

Abstract
Mutations in APP, PSEN1, MAPT and GRN are the most common genetic causes of dementia. The previous miss-assignment of pathogenicity to benign variants in these genes stresses the importance of discerning between disease causing mutations and benign variants with no pathogenic effect on the function of the respective protein. In this study we sequenced GRN and MAPT in 282 samples from the Centre d’Etude du Polymorphisme Humain - Human Genome Diversity Cell Line Panel, in order to identify benign variants that could otherwise be mistaken for pathogenic mutations. We found sixteen different non-synonymous changes, eleven of which are novel variants.
Keywords: GRN, MAPT, benign variants, pathogenicity
INTRODUCTION
The most common genetic lesions leading to dementia are mutations in the amyloid precursor protein (APP; MIM# 104760) and presenilin 1 genes (PSEN1; MIM# 104311), causing Alzheimer’s disease; and mutations in the progranulin (GRN; MIM# 138045) and microtubule-associated protein tau genes (MAPT; MIM# 157140), causing frontotemporal lobar degeneration. The cases linked to mutation of these genes typically have an earlier onset of the disease and aggregate within families, presenting with an autosomal dominant mode of inheritance [Rademakers, et al., 2003].
Hundreds of pathogenic mutations have been described in these four genes; however, it is often unclear whether identified variants are true disease causing mutations or rare benign changes. The dementia genes mentioned above were identified by positional cloning in different family based studies [Baker, et al., 2006; Cruts, et al., 2006; Goate, et al., 1991; Hutton, et al., 1998; Levy-Lahad, et al., 1995; Sherrington, et al., 1995]. These genes have subsequently been sequenced in other AD and FTLD patients and several novel mutations described. Frequently, pathogenicity has been assumed following screening for the variant in a relatively small number of control subjects. Most of the screening efforts have been done in an ad hoc manner in highly studied populations, such as Caucasians or East Asians and little has been done in other populations. The main result of this strategy is that variants have been reported and assumed to be pathogenic, sometimes with little supporting evidence. This is particularly problematic from a clinical genetic perspective, because it can lead to incorrect information being given to family members, but also from a fundamental scientific perspective, because it has the potential to mislead research on the basic mechanisms underlying the pathological process [Guerreiro, et al., 2008a].
We have previously reported a systematic reassessment of the pathogenicity of variants present in genes involved in AD. In that study, in addition to screening the presenilin and APP genes in a large series of early onset AD patients from Iberia, we performed sequencing analysis in a series of unrelated African individuals from seven different populations obtained from the Centre d’Etude du Polymorphisme Humain - Human Genome Diversity Cell Line Panel (CEPH-HGDP), as well as in Iberian controls. The study of these 130 African samples allowed us to identify five new variants in the screened genes [Guerreiro, et al., 2008a].
In order to extend these results to other genes frequently involved in dementia, we have performed a thorough assessment of common benign genetic variability in the GRN and MAPT loci in 282 samples from different regions of the world, including 87 African samples. Together with our previous report, we were able to generate a list of variants that may otherwise have been classified as pathogenic mutations.
MATERIALS AND METHODS
Samples
GRN and MAPT were sequenced in a series of 282 samples from different regions of the world (106 Asian, 84 European, 5 Middle Eastern and 87 African), obtained from the CEPH-HGDP [Cann, et al., 2002]. The number of samples from each population, geographic origin and region are detailed in Table 1.
Table 1.
