

Abstract
Transforming growth factor (TGF)-β signaling makes a significant contribution to the pathogenesis of breast cancer bone metastasis. In other tumor types, TGF-β has been shown to promote tumor vascularity. Here, we report that inhibition of TGF-β significantly reduces microvessel density in mammary tumor-induced bone lesions, mediated by decreased expression of both vascular endothelial growth factor (VEGF) and monocyte chemotactic protein (MCP)-1, both known angiogenic factors. Cathepsin G upregulation at the tumor-bone interface has been linked to increased TGF-β signaling, and we also report that inhibition of Cathepsin G reduced tumor vascularity, as well as VEGF and MCP-1 expression.
Keywords: TGF-β, Cathepsin G, Angiogenesis, Breast Cancer, Bone Metastasis
Introduction
Breast cancer is the most common form of non-skin cancer among women in the United States [1]. Prognosis for women diagnosed with breast cancer varies widely depending on the extent of the disease spread at the time of diagnosis, with a dramatic decrease in life expectancy as breast cancer progresses from primary tumor to local/regional spread to distant metastases. As breast cancer cells leave the primary site, they have a high propensity to metastasize to the bone, lung, liver, and brain, with bone being the most common site distant to the lymph nodes [2]. Bone metastases carry significant consequences including intractable pain, increased risk of pathologic fracture, anemia, and hypercalcemia. Together these consequences reduce both the quality of life and life expectancy of the patient. Thus, understanding the molecular mechanisms that allow mammary tumor cells to thrive in the bone microenvironment is important in designing new therapeutic interventions.
As breast cancer cells enter the bone microenvironment, a vicious cycle ensues that favors both tumor growth as well as bone destruction [3-5]. One component of this cycle is enhanced TGF-β signaling at the tumor-bone interface [6]. The downstream effects of TGF-β signaling are diverse, including chemoattraction of tumor cells, increased tumor cell proliferation, and enhanced osteoclast differentiation and activation [3, 6, 7]. Outside of the bone microenvironment TGF-β has been shown to be an angiogenic factor that increases the vascularity of tumors, allowing an increased nutrient and oxygen supply and subsequently increased tumor growth. TGF-β and hypoxia seem to work synergistically in driving angiogenesis. Hypoxia-inducible factor (HIF)-1 is increased under hypoxic conditions, and this factor has been shown to work cooperatively with TGF-β signaling in inducing the expression of vascular endothelial growth factor (VEGF) which drives angiogenesis [8].
In addition to the direct effects of VEGF, VEGF also appears to induce the expression of monocyte chemotactic protein (MCP)-1, which has been shown to be the primary mediator of TGF-β-induced angiogenesis [9]. VEGF induces the expression of MCP-1 by endothelial cells. In an angiogenesis assay, VEGF has been shown to increase tubule formation, and this effect is abrogated by specific inhibition of MCP-1 using a neutralizing antibody [10]. MCP-1 signaling results in recruitment of vascular smooth muscle cells and mesenchymal cells towards endothelial cells which are the primary producers of MCP-1. This ultimately appears to be the major downstream effect of TGF-β, as TGF-β has been shown to increase the formation of new blood vessels in a chorioallantoic membrane assay and this effect is abrogated by inhibition of MCP-1 [9]. In addition to recruiting smooth muscle cells, MCP-1 also appears to be a chemotactic agent for endothelial cells via signaling through CCR2 [11]. CCR2 is a chemokine receptor that binds members of the CC chemokine family which are identified by their adjacent cysteine residues. This family of chemokines demonstrates a diverse array of downstream effects, including chemoattraction of monocytes, macrophages, T cells, B cells, basophils, eosinophils, and natural killer cells. Interestingly, neutrophils have never been shown to respond to CC chemokines [12].
