-terminal Pro-Pro extension. Regul Pept. 2007;141:35–43. doi: 10.1016/j.regpep.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Clark A, Chargé S, Badman M, de Koning E. Islet amyloid in type 2 (non-insulin-dependent) diabetes. APMIS. 1996;104:12–18. doi: 10.1111/j.1699-0463.1996.tb00680.x. [DOI] [PubMed] [Google Scholar]
- Clemens T, Cormier S, Eichinger A, Endlich K, Fiaschi-Taesch N, Fischer E, et al. Parathyroid hormone-related protein and its receptors: nuclear functions and roles in the renal and cardiovascular systems, the placental trophoblasts and the pancreatic islets. Br J Pharmacol. 2001;134:1113–1136. doi: 10.1038/sj.bjp.0704378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangoor D, Rubinraut S, Fridkin M, Gozes I. Novel extended and branched N-terminal analogs of VIP. Regul Pept. 2006;137:42–49. doi: 10.1016/j.regpep.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Dangoor D, Rubinraut S, Fridkin M, Gozes I. Novel analogs of VIP with multiple C-terminal domains. Peptides. 2007;28:1622–1630. doi: 10.1016/j.peptides.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Dangoor D, Biondi B, Gobbo M, Vachutinski Y, Fridkin M, Gozes I, et al. Novel glycosylated VIP analogs: synthesis, biological activity, and metabolic stability. Journal of Peptide Science. 2008;14:321–328. doi: 10.1002/psc.932. [DOI] [PubMed] [Google Scholar]
- Delgado M, Reduta A, Sharma V, Ganea D. VIP/PACAP oppositely affects immature and mature dendritic cell expression of CD80/CD86 and the stimulatory activity for CD4(+) T cells. J Leukoc Biol. 2004;75:1122–1130. doi: 10.1189/jlb.1203626. [DOI] [PubMed] [Google Scholar]
- Delgado M, Varela N, Gonzalez-Rey E. Vasoactive intestinal peptide protects against beta-amyloid-induced neurodegeneration by inhibiting microglia activation at multiple levels. Glia. 2008;56:1091–1103. doi: 10.1002/glia.20681. [DOI] [PubMed] [Google Scholar]
- Denker PS, Dimarco PE. Exenatide (exendin-4)-induced pancreatitis. A case report. Diabetes Care. 2006;29:471. doi: 10.2337/diacare.29.02.06.dc05-2043. [DOI] [PubMed] [Google Scholar]
- Ding K, Zhong Q, Xie D, Chen H, Della-Fera M, Bollag R, et al. Effects of glucose-dependent insulinotropic peptide on behavior. Peptides. 2006;27:2750–2755. doi: 10.1016/j.peptides.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Doga M, Bonadonna S, Gola M, Mazziotti G, Giustina A. Growth hormone deficiency in the adult. Pituitary. 2006;9:305–311. doi: 10.1007/s11102-006-0410-y. [DOI] [PubMed] [Google Scholar]
- Drucker D. Glucagon-like peptides. Diabetes. 1998;47:159–169. doi: 10.2337/diab.47.2.159. [DOI] [PubMed] [Google Scholar]
- Dunn A, Berridge C. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- Dunphy J, Taylor R, Fuller P. Tissue distribution of rat glucagon receptor and GLP-1 receptor gene expression. Mol Cell Endocrinol. 1998;141:179–186. doi: 10.1016/s0303-7207(98)00096-3. [DOI] [PubMed] [Google Scholar]
- Edelman S. Optimizing diabetes treatment using an amylin analogue. The Diabetes Educator. 2008;34:4S. doi: 10.1177/0145721707313939. [DOI] [PubMed] [Google Scholar]
- Elliott-Hunt C, Kazlauskaite J, Wilde G, Grammatopoulos D, Hillhouse E. Potential signalling pathways underlying corticotrophin-releasing hormone-mediated neuroprotection from excitotoxicity in rat hippocampus. J Neurochem. 2002;80:416–425. doi: 10.1046/j.0022-3042.2001.00712.x. [DOI] [PubMed] [Google Scholar]
- Epand R, Orlowski R, Epand R. The biological potency of a series of analogues of human calcitonin correlates with their interactions with phospholipids. Biopolymers. 2004;76:258–265. doi: 10.1002/bip.20030. [DOI] [PubMed] [Google Scholar]
- Fahrenkrug J. PACAP—a multifaceted neuropeptide. Chronobiol Int. 2006;23:53–61. doi: 10.1080/07420520500464569. [DOI] [PubMed] [Google Scholar]
- Feinglos M, Saad M, Pi-Sunyert F, An B, Santiago O. Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycaemic control and bodyweight in subjects with type 2 diabetes. Diabetic Medicine. 2005;22:1016–1023. doi: 10.1111/j.1464-5491.2005.01567.x. [DOI] [PubMed] [Google Scholar]
- Felley C, Qian J, Mantey S, Pradhan T, Jensen R. Chief cells possess a receptor with high affinity for PACAP and VIP that stimulates pepsinogen release. Am J Physiol. 1992;263:G901–G907. doi: 10.1152/ajpgi.1992.263.6.G901. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr., Jensen R, Charlton C, Miller R, O’Donohue T, Moody T. Secretin: specific binding to rat brain membranes. J Neurosci. 1983;3:1620–1625. doi: 10.1523/JNEUROSCI.03-08-01620.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallai V, Sarchielli P, Floridi A, Franceschini M, Codini M, Glioti G, et al. Vasoactive peptides levels in the plasma of young migraine patients with and without aura assessed both interictally and ictally. Cephalalgia. 1995;15:384–390. doi: 10.1046/j.1468-2982.1995.1505384.x. [DOI] [PubMed] [Google Scholar]
- Gault V, Kerr B, Irwin N, Flatt P. C-terminal mini-PEGylation of glucose-dependent insulinotropic polypeptide exhibits metabolic stability and improved glucose homeostasis in dietary-induced diabetes. Biochem Pharmacol. 2008;75:2325–2333. doi: 10.1016/j.bcp.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Gautier J, Choukem S, Girard J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 2008;34(Suppl 2):S65–S72. doi: 10.1016/S1262-3636(08)73397-4. [DOI] [PubMed] [Google Scholar]