Number of samples studied per region, geographic origin and population
| Region | Number of samples | Geographic origin | Number of samples | Population | Number of samples |
|---|---|---|---|---|---|
| Asia | 106 | Cambodia | 10 | Cambodian | 10 |
| China | 48 | Dai | 2 | ||
| Daur | 2 | ||||
| Han | 15 | ||||
| Hezhen | 3 | ||||
| Lahu | 2 | ||||
| Miaozu | 4 | ||||
| Mongolia | 4 | ||||
| Naxi | 3 | ||||
| Orogen | 1 | ||||
| She | 3 | ||||
| Tu | 2 | ||||
| Tujia | 1 | ||||
| Xibo | 4 | ||||
| Yizo | 2 | ||||
| Japan | 10 | Japanese | 10 | ||
| Pakistan | 32 | Balochi | 15 | ||
| Pathan | 17 | ||||
| Siberia | 6 | Yakut | 6 | ||
| Europe | 84 | France | 30 | French | 15 |
| French Basque | 15 | ||||
| Italy | 15 | Sardinian | 15 | ||
| Italy (Bergamo) | 3 | North Italian | 3 | ||
| Orkney Islands | 6 | Orcadian | 6 | ||
| Russia | 15 | Russian | 15 | ||
| Russia Caucasus | 15 | Adygei | 15 | ||
| Middle East | 5 | Israel (Carmel) | 5 | Druze | 5 |
| North Africa | 10 | Algeria (Mzab) | 10 | Mozabite | 10 |
| Subsaharan Africa Congo | 77 | Democratic Republic of | 10 | Mbuti Pygmy | 10 |
| Kenya | 11 | Nantu N.E. | 11 | ||
| Namibia | 5 | San | 5 | ||
| Nigeria | 22 | Yoruba | 22 | ||
| Senegal | 21 | Mandenka | 21 | ||
| South Africa | 8 | Bantu S.E. Pedi | 1 | ||
| Bantu S.E. S. Sotho | 1 | ||||
| Bantu S.E. Tswana | 2 | ||||
| Bantu S.E. Zulu | 1 | ||||
| Bantu S.W. Herero | 2 | ||||
| Bantu S.W. Ovambo | 1 |
DNA Sequencing
The exonic regions of GRN (exons 1-12, plus noncoding exon 0; NM_002087.2) and MAPT (exons 1, 9-13; NM_005910.4), as well as the flanking intronic sequences, were PCR amplified using oligonucleotide primers (sequences available on request) and Roche FastStart PCR Master Mix polymerase (Roche Diagnostics Corp., IN). Each PCR product was sequenced using the same forward and reverse primers with Applied Biosystems BigDye terminator v3.1 sequencing chemistry and run on an ABI3730xl genetic analyzer as per manufacturer’s instructions (Applied Biosystems). The sequences were analyzed with Sequencher software, version 4.2 (Genecodes, VA).
RESULTS
The sequencing of MAPT and GRN in 282 control samples from the human genome diversity re-sequencing panel allowed us to identify sixteen different non-synonymous changes. Of these, three were new non-synonymous changes in MAPT (c.50C>T, p.T17M; c.88A>G, p.T30A; c.898G>A, p.V300I) and thirteen were found in the coding region of GRN. From these latter, eight are new changes (c.163A>T, p.R55W; c.205G>A, p.A69T; c.355_357del, p.N119del; c.662G>C, p.C221S; c.824G>A, p.P275L; c.1126C>T, p.D376N; c.1193G>A, p.S398L; and c.1691, p.R564H); four are already reported variants, until now classified with unclear pathogenicity (c.359G>T, p.S120Y; c.545G>A, p.T182M; c.1298G>A, p.R433Q; and c.1544G>C, p.G515A); and one is already known as a non-pathogenic change (c.55C>T, p.R19W).
All coding non-synonymous variants were observed in African and Asian individuals and, as expected, a greater percentage of African individuals presented coding changes (twenty nine out of thirty six subjects presenting coding non-synonymous variants) when compared to other populations (Table 2).
Table 2.