In this report, we evaluated the effect of inhibition of TGF-β on tumor vascularity in mammary tumor-induced bone lesions. We found tumor vascularity to be reduced and sought to delineate the mechanism by which this occurred. We then evaluated the expression of several angiogenic factors, including MCP-1 and VEGF which are known to be influenced by TGF-β signaling in other systems. We found both MCP-1 and VEGF expression to be reduced by inhibition of TGF-β. We have previously linked enhanced TGF-β signaling at the tumor-bone interface to upregulation in Cathepsin G expression. This appears to occur as increased expression of Cathepsin G results in increased activation of pro-MMP9 which also shows increased expression at the tumor-bone interface. Increased active MMP9 is then responsible for cleaving and activating latent TGF-β resulting in enhanced TGF-β signaling [13]. Based on this, we inhibited Cathepsin G in vivo and similarly found a decrease in tumor vascularity as well as reduced MCP-1 and VEGF expression. Thus, we have demonstrated that inhibition of Cathepsin G reduces TGF-β signaling which subsequently reduces tumor vascularity which is mediated by decreases in both MCP-1 and VEGF. This provides further evidence that Cathepsin G is a potential therapeutic target in the treatment of mammary tumor-induced osteolytic lesions.
Materials and Methods
Inhibition of TGF-β in vivo
TGF-β was inhibited in vivo in a murine bone invasion model as previously described [6]. 1 × 105 Cl66 cells were implanted onto the calvaria of female BALB/c mice. Tumor growth was monitored twice weekly. Mice were treated with neutralizing anti-TGF-β antibody (Clone 1D11; R&D Systems, Minneapolis, MN) at a dose of 2.5 mg/kg bodyweight three times per week. Mice were sacrificed and necropsied for examination of osteolytic lesions four weeks after implantation. At that time, the tumor and the underlying bone were divided into two pieces. One piece was used for separation of the tumor-bone interface from the tumor alone area for further analysis and the other piece was used for histology sections. All studies were done in accordance with the Institutional Animal Use and Care Committee of the University of Nebraska Medical Center.
Protein was extracted from the samples using T-PER tissue protein extractor solution (Pierce, Rockford, IL) following the manufacturer's provided protocol. Protein samples were quantified using a BCA protein assay kit (Pierce, Rockford, IL).
Total RNA was isolated using Trizol® reagent (Invitrogen, Carlsbad, CA).
Inhibition of Cathepsin G in vivo
Cathepsin G function was inhibited in vivo in a murine bone invasion model as previously described [14]. 1 × 105 Cl66 tumor cells were implanted onto the calvaria of female BALB/c mice. Tumor growth was monitored twice a week. Beginning seven days after tumor implantation, mice were injected subcutaneously with Na-Tosyl-Phe-chloromethylketone (TPCK; Sigma-Aldrich, St. Louis, MO) at 50 mg/kg/day or 50 μL DMSO for 21 days. Mice were sacrificed at day 31 post-implantation and necropsied for examination of osteolytic lesions.
Determination of microvessel density
Immunohistochemistry was performed for isolectin B4. Isolectin B4 is a glycoprotein expressed by endothelial cells which has previously been used to label microvessels in order to quantitate microvessel density [15-17]. Sections from TPCK-treated animals, anti-TGF-β treated animals, or control (DMSO)-treated animals were rehydrated using a series of xylenes and ethanols. Endogenous peroxidase activity was quenched using 3% H2O2 in methanol. Antigen retrieval was then performed by boiling sections in 10 mM sodium citrate buffer, pH 6.0, for 11 minutes. Sections were blocked using antibody diluent (BD Biosciences, San Jose, CA). Sections were then incubated for two hours at room temperature with biotinylated antibody directed against isolectin B4 (Vector Laboratories, Burlingame, CA) diluted 1:50 in blocking solution. After washing, sections were incubated with avidin-biotin complex (Vectastain ABC, Vector Laboratories) for 20 minutes at room temperature. Sections were then washed and developed using diaminobenzidine tetrahydrochloride (DAB) (Vector Laboratories) substrate. The sections were then counterstained with hematoxylin. Species specific IgG isotype was added in lieu of primary antibody as a negative control and these sections demonstrated no detectable staining.