- Gensure R, Shimizu N, Tsang J, Gardella T. Identification of a Contact Site for Residue 19 of Parathyroid Hormone (PTH) and PTH-Related Protein Analogs in Transmembrane Domain Two of the Type 1 PTH Receptor. Mol Endocrinol. 2003;17:2647–2658. doi: 10.1210/me.2003-0275. [DOI] [PubMed] [Google Scholar]
- Gómez J. Growth hormone and insulin-like growth factor-I as an endocrine axis in Alzheimer’s disease. Endocr Metab Immune Disord Drug Targets. 2008;8:143–151. doi: 10.2174/187153008784534367. [DOI] [PubMed] [Google Scholar]
- Gonzalez B, Basille M, Vaudry D, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide promotes cell survival and neurite outgrowth in rat cerebellar neuroblasts. Neuroscience. 1997;78:419–430. doi: 10.1016/s0306-4522(96)00617-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Varela N, Chorny A, Delgado M. Therapeutic approaches of vasoactive intestinal peptide as a pleiotropic immunomodulator. Curr Pharm Des. 2007;13:1113–1139. doi: 10.2174/138161207780618966. [DOI] [PubMed] [Google Scholar]
- Goodyer C, de Stéphano L, Lai W, Guyda H, Posner B. Characterization of insulin-like growth factor receptors in rat anterior pituitary, hypothalamus, and brain. Endocrinology. 1984;114:1187–1195. doi: 10.1210/endo-114-4-1187. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos D, Chrousos G. Functional characteristics of CRH receptors and potential clinical application of CRH-receptor antagonists. Trends Endocrinol Metab. 2002;13:436–444. doi: 10.1016/s1043-2760(02)00670-7. [DOI] [PubMed] [Google Scholar]
- Gray SL, Yamaguchi N, Vencova P, Sherwood NM. Temperature sensitive phenotype in mice lacking pituitary adenylate cyclase activating polypeptide. Endocrinology. 2002;143:3946–3954. doi: 10.1210/en.2002-220401. [DOI] [PubMed] [Google Scholar]
- Green B, Flatt P. Incretin hormone mimetics and analogues in diabetes. Best Pract Res Clin Endocrinol Metab. 2007;21:497–516. doi: 10.1016/j.beem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Grinevich V, Fournier A, Pelletier G. Effects of pituitary adenylate cyclase-activating polypeptide (PACAP) on corticotrophin-releasing hormone (CRH) gene expression in the rat hypothalamic paraventricular nucleus. Brain Res. 1997;773:190–196. doi: 10.1016/s0006-8993(97)01011-1. [DOI] [PubMed] [Google Scholar]
- Hansotia T, Drucker D. GIP and GLP-1 as incretin hormones: lessons from single and double incretin receptor knockout mice. Regul Pept. 2005;128:125–134. doi: 10.1016/j.regpep.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Hanstein R, Lu A, Wurst W, Holsboer F, Deussing JM, Clement AB, et al. Transgenic overexpression of corticotropin releasing hormone provides partial protection against neurodegeneration in an in vivo model of acute excitotoxic stress. Neuroscience. 2008;156:712–721. doi: 10.1016/j.neuroscience.2008.07.034. [DOI] [PubMed] [Google Scholar]
- Harmar A, Marston H, Shen S, Spratt C, West K, Sheward W, et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- Harvey S, Hayer S. Parathyroid hormone binding sites in the brain. Peptides. 1993;14:1187–1191. doi: 10.1016/0196-9781(93)90174-f. [DOI] [PubMed] [Google Scholar]
- Hashimoto H. Physiological significance of pituitary adenylate cyclase-activating polypeptide (PACAP) in the nervous system. Yakugaku Zasshi. 2002;122:1109–1121. doi: 10.1248/yakushi.122.1109. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Hashimoto H, Shintani N, Chiba S, Hattori S, Okada T, et al. Pituitary adenylate cyclase-activating polypeptide is associated with schizophrenia. Mol Psychiatry. 2007;12:1026–1032. doi: 10.1038/sj.mp.4001982. [DOI] [PubMed] [Google Scholar]
- Heuser I, Baronti F, Marin C, Ma N, Merriam G, Chase T, et al. Growth hormone secretion in Alzheimer’s disease: 24-hour profile of basal levels and response to stimulation and suppression studies. Neurobiol Aging. 1992;13:255–260. doi: 10.1016/0197-4580(92)90037-x. [DOI] [PubMed] [Google Scholar]
- Hikichi T, Yamamoto Y, Tsutsumi T, Isogawa K, Akiyoshi J. Conditioned fear freezing behavior suppression treated by CRH1 receptor antagonist. Jpn J Psychopharm. 1997;17:284. [Google Scholar]
- Hill JM, Hauser JM, Sheppard LM, Abebe D, Spivak-Pohis I, Kushnir M, et al. Blockade of VIP during mouse embryogenesis modifies adult behavior and results in permanent changes in brain chemistry. J Mol Neurosci. 2007;33:278–283. doi: 10.1385/jmn:31:03:185. [DOI] [PubMed] [Google Scholar]
- Hirasawa T, Nakamura T, Mizushima A, Morita M, Ezawa I, Miyakawa H, et al. Adverse effects of an active fragment of parathyroid hormone on rat hippocampal organotypic cultures. Br J Pharmacol. 2000;129:21–28. doi: 10.1038/sj.bjp.0702949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. 1996;17:187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- Holz G, Chepurny O. Glucagon-like peptide-1 synthetic analogs: new therapeutic agents for use in the treatment of diabetes mellitus. Curr Med Chem. 2003;10:2471–2483. doi: 10.2174/0929867033456648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath K, Stefanatos G, Sokoloski KN, Wachtel R, Nabors L, Tildon JT. Improved social and language skills after secretin administration in patients with autistic spectrum disorders. J Assoc Acad Minor Phys. 1998;9:9–15. [PubMed] [Google Scholar]