Variants, excluding synonymous changes, found in the coding region of GRN and MAPT and in exon 0 of GRN
| Gene | Variant | Frequency N (%) | Frequency by population (N) | Frequency by Geographic origin (N) | Frequency by region (N) | |
|---|---|---|---|---|---|---|
| DNA | Predicted protein | |||||
| GRN | g.96073G>A | EX0+49* | 1 (0.4%) | Adygei (1) | Russia Caucasus (1) | 1 Europe |
| GRN | g.96144A>G | EX0+120* | 1 (0.4%) | Russian (1) | Russia (1) | 1 Europe |
| GRN | g.96172G>T | EX0+148* | 5 (1.8%) | Bantu N.E. (1) Mandenka (3) Yoruba (1) |
Kenya (1) Senegal (3) Nigeria (1) |
Subsaharan Africa (5) |
| GRN | g.96199C>G | EX0+175* | 1 (0.4%) | 1 French | France (1) | Europe (1) |
| GRN | g.96206T>C | EX0+182* | 2 (0.7%) | Mandenka (2) | Senegal (2) | Subsaharan Africa (2) |
| GRN | g.96232C>G | EX0+208* | 1 (0.4%) | Adygei (1) | Russia Caucasus (1) | Europe (1) |
| GRN | c.55C>T | p.R19W | 8; 1 homozygous (2.8%) | Mandenka (1) Yoruba (3) Mbuti Pygmy (2) Mozabite (2) |
Senegal (1) Nigeria (3) Democratic Republic of Congo (2) Algeria Mzab (1) |
Sub-Saharan Africa (6) North Africa (2) |
| GRN | c.163A>T | p. R55W | 1 (0.4%) | Cambodia (1) | Cambodia (1) | Asia (1) |
| GRN | c.205G>A | p.A69T | 1 (0.4%) | Xibo (1) | China (1) | Asia (1) |
| GRN | c.355_357del | p.N119del | 1 (0.4%) | Mongolia (1) | China (1) | Asia (1) |
| GRN | c.359G>T | p.S120Y | 2 (0.7%) | Balochi (2) | Pakistan (2) | Asia (2) |
| GRN | c.545G>A | p.T182M | 8 (2.8%) | Bantu N.E. (1) Yoruba (1) Mbuti Pygmy (6) |
Kenya (1) Nigeria (1) Democratic Republic of Congo (6) |
Sub-Saharan Africa (8) |
| GRN | c.662G>C | p.C221S | 1 (0.4%) | Daur (1) | China (1) | Asia (1) |
| GRN | c.824G>A | p.P275L | 1 (0.4%) | Yoruba (1) | Nigeria (1) | Sub-Saharan Africa (1) |
| GRN | c.1126C>T | p.D376N | 2 (0.7%) | Bantu N.E. (1) Mbuti Pygmy (1) |
Kenya (1) Democratic Republic of Congo (1) |
Sub-Saharan Africa (2) |
| GRN | c.1193G>A | p.S398L | 1 (0.4%) | Pathan (1) | Pakistan (1) | Asia (1) |
| GRN | c.1298G>A | p.R433Q | 2 (0.7%) | Mandenka (1) Yoruba (1) |
Senegal (1) Nigeria (1) |
Sub-Saharan Africa (2) |
| GRN | c.1544G>C | p.G515A | 4 (1.4%) | Bantu S.E. Tswana (1) Bantu N.E. (1) Yoruba (1) Mozabite (1) |
South Africa (1) Kenya (1) Nigeria (1) Algeria Mzab (1) |
Sub-Saharan Africa (3) North Africa (1) |
| GRN | c.1691G>A | p.R564H | 1 (0.4%) | Bantu N.E. (1) | Kenya (1) | Sub-Saharan Africa (1) |
| MAPT | c.50C>T | p.T17M | 1 (0.4%) | Yoruba (1) | Nigeria (1) | Sub-Saharan Africa (1) |
| MAPT | c.88A>G | p.T30A | 1 (0.4%) | Bantu S.E. Tswana (1) | South Africa (1) | Sub-Saharan Africa (1) |
| MAPT | c.898G>A | p.V300I | 1 (0.4%) | Mozabite (1) | Algeria Mzab (1) | North Africa (1) |
Nucleotide numbering (“c.”) reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequences (GRN: NM_002087.2; MAPT: NM_005910.4), according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Genomic numbering (“g.”) refers to sequence GRN: NG_007886.1. Protein numbering (“p.”) refers to sequences GRN: NP_002078.1 and MAPT: NP_058519.2.
Refers to the nomenclature employed by van der Zee and colleagues for an easier comparison.
A Mbuti Pygmy sample harbored two non-synonymous changes in GRN (p.R19W and p.T182M). Two samples harbored a GRN change and a MAPT change: a Mozabite individual presented the GRN p.R19W and the MAPT p.V300I variants, and a Bantu SE Tswana presented the p.G515A change in GRN and the p.T30A in MAPT.
Six different changes were found in exon 0 of GRN: +49G>A, +120A>G, +148G>T, +175C>G, +182T>C and +208C>G (nomenclature employed by van der Zee and colleagues is used here for easier comparison; for genomic nomenclature please refer to Table 2). From these, only the EX0+148G>T has been previously described in a Belgian FTLD patient [van der Zee, et al., 2007].