The microvessel hot spot technique was used to quantify tumor vascularity [18-20]. Using a light microscope under low power, the three areas of highest microvessel density in each section were selected. In the center of each hot spot, the microscope was switched to high power (40x objective) and the number of vessels with a clearly defined lumen was counted using a 5×5 reticle grid (Klarmann Rulings, Litchfield, NH), giving the microvessel density as the number of vessels per high power field.
Real-time polymerase chain reaction analysis of angiogenic factors
For real-time quantitative reverse transcription based polymerase chain reaction (qRT-PCR) analysis, 5 μg of total RNA from the tumor-bone interface of TPCK-treated, anti-TGF-β treated, and control (DMSO)-treated mice was used for reverse transcription. First strand cDNA was generated using oligo (dT)18 (Fermentas, Hanover, MD) and Superscript II RT (Invitrogen). 2 μL of the resulting cDNA (1:10 dilution) were used in the real-time reactions with gene specific primers for vascular endothelial growth factor (VEGF), monocyte chemotactic protein-1 (MCP-1), fibroblast growth factor-2 (FGF-2), platelet derived growth factor-α (PDGF-α), and glyceraldehyde 3 phosphate dehydrogenase (GAPDH). qRT-PCR reactions were carried out using FastStart SYBR Green Master mix (Roche, Indianapolis, IN) and a MyIQ iCycler (Bio-Rad, Hercules, CA). Fluorescence intensity was measured at the end of each elongation step as a means to evaluate the amount of formed PCR product. GAPDH was used as a reference in order to normalize the samples.
Western blot analysis of MCP-1 and VEGF
75 μg of protein from the tumor-bone interface from control-treated, anti-TGF-β-treatead, and TPCK-treated mice was separated on a 12% SDS-polyacrylamide gel and then was transferred to a PVDF membrane (GE Healthcare, Piscataway, NJ). The membranes were immunoblotted using 1:100 anti-VEGF antibody (R&D Systems), 1:500 anti-MCP-1 antibody (PeproTech, Rocky Hill, NJ) and 1:2000 anti-β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and developed using an ECL Plus Western Blotting Detection System (GE Healthcare) per manufacturer's protocol and imaged using a Typhoon 9410 Variable Mode Imager (GE Healthcare). The bands for MCP-1, VEGF, and β-actin were then quantified and compared using ImageQuant® 5.1 (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis
For all in vivo studies, the Mann-Whitney U-test was used for statistical comparison. A p < 0.05 was considered significant.
Results
Inhibition of TGF-β reduces microvessel density
We first examined the effects of TGF-β signaling on the vascularity of mammary tumor-induced bone lesions. We utilized an anti-TGF-β antibody in vivo in order to block TGF-β signaling and evaluated microvessel density using isolectin B4 immunostaining in both anti-TGF-β treated animals and control-treated animals. Microvessel density was reduced in anti-TGF-β treated animals compared to control (Figure 1A-B).
Figure 1. Inhibition of TGF-β reduces vascularity in mammary tumor-induced bone lesions.

A.i. Immunohistochemistry for isolectin B4 labeling microvessels in a section obtained from a control-treated animal. Scale bar represents 32 nm. ii. Immunohistochemistry for isolectin B4 labeling microvessels in a section obtained from an anti-TGF-β-treated animal. Scale bar represents 32 nm. B. Treatment with anti-TGF-β antibody significantly reduced the microvessel density compared to control-treatment. Error bars represent standard deviation. **, p < 0.01
MCP-1 and VEGF expression are reduced in anti-TGF-β treated mice
qRT-PCR was then used to evaluate expression of angiogenic factors including FGF-2, PDGF-α, VEGF, and MCP-1. Anti-TGF-β treated animals demonstrated similar expression of both FGF-2 (Figure 2A) and PDGF-α (Figure 2B). Treatment with anti-TGF-β antibody significantly reduced expression of VEGF (Figure 2C) and MCP-1 (Figure 2D).
Figure 2. Inhibition of TGF-β reduces the expression of VEGF and MCP-1 mRNA.