- Huang C, Sun L, Moonga B, Zaidi M. Molecular physiology and pharmacology of calcitonin. Cell Mol Biol. 2006;52:33–43. [PubMed] [Google Scholar]
- Ionescu M, Frohlman LA. Pulsatile secretion of growth hormone (GH) persists during continuous stimulation by CJC-1295, a long-acting GH-releasing hormone analog. J Clin Endocrinol Metab. 2006;91:4792–4797. doi: 10.1210/jc.2006-1702. [DOI] [PubMed] [Google Scholar]
- Irwin D. Molecular evolution of proglucagon. Regul Pept. 2001;98:1–12. doi: 10.1016/s0167-0115(00)00232-9. [DOI] [PubMed] [Google Scholar]
- Irwin N, Green B, Mooney M, Greer B, Harriott P, Bailey C, et al. A novel, long-acting agonist of glucose-dependent insulinotropic polypeptide suitable for once-daily administration in type 2 diabetes. J Pharmacol Exp Ther. 2005;314:1187–1194. doi: 10.1124/jpet.105.086082. [DOI] [PubMed] [Google Scholar]
- Isogawa K, Akiyoshi J, Hikichi T, Yamamoto Y, Tsutsumi T, Nagayama H. Effect of corticotrophin releasing factor receptor 1 antagonist on extracellular norepinephrine, dopamine and serotonin in hippocampus and prefrontal cortex of rats in vivo. Neuropeptides. 2000;34:234–239. doi: 10.1054/npep.2000.0806. [DOI] [PubMed] [Google Scholar]
- Izdebski J, Pinski J, Horvath J, Halmos G, Groot K, Schally A. Synthesis and biological evaluation of superactive agonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A. 1995;92:4872–4876. doi: 10.1073/pnas.92.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhamanadas J, MacTavish D. Antagonist of the amylin receptor blocks beta-amyloid toxicity in rat cholinergic basal forebrain neurons. J Neurosci. 2004;24:5579–5584. doi: 10.1523/JNEUROSCI.1051-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastin A, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood brain barrier. J Mol Neurosci. 2002;18:7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- Kato J, Tsuruda T, Kitamura K, Eto T. Adrenomedullin: a possible autocrine or paracrine hormone in the cardiac ventricles. Hypertens Res. 2003;26(Suppl):S113–S119. doi: 10.1291/hypres.26.s113. [DOI] [PubMed] [Google Scholar]
- Kawase T, Okuda K, Burns D. Immature osteoblastic MG63 cells possess two calcitonin gene-related peptide receptor subtypes that respond differently to [Cys(Acm)2,7] calcitonin gene-related peptide and CGRP8-37. Am J Physiol Cell Physiol. 2005;289:811–818. doi: 10.1152/ajpcell.00504.2004. [DOI] [PubMed] [Google Scholar]
- Kern J, Espinoza E, Trivedi M. The effectiveness of secretin in the management of autism. Expert Opin Pharmacother. 2004;5:379–387. doi: 10.1517/14656566.5.2.379. [DOI] [PubMed] [Google Scholar]
- Kiaris H, Schally A, Varga J. Antagonists of growth hormone-releasing hormone inhibit the growth of U-87MG human glioblastoma in nude mice. Neoplasia. 2000;2:242–250. doi: 10.1038/sj.neo.7900074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kineman R. Antitumorigenic actions of growth hormone-releasing hormone antagonists. Proc Natl Acad Sci U S A. 2000;97:532–534. doi: 10.1073/pnas.97.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun. 1993;192:553–560. doi: 10.1006/bbrc.1993.1451. [DOI] [PubMed] [Google Scholar]
- Knopp J, Diner B, Blitz M, Lyritis G, Rowe B. Calcitonin for treating acute pain of osteoporotic vertebral compression fractures: a systematic review of randomized, controlled trials. Osteoporos Int. 2005;16:1281–1290. doi: 10.1007/s00198-004-1798-8. [DOI] [PubMed] [Google Scholar]
- Knudsen L, Nielsen P, Huusfeldt P, Johansen N, Madsen K, Pedersen F, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, Kodra JT, et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A. 2007;104:937–942. doi: 10.1073/pnas.0605701104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köves K, Arimura A, Somogyvári-Vigh A, Vigh S, Miller J. Immunohistochemical demonstration of a novel hypothalamic peptide, pituitary adenylate cyclase-activating polypeptide, in the ovine hypothalamus. Endocrinology. 1990;127:264–271. doi: 10.1210/endo-127-1-264. [DOI] [PubMed] [Google Scholar]
- Kruger D, Gatcomb P, Owen S. Clinical implications of amylin and amylin deficiency. Diabetes Educ. 1999;25:389–397. doi: 10.1177/014572179902500310. [DOI] [PubMed] [Google Scholar]
- LaBuda C, Usdin T. Tuberoinfundibular peptide of 39 residues decreases pain-related affective behavior. Neuroreport. 2004;15:1779–1782. doi: 10.1097/01.wnr.0000134849.63755.15. [DOI] [PubMed] [Google Scholar]
- Lanfranco F, Gianotti L, Giordano R, Pellegrino M, Maccario M, Arvat E. Ageing, growth hormone and physical performance. J Encrinol Invest. 2003;26:861–872. doi: 10.1007/BF03345237. [DOI] [PubMed] [Google Scholar]
- Ladram A, Besne I, Breton L, Lcharriere O, Nicolas P, Amiche M. Pharmacologic study of C-terminal fragments of frog skin calcitonin gene-related peptide. Peptides. 2008;29:1150–1156. doi: 10.1016/j.peptides.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Lange M, Müller J, Svendsen O, Kastrup K, Juul A, Feldt-Rasmussen U. The impact of idiopathic childhood-onset growth hormone deficiency (GHD) on bone mass in subjects without adult GHD. Clin Endocrinol (Oxf) 2005;62:18–23. doi: 10.1111/j.1365-2265.2004.02164.x. [DOI] [PubMed] [Google Scholar]