Several synonymous changes were found in the GRN gene, including nine new variants (c.228C>T, p.T76T; c.267C>T, p.A89A; c.402G>A, p.P134P; c.507C>G, p.A169A; c.546G>A, p.T182T; c.786C>T, p.S262S; c.1227G>A, p.T409T; c.1515C>T, p.A505A; and c.1554C>T, p.D518D) and two variants until now classified with unclear pathogenicity (c.42G>A, p.L14L; and c.1695C>T, p.C565C). In the MAPT gene, seven synonymous changes were found, from which three were new (c.42C>T, p.H14H; c.600C>T, p.P200P; and c.798G>A, p.L266L). Although the vast majority of the synonymous changes were observed in African individuals, this type of variations was found in samples from all the regions (Supp. Table S1).
In order to predict the impact in protein function of the non-synonymous variants found in GRN and MAPT, we performed an in silico analysis using two software - SIFT [Ng and Henikoff, 2003] and PolyPhen [Ramensky, et al., 2002; Sunyaev, et al., 2001] (Table 3). From the three variants found in MAPT, one was considered to possibly affect the protein function (p.T17M) while the other two were predicted to be benign. However, the confidence in the prediction for the effect of p.T17M was considered to be low, by the software (SIFT), since the sequences used were not diverse enough.
Table 3.
GRN and MAPT coding variants altering the amino acid sequence and respective PolyPhen and SIFT predictions of the impact of each variant
| Gene | Variant | Number of individuals | PolyPhen | SIFT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Location in the gene | DNA change | Location in the protein | Predicted protein change | New variant? | PSIC score | Result | Score | Result | ||
| GRN | Ex 1 | c.55C>T | ParaGran | p.R19W | Already described | 5 | 0.447 | Benign | 0.03 | Affect protein function* |
| GRN | Ex 2 | c.163A>T | InterPara Gran | p.R55W | New | 1 | 1.622 | Possibly damaging | 0.14 | Tolerated |
| GRN | Ex 2 | c.205G>A | Gran G | p.A69T | New | 1 | 0.781 | Benign | 0.30 | Tolerated |
| GRN | Ex 4 | c.355_357del | InterGF | p.N119del | New | 1 | n.a. | n.a. | n.a. | n.a. |
| GRN | Ex 4 | c.359G>T | InterGF | p.S120Y | Already described | 2 | 1.627 | Possibly damaging | 0.09 | Tolerated |
| GRN | Ex 5 | c.545G>A | InterFB | p.T182M | Already described | 8 | 1.556 | Possibly damaging | 0.04 | Affect protein function |
| GRN | Ex 6 | c.662G>C | Gran B | p.C221S | New | 1 | 3.713 | Probably damaging | 0.01 | Affect protein function |
| GRN | Ex 7 | c.824G>A | InterBA | p.P275L | New | 1 | 3.146 | Probably damaging | 0.01 | Affect protein function |
| GRN | Ex 9 | c.1126C>T | Gran C | p.D376N | New | 2 | 0.32 | Benign | 1.00 | Tolerated |
| GRN | Ex 10 | c.1193G>A | Gran C | p.S398L | New | 1 | 0.895 | Benign | 0.14 | Tolerated |
| GRN | Ex 10 | c.1298G>A | InterCD | p.R433Q | Already described | 2 | 0.395 | Benign | 0.43 | Tolerated |
| GRN | Ex 11 | c.1544G>C | InterDE | p.G515A | Already described | 4 | 1.12 | Benign | 0.47 | Tolerated |
| GRN | Ex 12 | c.1691G>A | Gran E | p.R564H | New | 1 | 0.592 | Benign | 0.31 | Tolerated |
| MAPT | Ex 1 | c.50C>T | N-terminal | p.T17M | New | 1 | 1.557 | Possibly damaging | 0.02 | Affect protein function* |
| MAPT | Ex 1 | c.88A>G | N-terminal | p.T30A | New | 1 | 0.512 | Benign | 0.13 | Tolerated |
| MAPT | Ex 10 | c.898G>A | R2 | p.V300I | New | 1 | 0.114 | Benign | 0.07 | Tolerated |
This is a low confidence prediction (this substitution may have been predicted to affect function just because the sequences used were not evolutionarily diverse enough - for specific details please see http://sift.jcvi.org/).
Nucleotide numbering (“c.”) reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequences (GRN: NM_002087.2; MAPT: NM_005910.4), according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Protein numbering (“p.”) refers to sequences GRN: NP_002078.1 and MAPT: NP_058519.2.
From the twelve missense variations found in GRN, three were predicted to be possibly damaging and two to be probably damaging by the PolyPhen software. The SIFT program predicted four of these twelve changes to affect protein function. Both programs predicted p.T182M, p.C221S and p.P275L to be possibly damaging to the protein.