A. FGF-2 mRNA expression was similar in anti-TGF-β-treated mice and control-treated mice. B. PDGF-α mRNA expression was similar in anti-TGF-β-treated mice and control-treated mice. C. VEGF mRNA expression was significantly reduced by treatment with anti-TGF-β antibody. D. MCP-1 mRNA expression was significantly reduced in anti-TGF-β-treated mice compared to control-treated mice. Error bars represent standard deviation. **, p < 0.01; ***, p < 0.001, Control treatment group n = 3. Anti-TGF-β treatment group n = 2.
MCP-1 and VEGF are reduced at the protein level in anti-TGF-β treated mice
Western blot analysis confirmed that treatment with anti-TGF-β antibody reduces expression of both MCP-1 (Figure 3A-B) and VEGF (Figure 3C-D) at the protein level compared to control-treatment.
Figure 3. Inhibition of TGF-β reduces the expression of VEGF and MCP-1 protein.

A.-B. Western blot analysis using β-actin to normalize samples showed reduced protein expression of MCP-1 in anti-TGF-β-treated mice compared to control-treated mice. C.-D. VEGF protein expression was reduced by treatment with anti-TGF-β antibody by western blot analysis using β-actin to normalize samples. **, p < 0.01; ***, p < 0.001
Inhibition of Cathepsin G reduces microvessel density
Cl66 tumor-bearing mice were treated with TPCK in order to inhibit Cathepsin G. Again, isolectin B4 immunostaining was used in order to label blood vessels and the microvessel density of TPCK-treated mice was compared to control-treated mice. TPCK-treated animals showed reduced tumor vascularity in comparison to control-treated animals (Figure 4A-B).
Figure 4. Inhibition of Cathepsin G reduces vascularity in mammary tumor-induced bone lesions.
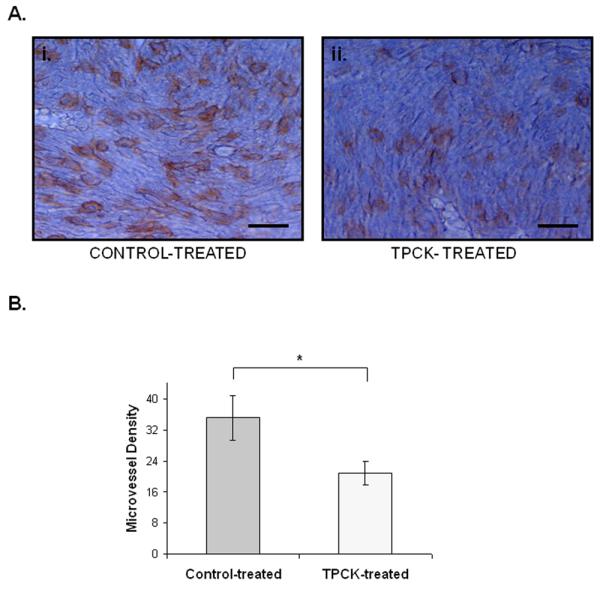
A. i. Immunohistochemistry for isolectin B4 labeling microvessels in a section obtained from a control-treated animal. Scale bar represents 32 nm. ii. Immunohistochemistry for isolectin B4 labeling microvessels in a section obtained from a TPCK-treated animal. Scale bar represents 32 nm. B. Treatment with TPCK significantly reduced the microvessel density compared to control-treatment. Error bars represent standard deviation. *, p < 0.05
MCP-1 and VEGF expression are reduced at the tumor-bone interface of TPCK-treated mice
We then evaluated known angiogenic factors that could potentially be modified by inhibition of Cathepsin G. We evaluated VEGF, MCP-1, FGF-2, and PDGF-α. We found that FGF-2 (Figure 5A) and PDGF-α (Figure 5B) expression were unchanged in TPCK-treated animals compared to control. We did observe decreased expression of both VEGF (Figure 5C) and MCP-1 (Figure 5D) at the mRNA level in TPCK-treated animals compared to control.