- Langer I, Gregoire F, Nachtergaol I, De Neef P, Vertongen P, Robberecht P. Hexanoylation of a VPAC2 receptor-preferring ligand markedly increased its selectivity and potency. Peptides. 2004;25:275–278. doi: 10.1016/j.peptides.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Langlet C, Gaspard N, Nachtergael I, Robberecht P, Langer I. Comparative efficacy of VIP and analogs on activation and internalization of the recombinant VPAC2 receptor expressed in CHO cells. Peptides. 2004;25:2079–2086. doi: 10.1016/j.peptides.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Lassen L, Jacobsen V, Haderslev P, Sperling B, Iversen H, Olesen J, et al. Involvement of calcitonin gene-related peptide in migraine: regional cerebral blood flow and blood flow velocity in migraine patients. J Headache Pain. 2008;9:151–157. doi: 10.1007/s10194-008-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Lee S, Youn Y, Na D, Chae S, Byun Y, et al. Synthesis, characterization, and pharmacokinetic studies of PEGylated glucagon-like peptide-1. Bioconjugate Chem. 2005;16:377–382. doi: 10.1021/bc049735+. [DOI] [PubMed] [Google Scholar]
- Lelievre V, Seksenyan A, Nobuta H, Yong WH, Chhith S, et al. Disruption of the PACAP gene promotes medulloblastoma in ptc1 mutant mice. Dev Biol. 2008;313:359–370. doi: 10.1016/j.ydbio.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch K, Ihl R, Frölich L, Rupprecht R, Müller U, Schulte H, et al. Endocrine responses to growth hormone releasing hormone and corticotrophin releasing hormone in early-onset Alzheimer’s disease. Psychiatry Res. 1990;33:107–112. doi: 10.1016/0165-1781(90)90063-b. [DOI] [PubMed] [Google Scholar]
- Lezoualc’h F, Engert S, Berning B, Behl C. Corticotropin-releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid beta precursor protein and with the suppression of nuclear factor-kappaB. Mol Endocrinol. 2000;14:147–159. doi: 10.1210/mend.14.1.0403. [DOI] [PubMed] [Google Scholar]
- Li S, Grinevich V, Fournier A, Pelletier G. Effects of pituitary adenylate cyclase-activating polypeptide (PACAP) on gonadotropin-releasing hormone and somatostatin gene expression in the rat brain. 1996;41:157–162. doi: 10.1016/0169-328x(96)00086-1. [DOI] [PubMed] [Google Scholar]
- Liaw C, Lovenberg T, Barry G, Oltersdorf T, Grigoriadis D, de Souza E. Cloning and characterization of the human corticotropin-releasing factor-2 receptor complementary deoxyribonucleic acid. Endocrinology. 1996;137:72–77. doi: 10.1210/endo.137.1.8536644. [DOI] [PubMed] [Google Scholar]
- Likar R, Mousa SA, Steinkellner H, Koppert W, Phillippitsch G, Stein C, et al. Involvement of intra-ocular corticotropin-releasing hormone in postoperative pain modulation. Clin J Pain. 2007;23:136–142. doi: 10.1097/01.ajp.0000210954.93878.0d. [DOI] [PubMed] [Google Scholar]
- Loh D, Abad C, Colwell C, Waschek J. Vasoactive intestinal polypeptide is critical for circadian regulation of glucocorticoids. Neuroendocrinology. 2008;88:246–255. doi: 10.1159/000140676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love JW. Peptic ulceration may be a hormonal deficiency disease. Med Hypotheses. 2008;70:1103–1107. doi: 10.1016/j.mehy.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Lund P, Goodman R, Dee P, Habener J. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci USA. 1982;79:345–349. doi: 10.1073/pnas.79.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz-Bucher B, Monnier D, Koch B. Evidence for the presence of receptors for pituitary adenylate cyclase-activating polypeptide in the neurohypophysis that are positively coupled to cyclic AMP formation and neurohypophyseal hormone secretion. Neuroendocrinology. 1996;64:153–161. doi: 10.1159/000127113. [DOI] [PubMed] [Google Scholar]
- Macica CM, Broadus AE. PTHrP regulates cerebral blood flow and is neuroprotective. Am J Physiol Regul Integr Comp Physiol. 2003;284:1019–1020. doi: 10.1152/ajpregu.00001.2003. [DOI] [PubMed] [Google Scholar]
- Malhotra R, Wakade T, Wakade A. Vasoactive intestinal polypeptide and muscarine mobilize intracellular Ca2+ through breakdown of phosphoinositides to induce catecholamine secretion. Role of IP3 in exocytosis. J Biol Chem. 1988;263:2123–2126. [PubMed] [Google Scholar]
- Mangin M, Ikeda K, Broadus A. Structure of the mouse gene encoding parathyroid hormone-related protein. Gene. 1990;95:195–202. doi: 10.1016/0378-1119(90)90362-u. [DOI] [PubMed] [Google Scholar]
- Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolec Med. 2005;7:3–36. doi: 10.1385/nmm:7:1-2:003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Golden E, Keselman A, Stone M, Mattson MP, Egan JM, et al. Therapeutic perspectives for the treatment of Huntington’s disease: treating the whole body. Histol Histopathol. 2008;23:237–250. doi: 10.14670/hh-23.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies and extends survival in a mouse model of Huntington’s disease. Diabetes. 2009;58:318–328. doi: 10.2337/db08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuo Y, Ohtaki T, Masuda Y, Tsuda M, Fujino M. Binding sites for pituitary adenylate cyclase activating polypeptide (PACAP): comparison with vasoactive intestinal polypeptide (VIP) binding site localization in rat brain sections. Brain Res. 1992;575:113–123. doi: 10.1016/0006-8993(92)90430-h. [DOI] [PubMed] [Google Scholar]