Considering all the gene variants present in the AD/FTD database (http://www.molgen.ua.ac.be/ADMutations and http://www.molgen.ua.ac.be/FTDMutations), GRN contains a higher percentage of non-pathogenic variants and variants of unclear pathogenicity (53%). For the MAPT gene, the pathogenicity of a variant seems to be closely related to the location of that variant: until now all the coding variants found in the three last exons of the gene are pathogenic (Figure 1 and Supp. Figure S1).
Figure 1.
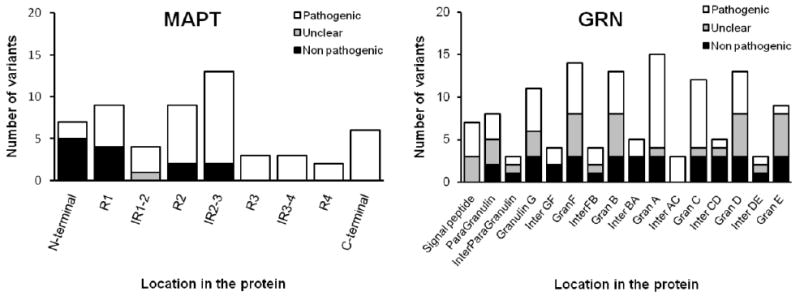
Location of non pathogenic, with unclear pathogenicity and pathogenic variants in functional protein domains found in GRN and MAPT according to the AD/FTD database (assessed on March 15th 2009) and including the variants found in the present study.
In GRN the vast majority of non-pathogenic changes and variants with unclear pathogenicity (described in the AD/FTD database and found in the present study) are missense variants. Of the 72 exonic substitutions described in GRN, only 5 (7%) are classified as pathogenic mutations (Figure 2 and Supp. Figure S2).
Figure 2.

Location of coding substitutions in functional protein domains, classified as non pathogenic, unclear pathogenicity and pathogenic.
DISCUSSION
The question of how to distinguish between sequence variants that have an effect in the stability or function of a protein, ultimately leading to a disease; and a neutral variation with no influence over the phenotype, is very difficult to answer. Several programs (the most widely used: PolyPhen and SIFT) have been developed in order to address this problem and the in silico interpretation of sequence variants is becoming more and more popular. However, some studies have shown that these in silico analysis do not account for all the variables involved (such as supramolecular interactions with other molecules or regions involved in control or signaling, and the existence of linkage disequilibrium) and remain flawed [Chan, et al., 2007; Tchernitchko, et al., 2004]. In this way, although these are consistent and easy analysis to perform, they should not replace functional assays or unequivocal linkage/association. We tested all the variants found in GRN and MAPT genes using SIFT and PolyPhen, expecting that all variants would be predicted to be well tolerated. This did not happen and discordant results between software were obtained for three of the fifteen predictions.
The variant found in exon 10 of MAPT gene is the first missense change in a microtubule binding domain to be considered as non pathogenic. Although we cannot completely exclude the fact that this Mozabite individual will come to develop dementia in the future, this is unlikely to occur. The p.V300I change is located in the second microtubule binding domain of the protein. Even though this is a highly conserved residue between different species, it is only partially conserved among the four microtubule binding domains of the protein. This alteration in the encoded amino acid sequence (valine to isoleucine) is a relatively conservative change, since these are both non polar, neutral amino acids with similar hydropathy indexes (4,2 and 4,5, respectively). Additionally, both software used to interpret the functional effects of sequence variants, predicted this to be a benign and well-tolerated variant.