Figure 5. Inhibition of Cathepsin G reduces the expression of VEGF and MCP-1 mRNA.

A. FGF-2 mRNA expression was similar in TPCK-treated mice and control-treated mice. B. PDGF-α mRNA expression was similar in TPCK-treated mice and control-treated mice. C. VEGF mRNA expression was significantly reduced by treatment with TPCK. D. MCP-1 mRNA expression was significantly reduced in TPCK-treated mice compared to control-treated mice. Error bars represent standard deviation. ***, p < 0.001, Control treatment group n = 4. TPCK treatment group n = 4.
MCP-1 and VEGF are reduced at the protein level in TPCK-treated mice
We then sought to confirm downregulation of VEGF and MCP-1 at the protein level using western blot analysis. We observed significantly reduced expression of both MCP-1 (Figure 6A-B) as well as VEGF (Figure 6C-D) at the protein level in TPCK-treated animals compared to control.
Figure 6. Inhibition of Cathepsin G reduces the expression of VEGF and MCP-1 protein.

A.-B. Western blot analysis using β-actin to normalize samples showed reduced protein expression of MCP-1 in TPCK-treated mice compared to control-treated mice. C.-D. VEGF protein expression was reduced by treatment with TPCK by western blot analysis using β-actin to normalize samples. **, p < 0.01
Discussion
As tumors grow, there is a steady increase in nutrient and oxygen demand requiring angiogenesis to meet these needs. Without continued angiogenesis, continued tumor growth cannot occur. As a result, research has been focused on recognizing angiogenic factors in various tumors and subsequent inhibition of these angiogenic factors. The first anti-angiogenic drug made available in the United States is bevacizumab which has contributed to the treatment of a variety of tumor types [21]. One signaling molecule that has been shown to be important to angiogenesis in a variety of models is TGF-β [8, 9, 22]. TGF-β has been shown to be important in the pathogenesis of mammary tumor-induced osteolytic bone lesions and TGF-β has been shown to be enhanced at the tumor-bone interface of these lesions [4-7, 23]. However, the downstream effects of enhanced TGF-β in these lesions with regard to tumor vascularity have never been studied.
We began by examining the effect of TGF-β signaling on angiogenesis in mammary tumor-induced osteolytic lesions. We utilized isolectin B4 immunostaining in order to compare microvessel density in mammary tumor-induced bone lesions treated with an anti-TGF-β antibody to lesions treated with a negative control. Microvessel density was significantly reduced in mice treated with anti-TGF-β antibody. This suggested that increased TGF-β signaling does likely contribute to increased tumor vascularity. Mice treated with anti-TGF-β antibody had significantly smaller tumors [6] and reduced angiogenesis and overall tumor vascularity may contribute to the observed reduction in tumor size.
In other tumor types, TGF-β-induced angiogenesis has been shown to be mediated by transcriptional upregulation of both VEGF and MCP-1. The relationship between TGF-β, VEGF, and MCP-1 is complex. Hypoxia via hypoxia-inducible factor (HIF)-1 appears to work synergistically with TGF-β to increase transcription of VEGF, a known angiogenic factor [8]. At least part of the downstream angiogenic effect of VEGF appears to be mediated by upregulation of MCP-1 production by endothelial cells [10]. In fact, in one in vitro assay, the ability of VEGF to function as an angiogenic agent was significantly reduced by neutralizing MCP-1 [10]. MCP-1 also has a known role as an angiogenic factor. MCP-1 serves as a chemoattractant for vascular smooth muscle cells as well as mesenchymal cells promoting new vessel formation [9, 11]. One mechanism for MCP-1 induced angiogenesis appears to be upregulation of a transcription factor known as MCP-1-induced protein (MCPIP) and inhibition of this transcription factor abrogates the ability of MCP-1 to serve as an angiogenic factor [24]. Interestingly, while VEGF has been shown to induce the expression of MCP-1, MCP-1 has also been shown to induce the expression of VEGF and this has been postulated as one of the mechanisms by which MCP-1 serves as an angiogenic factor. MCP-1 has been shown to increase the expression of HIF-1 which in turn increases the expression of VEGF [25]. Thus, the relationship between TGF-β, VEGF, and MCP-1 is complex but the end result of TGF-β signaling appears to be increased VEGF and MCP-1 resulting in enhanced angiogenesis.