- Mattson M, Maudsley S, Martin B. A neural signaling triumvirate that influences ageing and age-related disease: insulin/IGF-1, BDNF and serotonin. Ageing Res Rev. 2004;3:445–464. doi: 10.1016/j.arr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in G protein-coupled receptor signaling. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo K, Vale W, Rivier J, Rosenfeld M, Evans R. Expression-cloning and sequence of a cDNA encoding human growth hormone-releasing factor. Nature. 1983;306:86–88. doi: 10.1038/306086a0. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I. Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides. 1984;5:53–69. doi: 10.1016/0196-9781(84)90265-1. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Vigh S, Schally A, Petrusz P. Immunocytochemical localization of growth hormone-releasing factor in the rat hypothalamus. Endocrinology. 1984;114:1082–1085. doi: 10.1210/endo-114-4-1082. [DOI] [PubMed] [Google Scholar]
- McLean M, Smith R. Corticotropin-releasing hormone and human parturition. Reproduction. 2001;121:493–501. doi: 10.1530/rep.0.1210493. [DOI] [PubMed] [Google Scholar]
- McQueen J, Heck A. Secretin for the treatment of autism. Ann Pharmacother. 2002;36:305–311. doi: 10.1345/aph.19113. [DOI] [PubMed] [Google Scholar]
- Millar RP, Lu ZL, Pawson AJ, Flanaga CA, Morgan K, Maudsley S. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235–275. doi: 10.1210/er.2003-0002. [DOI] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl R, Minamino N, Uehara A, Jiang L, et al. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulate adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164:567–574. doi: 10.1016/0006-291x(89)91757-9. [DOI] [PubMed] [Google Scholar]
- Murase T, Kondo K, Otake K, Oiso Y. Pituitary adenylate cyclase-activating polypeptide stimulates arginine vasopressin release in conscious rats. Neuroendocrinology. 1993;57:1092–1096. doi: 10.1159/000126475. [DOI] [PubMed] [Google Scholar]
- Mutt V. Chemistry, isolation and purification of gastrointestinal hormones. Biochem Soc Trans. 1980;8:11–14. doi: 10.1042/bst0080011. [DOI] [PubMed] [Google Scholar]
- Nemeroff C, Krishnan K, Belkin B, Ritchie J, Clark C, Vale W, et al. Growth hormone response to growth hormone releasing factor in Alzheimer’s disease. Neuroendocrinology. 1989;50:663–666. doi: 10.1159/000125296. [DOI] [PubMed] [Google Scholar]
- Nishijima I, Yamagata T, Spencer C, Weeber E, Alekseyenko O, Sweatt J, et al. Secretin receptor-deficient mice exhibit impaired synaptic plasticity and social behavior. Hum Mol Genet. 2006;15:3241–3250. doi: 10.1093/hmg/ddl402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi T, Matsuoka H. Cardiac adrenomedullin: its role in cardiac hypertrophy and heart failure. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:231–242. doi: 10.2174/1568016054368241. [DOI] [PubMed] [Google Scholar]
- Nyberg J, Anderson M, Meister B, Alborn A, Ström A, Brederlau A, et al. Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J Neurosci. 2005;25:1816–1825. doi: 10.1523/JNEUROSCI.4920-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg J, Jacobsson C, Anderson M, Eriksson P. Immunohistochemical distribution of glucose-dependent insulinotropic polypeptide in the adult rat brain. J Neurosci Res. 2007;85:2099–2119. doi: 10.1002/jnr.21349. [DOI] [PubMed] [Google Scholar]
- Odegard PS, Setter SM, Iltz JL. Update in the pharmacologic treatment of Diabetes Mellitus: focus on pramlintide and exentide. Diabetes Educator. 2006;32:693–705. doi: 10.1177/0145721706294003. [DOI] [PubMed] [Google Scholar]
- Offen D, Sherki Y, Melamed E, Fridkin M, Brenneman D, Gozes I. Vasoactive intestinal peptide (VIP) prevents neurotoxicity in neuronal cultures: relevance to neuroprotection in Parkinson’s disease. Brain Res. 2000;854:257–262. doi: 10.1016/s0006-8993(99)02375-6. [DOI] [PubMed] [Google Scholar]
- O’Hartem F, Hunter K, Gault V, Irwin N, Green B, Greer B, et al. Antagonistic effects of two novel GIP analogs, (Hyp3)GIP and (Hyp3)GIPLys16PAL, on the biological actions of GIP and longer-term effects in diabetic ob/ob mice. Am J Physiol Endocrinol Metab. 2007;292:1674–1682. doi: 10.1152/ajpendo.00391.2006. [DOI] [PubMed] [Google Scholar]
- Okazaki R. Parathyroid disorders and bone metabolism. Clin Calcim. 2005;15:66–72. [PubMed] [Google Scholar]
- Okazaki M, Ferrandon S, Vilardaga J, Bouxsein M, Potts J, Gardella T. Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation. PNAS. 2008;105:16525–16530. doi: 10.1073/pnas.0808750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoue S, Yamada S, Yajima T. Bioactive analogues and drug delivery systems of vasoactive intestinal peptide (VIP) for the treatment of asthma/COPD. Peptides. 2007;28:1640–1650. doi: 10.1016/j.peptides.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Onoue S, Misaka S, Yamada S. Structure-activity relationship of vasoactive intestinal peptide (VIP): potent agonists and potential clinical applications. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:579–590. doi: 10.1007/s00210-007-0232-0. [DOI] [PubMed] [Google Scholar]
- Otto-Buczkowska E, Mazur-Dworzecka U, Dworzecki T. Role of amylin in glucose homeostasis and its perspective use in diabetes management. Przegl Lek. 2008;65:135–139. [PubMed] [Google Scholar]