Frameshift and nonsense GRN mutations have been clearly demonstrated to induce degradation of the mutant RNA by a nonsense mediated decay process. Conversely, the role of missense mutations in this gene remains unclear. From the 72 missense synonymous and non-synonymous variants in the GRN gene, present in the AD/FTD database and found in this study, only five (7%) remain classified as pathogenic. Three of these are point mutations in the translation initiation codon predicted to result in reduced mRNA levels [p.M1 (g.1A>G, g.2T>C, g.3G>A)] [Baker, et al., 2006; Cruts, et al., 2006; Gass, et al., 2006; Le Ber, et al., 2008]. One, (p.A9D) has been reported several times [Gass, et al., 2006; Mukherjee, et al., 2006; Spina, et al., 2007] as a pathogenic mutation affecting the GRN signal peptide. This mutation is believed to prevent N-glycosylation and subsequently cause the mislocation of the protein with accumulation of the mutant protein in the Golgi apparatus and prevention of its secretion [Mukherjee, et al., 2008; Shankaran, et al., 2008]. Additionally this variant greatly reduces GRN expression due to inefficient translation or degradation [Shankaran, et al., 2008]. And the last one (p.C521Y) was found in a Spanish family presenting with progressive nonfluent aphasia [Cruchaga, et al., 2008]. While there are strong evidences for the pathogenicity of the first four mutations, this is not the case for p.C521Y. The carriers of this variant showed significant lower scores than controls in word generation and word learning tests, indicating that they could be at a prodromal stage of the disease. This genetic change was found in seven out of 10 family members: two of these presented dementia, one presented primary progressive aphasia and four were non-affected members. This can indicate, as the authors suggest, that p.C521Y may cause disease with reduced penetrance, but a more conservative interpretation is that this variant is a benign change not related to disease.
Several other point mutations have been proposed to be pathogenic, including p.P248R, p.R432C [van der Zee, et al., 2007], p.C139R and p.P451L [Brouwers, et al., 2008] (Figure 3).
Figure 3.

Progranulin sequence alignment of individual granulin domains with conserved cysteine residues highlighted in blue. Pathogenic missense mutations are represented in red, mutations reported as pathogenic [Brouwers, et al., 2008; van der Zee, et al., 2007] or considered as pathogenic in the AD/FTD database [Cruchaga, et al., 2008] are represented in yellow and the variants found in this study are represented in green.
No segregation data is available for any of these variants and the assumption of pathogenicity has mainly relied on in silico analysis (SIFT, protein sequence conservation and protein modeling). From these, only p.Pro248Leu and p.Arg432Cys have been studied in vitro. These are significantly expressed but cause deficient transport of the protein through the secretory pathway, resulting in degradation and reduction of GRN secretion (p.P248L presented a reduced secretion by about 70% and p.R432C by about 45%) [Shankaran, et al., 2008]. The p.P248L variant occurs in a residue adjacent to a cysteine in granulin B and p.R432C is located in the linker region between granulins C and D. In our study we have found three variants predicted by SIFT and PolyPhen to affect protein function (p.T182M, p.C221S and p.P275L). The p.T182M variant was found in 8 individuals and is clearly a benign variant, while p.C221S and p.P275L were found in one subject, each. The p.C221S change alters a cysteine residue in granulin B that is 100% conserved between all granulins (as does p.C139R and p.C521Y), while p.P275L is located in the linker region between granulins B and A. Thus the variants found in the present study are difficult to distinguish from these previously described as pathogenic and represented in yellow in Figure 3.
Although it remains possible that FTLD cases may exist within the HGDP samples, this is unlikely and if there were FTLD cases among HGDP samples we might expect to see some nonsense GRN mutations. Instead, we found a large number of probably benign missense variants. Our data supports the hypothesis that GRN point variants, with rare exceptions, are not pathogenic and should not be considered as definitely pathogenic unless segregation data is available. Additionally, all clearly pathogenic mutations act by abolishing the expression of the protein, while the definite outcome of a deficient GRN secretion, despite linked to a plausible mechanism, is still difficult to understand and should be viewed with caution.
It is important to discriminate between a genetically normal variant and a pathogenic mutation. Examples of the precedent of misassignment of pathogenicity are the mutations p.E318G and p.InsR352 within PSEN1. Both variants were initially reported to be pathogenic and were later found to either be normal coding variants (p.E318G) or a rare non-pathogenic mutation (p.InsR352) [Boeve, et al., 2006; Mattila, et al., 1998]. Such errors may lead to incorrect and potentially damaging information being given to family members. Several variants found in this study have been previously described in patients. The p.S120Y change in GRN has been found before in an ALS-FTLD patient [Schymick, et al., 2007]. The p.T182M variant was reported in one FTLD patient [Bronner, et al., 2007], in a single case of limb onset sporadic ALS [Schymick, et al., 2007] and in two of three affected siblings presenting early onset FTLD [Guerreiro, et al., 2008b]. Although this variant has never been found in controls before, its pathogenicity has always been considered unclear. The p.R433Q change was previously found in an African American patient with FTLD. The pathogenicity of this variant was reported as unclear since no family history of FTLD was reported for the affected individual and no other DNA samples were available for segregation analysis. Additionally, another variant at the same codon (p.R433W) had been previously reported in a neurologically normal individual. The p.G515A variant has been previously found in two AD patients [Brouwers, et al., 2008], in four FTLD patients and two healthy control individuals [Gass, et al., 2006; van der Zee, et al., 2007].