Based on this information, we examined the expression of the known angiogenic factors VEGF, MCP-1, PDGF-α, and FGF-2 in anti-TGF-β treated mice compared to control-treated mice to attempt to elucidate the mechanism of TGF-β-induced angiogenesis in this model. We found both VEGF and MCP-1 transcript levels to be higher in control-treated mice compared to anti-TGF-β-treated mice. As a control, we did not find any significant difference in PDGF-α or FGF-2 expression. We then sought to confirm these results with protein level analysis. Western blot analysis confirmed decreased expression of both VEGF and MCP-1 in anti-TGF-β treated animals. Together these results suggest that in mammary tumor-induced osteolytic lesions, TGF-β-induced angiogenesis is mediated by increased expression of both MCP-1 and VEGF, known angiogenic factors.
We have shown that one mechanism by which TGF-β signaling is increased at the tumor-bone interface is Cathepsin-G mediated activation of pro-MMP9 with subsequent activation of latent TGF-β by active MMP9 (personal communication). As a result, we sought to determine if inhibition of Cathepsin G results in a similar reduction in tumor vascularity as well as MCP-1 and VEGF expression. We utilized TPCK, an inhibitor of Cathepsin G, in vivo and evaluated tumor vascularity. Tumors in TPCK-treated mice demonstrated significantly reduced microvessel density compared to control-treated animals. Similar to what we observed when we inhibited TGF-β, TPCK-treatment led to a reduction in the expression of both VEGF and MCP-1 at both the mRNA and protein levels. We did not observe any change in expression of PDGF-α or FGF-2.
Inhibition of Cathepsin G shows similar changes to inhibition of TGF-β in tumor vascularity as well as in expression of MCP-1 and VEGF. We believe this is ultimately due to the fact that inhibition of Cathepsin G results in reduced TGF-β signaling (personal communication). These studies provide further evidence for the link between Cathepsin G and TGF-β. Moreover, these studies add to the growing evidence that Cathepsin G makes an important contribution to mammary tumor-induced osteolytic lesions. Cathepsin G appears to be a viable therapeutic target with inhibition both reducing osteolysis [14] as well as reducing tumor vascularity.
Acknowledgements
We thank Michelle Varney for careful review and critique of this manuscript. This work was supported in part by Susan G. Komen for the Cure grant KG090860, Cancer Glycobiology Program from Nebraska Research Initiative and by grant CA72781 (R.K.S.) and Cancer Center Support Grant (P30CA036727) from National Cancer Institute, National Institutes of Health, the Department of Defense Predoctoral Fellowship (K.C.N.), and the Howard Hughes Medical Institute Research Training Fellowship (T.J.W).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement.
All authors declare no conflicts of interest and acknowledge no financial or personal relationships with other people or organizations that could inappropriately influence (bias) their work.