- Owens M, Nemeroff C. Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- Parkinson D, Thakker R. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet. 1992;1:149–152. doi: 10.1038/ng0592-149. [DOI] [PubMed] [Google Scholar]
- Patel NC, Yeh JY, Shepherd MD, Crismon ML. Secretin treatment for autistic disorder: a critical analysis. Pharmacotherapy. 2002;22:905–914. doi: 10.1592/phco.22.11.905.33622. [DOI] [PubMed] [Google Scholar]
- Pedersen W, McCullers D, Culmsee C, Haughey N, Herman J, Mattson M. Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2001;8:492–503. doi: 10.1006/nbdi.2001.0395. [DOI] [PubMed] [Google Scholar]
- Poole K, Reeve J. Parathyroid hormone - a bone anabolic and catabolic agent. Curr Opin Pharmacol. 2005;5:612–617. doi: 10.1016/j.coph.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Pomara N, Singh R, Deptula D, LeWitt P, Bissette G, Stanley M, et al. CSF corticotropin-releasing factor (CRF) in Alzheimer’s disease: its relationship to severity of dementia and monoamine metabolites. Biol Psychiatry. 1989;26:500–504. doi: 10.1016/0006-3223(89)90071-1. [DOI] [PubMed] [Google Scholar]
- Pratley R, Gilbert M. Targeting incretins in type 2 diabetes: role of GLP-1 receptor agonists and DPP-4 inhibitors. Rev Diabet Stud. 2008;5:73–94. doi: 10.1900/RDS.2008.5.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Yang F. Calcitonin may be a useful therapeutic agent for osteoclastogenesis syndromes involving premature eruption of the tooth. Med Hypotheses. 2008;70:1048–1050. doi: 10.1016/j.mehy.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Reed H, Meyer-Spasche A, Cutler D, Coen C, Piggins H. Vasoactive intestinal polypeptide (VIP) phase-shifts the rat suprachiasmatic nucleus clock in vitro. Eur J Neurosci. 2001;13:839–843. doi: 10.1046/j.0953-816x.2000.01437.x. [DOI] [PubMed] [Google Scholar]
- Reglödi D, Lubics A, Kiss P, Lengvári I, Gaszner B, Tóth G, et al. Effect of PACAP in 6-OHDA-induced injury of the substantia nigra in intact young and ovariectomized female rats. Neuropeptides. 2006;40:265–274. doi: 10.1016/j.npep.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Somogyvari-Vigh A, Szanto Z, Kertes E, Lenard L, et al. Effects of pretreatment with PACAP on the infarct size and functional outcome in rat permanent focal cerebral ischemia. Peptides. 2002;23:2227–2234. doi: 10.1016/s0196-9781(02)00262-0. [DOI] [PubMed] [Google Scholar]
- Reichlin S. Neuroendocrine significance of vasoactive intestinal polypeptide. Ann N Y Acad Sci. 1988;527:431–449. doi: 10.1111/j.1749-6632.1988.tb26998.x. [DOI] [PubMed] [Google Scholar]
- Rekasi Z, Varga J, Schally A, Halmos G, Armatis P, Groot K, et al. Antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide inhibit tumor proliferation by different mechanisms: evidence from in vitro studies on human prostatic and pancreatic cancers. Endocrinology. 2000;141:2120–2128. doi: 10.1210/endo.141.6.7511. [DOI] [PubMed] [Google Scholar]
- Riepl R, Lehnert P. The mediators of bile action on the exocrine pancreas. Scand J Gastroenterol. 1993;28:369–374. doi: 10.3109/00365529309098234. [DOI] [PubMed] [Google Scholar]
- Roe S, McGowan E, Rothwell N. Evidence for the involvement of corticotrophin-releasing hormone in the pathogenesis of traumatic brain injury. Eur J Neurosci. 1998;10:553–559. doi: 10.1046/j.1460-9568.1998.00064.x. [DOI] [PubMed] [Google Scholar]
- Rosenfeld R, Ceda G, Wilson D, Dollar L, Hoffman A. Characterization of high affinity receptors for insulin-like growth factors I and II on rat anterior pituitary cells. Endocrinology. 1984;114:1571–1575. doi: 10.1210/endo-114-5-1571. [DOI] [PubMed] [Google Scholar]
- Sawchenko P, Swanson L, Rivier J, Vale W. The distribution of growth-hormone-releasing factor (GRF) immunoreactivity in the central nervous system of the rat: an immunohistochemical study using antisera directed against rat hypothalamic GRF. J Comp Neurol. 1985;237:100–115. doi: 10.1002/cne.902370108. [DOI] [PubMed] [Google Scholar]
- Schmitz O, Brock B, Rungby J. Amylin agonists: a novel approach in the treatment of diabetes. Diabetes. 2004;53:S233–S238. doi: 10.2337/diabetes.53.suppl_3.s233. [DOI] [PubMed] [Google Scholar]
- Schiöth HB, Fredriksson R. The GRAFS classification system of G protein coupled receptors in comparative perspective. Gen Comp Endocrinol. 2005;142:94–101. doi: 10.1016/j.ygcen.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Seki Y, Suzuki Y, Baskaya M, Kano T, Saito K, Takayasu M, et al. The effects of pituitary adenylate cyclase-activating polypeptide on cerebral arteries and vertebral artery blood flow in anesthetized dogs. Eur J Pharmacol. 1995;275:259–266. doi: 10.1016/0014-2999(95)00011-9. [DOI] [PubMed] [Google Scholar]
- Selim A, Mahon M, Juppner H, Bringhurst F, Divieti P. Role of calcium channels in carboxyl-terminal parathyroid hormone receptor signaling. Am J Physiol Cell Physiol. 2006;291:C114–C121. doi: 10.1152/ajpcell.00566.2005. [DOI] [PubMed] [Google Scholar]
- Severs J, Froland W. Dimerization of PACAP peptide analogue in DMSO via asparagine and aspartic acid residues. J Pharm Sci. 2008;97:1246–1256. doi: 10.1002/jps.21116. [DOI] [PubMed] [Google Scholar]