Our study adds to the classification of variants within MAPT and GRN, and, identifies novel variants that could easily be mistaken as pathogenic mutations in future studies. Overall, our results stress the need for a coordinated effort to resequence the genes involved in neurological disorders in large worldwide samples.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services (Z01 AG000951-06) and the Portuguese Fundacao para a Ciencia e Tecnologia grant (SFRH/BD/27442/2006).
Contract grant sponsor: Intramural Research Program, NIA; Contract grant number: Z01 AG000951-06.
References
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Baker M, Dickson DW, Parisi JE, Giannini C, Josephs KA, Hutton M, Pickering-Brown SM, Rademakers R, Tang-Wai D, et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G->A mutation in progranulin: a clinicopathologic study. Brain. 2006;129(Pt 11):3103–14. doi: 10.1093/brain/awl268. [DOI] [PubMed] [Google Scholar]
- Bronner IF, Rizzu P, Seelaar H, van Mil SE, Anar B, Azmani A, Kaat LD, Rosso S, Heutink P, van Swieten JC. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. 2007;15(3):369–74. doi: 10.1038/sj.ejhg.5201772. [DOI] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, van der Zee J, Pickut BA, Van den Broeck M, Mattheijssens M, Peeters K, et al. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. 2008;71(9):656–64. doi: 10.1212/01.wnl.0000319688.89790.7a. [DOI] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, et al. A human genome diversity cell line panel. Science. 2002;296(5566):261–2. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nystrom M, Grimm AJ, et al. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR) Hum Mutat. 2007;28(7):683–93. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Fernandez-Seara MA, Seijo-Martinez M, Samaranch L, Lorenzo E, Hinrichs A, Irigoyen J, Maestro C, Prieto E, Marti-Climent JM, et al. Cortical Atrophy and Language Network Reorganization Associated with a Novel Progranulin Mutation. Cereb Cortex. 2008 doi: 10.1093/cercor/bhn202. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15(20):2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Guerreiro RJ, Baquero M, Blesa R, Boada M, Bras JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2008a doi: 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Santana I, Bras JM, Revesz T, Rebelo O, Ribeiro MH, Santiago B, Oliveira CR, Singleton A, Hardy J. Novel progranulin mutation: screening for PGRN mutations in a Portuguese series of FTD/CBS cases. Mov Disord. 2008b;23(9):1269–73. doi: 10.1002/mds.22078. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, Hahn-Barma V, van der Zee J, Clot F, Bakchine S, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131(Pt 3):732–46. doi: 10.1093/brain/awn012. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–7. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Mattila KM, Forsell C, Pirttila T, Rinne JO, Lehtimaki T, Roytta M, Lilius L, Eerola A, St George-Hyslop PH, Frey H, et al. The Glu318Gly mutation of the presenilin-1 gene does not necessarily cause Alzheimer’s disease. Ann Neurol. 1998;44(6):965–7. doi: 10.1002/ana.410440617. [DOI] [PubMed] [Google Scholar]
- Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60(3):314–22. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S, Kauwe JS, Norton J, Levitch D, Bigio EH, et al. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29(4):512–21. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. ScientificWorldJournal. 2003;3:497–519. doi: 10.1100/tsw.2003.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P, Elder J, Chio A, Restagno G, Robberecht W, et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry. 2007;78(7):754–6. doi: 10.1136/jnnp.2006.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283(3):1744–53. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Grafman J, Ghetti B. Corticobasal syndrome associated with the A9D Progranulin mutation. J Neuropathol Exp Neurol. 2007;66(10):892–900. doi: 10.1097/nen.0b013e3181567873. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10(6):591–7. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Tchernitchko D, Goossens M, Wajcman H. In silico prediction of the deleterious effect of a mutation: proceed with caution in clinical genetics. Clin Chem. 2004;50(11):1974–8. doi: 10.1373/clinchem.2004.036053. [DOI] [PubMed] [Google Scholar]
- van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D, et al. Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat. 2007;28(4):416. doi: 10.1002/humu.9484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.