- Sherwood N, Krueckl S, McRory J. The origin and function of the pituitary adenylate cyclase-activating polypeptide (PACAP)/glucagon superfamily. Endocr Rev. 2000;21:619–670. doi: 10.1210/edrv.21.6.0414. [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Hotta M, Masuda A, Imaki T, Obara N, Hizuka N, et al. Studies on the response of growth hormone (GH) secretion to GH-releasing hormone, thyrotropin-releasing hormone, gonadotropin-releasing hormone, and somatostatin in acromegaly. J Clin Endocrinol Metab. 1986;63:167–173. doi: 10.1210/jcem-63-1-167. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Dean T, Khatri A, Gardella T. Amino-terminal parathyroid hormone fragment analogs containing alpha, alpha-di-alkyl amino acids at positions 1 and 3. J Bone Miner Res. 2004;19:2078–2086. doi: 10.1359/JBMR.040914. [DOI] [PubMed] [Google Scholar]
- Shin YK, Martin B, Golden E, Dotson CD, Maudsley S, Kim W, et al. Modulation of taste sensitivity by GLP-1 signaling. J Neurochem. 2008;106:455–463. doi: 10.1111/j.1471-4159.2008.05397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani N, Mori W, Hashimoto H, Imai M, Tanaka K, Tomimoto S, et al. Defects in reproductive functions in PACAP-deficient female mice. Regul Pept. 2002;109:45–48. doi: 10.1016/s0167-0115(02)00169-6. [DOI] [PubMed] [Google Scholar]
- Siegel EG, Scharf G, Gallwitz B, Mentlein R, Morys-Wortmann C, Folsch UR, et al. Comparison of the effect of native glucagon-like peptide 1 and dipeptidyl peptidase IV-resistant analogues on insulin release from rat pancreatic islets. Eur J Clin Invest. 1999;29:610–614. doi: 10.1046/j.1365-2362.1999.00440.x. [DOI] [PubMed] [Google Scholar]
- Siminoski K, Josse R. Prevention and management of osteoporosis: consensus statements from the Scientific Advisory Board of the Osteoporosis Society of Canada. 9. Calcitonin in the treatment of osteoporosis. CMAJ. 1996;155:962–965. [PMC free article] [PubMed] [Google Scholar]
- Somogyvári-Vigh A, Reglodi D. Pituitary adenylate cyclase activating polypeptide: a potential neuroprotective peptide. Curr Pharm Des. 2004;10:2861–2889. doi: 10.2174/1381612043383548. [DOI] [PubMed] [Google Scholar]
- Sorg O, Magistretti P. Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J Neurosci. 1992;12:4923–4931. doi: 10.1523/JNEUROSCI.12-12-04923.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyroulias G, Papazacharias S, Pairas G, Cordopatis P. Monitoring the structural consequences of Phe12→d-Phe and Leu15→Aib substitution in human/rat corticotropin releasing hormone. Implications for design of CRH antagonists. Eur J Biochem. 2002;269:6009–6019. doi: 10.1046/j.1432-1033.2002.03278.x. [DOI] [PubMed] [Google Scholar]
- Staines DR. Do vasoactive neuropeptide autoimmune disorders explain pyridostigmine’s association with Gulf War syndrome? Med Hypotheses. 2005;65:591–594. doi: 10.1016/j.mehy.2005.02.036. [DOI] [PubMed] [Google Scholar]
- Staines DR. Is Parkinson’s disease an autoimmune disorder of endogenous vasoactive neuropeptides? Med Hypotheses. 2007;69:1208–1211. doi: 10.1016/j.mehy.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Staines DR. Are multiple sclerosis and amyotrophic lateral sclerosis autoimmune disorders of endogenous vasoactive neuropeptides? Med Hypotheses. 2008;70:413–418. doi: 10.1016/j.mehy.2007.04.038. [DOI] [PubMed] [Google Scholar]
- Stinus L, Cador M, Zorrilla EP, Koob GF. Buprenorphine and a CRF1 antagonist block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology. 2005;30:90–98. doi: 10.1038/sj.npp.1300487. [DOI] [PubMed] [Google Scholar]
- Ströhle A, Holsboer F. Stress responsive neurohormones in depression and anxiety. Pharmacopsychiatry. 2003;36:S207–S214. doi: 10.1055/s-2003-45132. [DOI] [PubMed] [Google Scholar]
- Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol Cell Endocrinol. 1994;105:131–137. doi: 10.1016/0303-7207(94)90162-7. [DOI] [PubMed] [Google Scholar]
- Swanson L, Simmons D. Differential steroid hormone and neural influences on peptide mRNA levels in CRH cells of the paraventricular nucleus: a hybridization histochemical study in the rat. J Comp Neurol. 1989;285:413–435. doi: 10.1002/cne.902850402. [DOI] [PubMed] [Google Scholar]
- Teichman S, Neale A, Lawrence B, Gagnon C, Castaigne J-P, Frohman L. Prolonged stimulation of growth hormone (GH) and insulin-like growth factor I secretion by CJC-1295, a long-acting analog of GH-releasing hormone, in healthy adults. J Clin Endocrin and Metab. 2006;91:799–805. doi: 10.1210/jc.2005-1536. [DOI] [PubMed] [Google Scholar]
- Thorner M, Chapman I, Gaylinn B, Pezzoli S, Hartman M. Growth hormone-releasing hormone and growth hormone-releasing peptide as therapeutic agents to enhance growth hormone secretion in disease and aging. Recent Prog Horm Res. 1997;52:215–44. discussion 244-246. [PubMed] [Google Scholar]
- Tibaduiza EC, Chen C, Beinborn M. A small molecule ligand of the glucagon-like peptide 1 receptor targets its amino-terminal hormone binding domain. J Biol Chem. 2001;276:37787–37793. doi: 10.1074/jbc.M106692200. [DOI] [PubMed] [Google Scholar]
- Usdin T, Hoare S, Wang T, Mezey E, Kowalak J. TIP39: a new neuropeptide and PTH2-receptor agonist from hypothalamus. Nat Neurosci. 1999;2:941–943. doi: 10.1038/14724. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotrophin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Varga J, Schally A, Csernus V, Zarándi M, Halmos G, Rékási Z. Synthesis and biological evaluation of antagonists of growth hormone-releasing hormone with high and protracted in vivo activities. Proc Natl Acad. Sci U S A. 1999;96:692–697. doi: 10.1073/pnas.96.2.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Gonzalez B, Basille M, Pamantung T, Fontaine M, Fournier A, et al. The neuroprotective effect of pituitary adenylate cyclase-activating polypeptide on cerebellar granule cells is mediated through inhibition of the CED3-related cysteine protease caspase-3/CPP32. Proc Natl Acad Sci U S A. 2000;97:13390–13395. doi: 10.1073/pnas.97.24.13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vgontzas AN, Bixler EO, Wittman AM, Zachman K, Lin H-M, Vela-Bueno A, et al. Middle-aged men show higher sensitivity of sleep to the arousing effects of corticotropin-releasing hormone than young men: clinical implications. J Clin Endocrinol Metab. 2001;86:1489–1495. doi: 10.1210/jcem.86.4.7370. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilsbøll T, Krarup T, Madsbad S, Holst J. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept. 2003a;114:115–121. doi: 10.1016/s0167-0115(03)00111-3. [DOI] [PubMed] [Google Scholar]
- Vilsbøll T, Knop F, Krarup T, Johansen A, Madsbad S, Larsen S, et al. The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J Clin Endocrinol Metab. 2003b;88:4897–4903. doi: 10.1210/jc.2003-030738. [DOI] [PubMed] [Google Scholar]
- Volpi R, Caffarra P, Scaglioni A, Boni S, Saginario A, Chiodera P, et al. Defective 5-HT 1-receptor-mediated neurotransmission in the control of growth hormone secretion in Parkinson’s disease. Neuropsychobiology. 1997;35:79–83. doi: 10.1159/000119395. [DOI] [PubMed] [Google Scholar]
- Vrbikova J, Hill M, Bendlova B, Grimmichova T, Dvorakova K, Vondra K, et al. Incretin levels in polycystic ovary syndrome. Endocrinol. 2008;159:121–127. doi: 10.1530/EJE-08-0097. [DOI] [PubMed] [Google Scholar]
- Wagner C, Caplan SR, Tannenbaum GS. Interactions of ghrelin signaling pathways with the growth hormone neuroendocrine axis: a new and experimentally tested model. J Mol Endocrinol. 2009 doi: 10.1677/JME-09-0023. DOI: 10.1677/JME-09-0023. [DOI] [PubMed] [Google Scholar]
- Wajcberg E, Tavaria A. Exenatide: clinical aspects of the first incretin-mimetic for the treatment of type 2 diabetes mellitus. Expert Opin Pharmacother. 2009;10:135–142. doi: 10.1517/14656560802611832. [DOI] [PubMed] [Google Scholar]
- Wapnir R, Teichberg S. Regulation mechanisms of intestinal secretion: implications in nutrient absorption. J Nutr Biochem. 2002;13:190–199. doi: 10.1016/s0955-2863(02)00181-x. [DOI] [PubMed] [Google Scholar]
- Weaver D, Deeds J, Lee K, Segre G. Localization of parathyroid hormone-related peptide (PTHrP) and PTH/PTHrP receptor mRNAs in rat brain. Brain Res Mol Brain Res. 1995;28:296–310. doi: 10.1016/0169-328x(94)00222-z. [DOI] [PubMed] [Google Scholar]
- Weir E, Brines M, Ikeda K, Burtis W, Broadus A, Robbins R. Parathyroid hormone-related peptide gene is expressed in the mammalian central nervous system. Proc Natl Acad Sci U S A. 1990;87:108–112. doi: 10.1073/pnas.87.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimalawansa S. Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Crit Rev Neurobiol. 1997;11:167–239. doi: 10.1615/critrevneurobiol.v11.i2-3.40. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Giguere J, Parisien M, Jeng W, St-Pierre SA, Brubaker PL, et al. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry. 2001;40:2860–2869. doi: 10.1021/bi0014498. [DOI] [PubMed] [Google Scholar]
- Xie D, Cheng H, Hamrick M, Zhong Q, Ding K, Correa D, et al. Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone. 2005;37:759–769. doi: 10.1016/j.bone.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Xie D, Zhong Q, Ding K, Cheng H, Williams S, Correa D, et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone. 2007;40:1352–1360. doi: 10.1016/j.bone.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Yamagata T, Urano H, Weeber E, Nelson D, Nishijima I. Impaired hippocampal synaptic function in secretin deficient mice. Neuroscience. 2008;154:1417–1422. doi: 10.1016/j.neuroscience.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Melmed S. Insulin-like growth factor I action on rat anterior pituitary cells: suppression of growth hormone secretion and messenger ribonucleic acid levels. Endocrinology. 1986;118:176–182. doi: 10.1210/endo-118-1-176. [DOI] [PubMed] [Google Scholar]
- Young A. Inhibition of glucagon secretion. Adv Pharmacol. 2005;52:151–171. doi: 10.1016/S1054-3589(05)52008-8. [DOI] [PubMed] [Google Scholar]
- Yung W, Leung P, Ng S, Zhang J, Chan S, Chow B. Secretin facilitates GABA transmission in the cerebellum. J Neurosci. 2001;15:7063–7068. doi: 10.1523/JNEUROSCI.21-18-07063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi M, Moonga B, Bevis P, Bascal Z, Breimer L. The calcitonin gene peptides: biology and clinical relevance. Crit Rev Clin Lab Sci. 1990;28:109–174. doi: 10.3109/10408369009105900. [DOI] [PubMed] [Google Scholar]
- Zhang K, Aruva M, Shanthly N, Cardi C, Patel C, Rattan S, et al. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase activating peptide (PACAP) receptor specific analogues for PET imaging of breast cancer: in vitro/in vivo evaluation. Regul Pept. 2007;144:91–100. doi: 10.1016/j.regpep.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]