Reference List
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J. Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Sanuki-Fujimoto N, Takeda A, Amemiya A, Ofuchi T, Ono M, Yamagami R, Hatayama J, Kunieda E, Shigematsu N. Pattern of tumor recurrence in initially nonmetastatic breast cancer patients: distribution and frequency of metastases at unusual sites. Cancer. 2008;113:677–682. doi: 10.1002/cncr.23612. [DOI] [PubMed] [Google Scholar]
- 3.Wilson TJ, Singh RK. Proteases as modulators of tumor-stromal interaction: Primary tumors to bone metastases. Biochim. Biophys. Acta. 2007 doi: 10.1016/j.bbcan.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roodman GD. Mechanisms of bone metastasis. N. Engl. J. Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 5.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat. Rev. Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 6.Futakuchi M, Nannuru KC, Varney ML, Sadanandam A, Nakao K, Asai K, Shirai T, Sato SY, Singh RK. Transforming growth factor-beta signaling at the tumor-bone interface promotes mammary tumor growth and osteoclast activation. Cancer Sci. 2009;100:71–81. doi: 10.1111/j.1349-7006.2008.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massague J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 9.Ma J, Wang Q, Fei T, Han JD, Chen YG. MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood. 2007;109:987–994. doi: 10.1182/blood-2006-07-036400. [DOI] [PubMed] [Google Scholar]
- 10.Yamada M, Kim S, Egashira K, Takeya M, Ikeda T, Mimura O, Iwao H. Molecular mechanism and role of endothelial monocyte chemoattractant protein-1 induction by vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2003;23:1996–2001. doi: 10.1161/01.ATV.0000096208.80992.63. [DOI] [PubMed] [Google Scholar]
- 11.Salcedo R, Ponce ML, Young HA, Wasserman K, Ward JM, Kleinman HK, Oppenheim JJ, Murphy WJ. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. [PubMed] [Google Scholar]
- 12.Singh S, Sadanandam A, Singh RK. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007;26:453–467. doi: 10.1007/s10555-007-9068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson TJ, Nannuru KC, Singh RK. Cathepsin G-mediated Activation of Pro-MMP9 at the Tumor-Bone Interface Promotes TGF-â Signaling and Bone Destruction. Molecular Cancer Research. 2009 doi: 10.1158/1541-7786.MCR-09-0028. In Press. [DOI] [PubMed] [Google Scholar]
- 14.Wilson TJ, Nannuru KC, Futakuchi M, Sadanandam A, Singh RK. Cathepsin G enhances mammary tumor-induced osteolysis by generating soluble receptor activator of nuclear factor-kappaB ligand. Cancer Res. 2008;68:5803–5811. doi: 10.1158/0008-5472.CAN-07-5889. [DOI] [PubMed] [Google Scholar]
- 15.Kopatz K, Distler C. Astrocyte invasion and vasculogenesis in the developing ferret retina. J. Neurocytol. 2000;29:157–172. doi: 10.1023/a:1026594721760. [DOI] [PubMed] [Google Scholar]
- 16.Herve MA, Buteau-Lozano H, Vassy R, Bieche I, Velasco G, Pla M, Perret G, Mourah S, Perrot-Applanat M. Overexpression of vascular endothelial growth factor 189 in breast cancer cells leads to delayed tumor uptake with dilated intratumoral vessels. Am. J. Pathol. 2008;172:167–178. doi: 10.2353/ajpath.2008.070181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubota Y, Hirashima M, Kishi K, Stewart CL, Suda T. Leukemia inhibitory factor regulates microvessel density by modulating oxygen-dependent VEGF expression in mice. J. Clin. Invest. 2008;118:2393–2403. doi: 10.1172/JCI34882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jong JS, van Diest PJ, Baak JP. Hot spot microvessel density and the mitotic activity index are strong additional prognostic indicators in invasive breast cancer. Histopathology. 2000;36:306–312. doi: 10.1046/j.1365-2559.2000.00850.x. [DOI] [PubMed] [Google Scholar]
- 19.Hansen S, Grabau DA, Rose C, Bak M, Sorensen FB. Angiogenesis in breast cancer: a comparative study of the observer variability of methods for determining microvessel density. Lab Invest. 1998;78:1563–1573. [PubMed] [Google Scholar]
- 20.Belien JA, Somi S, de Jong JS, van Diest PJ, Baak JP. Fully automated microvessel counting and hot spot selection by image processing of whole tumour sections in invasive breast cancer. J. Clin. Pathol. 1999;52:184–192. doi: 10.1136/jcp.52.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Los M, Roodhart JM, Voest EE. Target practice: lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist. 2007;12:443–450. doi: 10.1634/theoncologist.12-4-443. [DOI] [PubMed] [Google Scholar]
- 22.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 23.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 24.Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP) J. Biol. Chem. 2008;283:14542–14551. doi: 10.1074/jbc.M802139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong KH, Ryu J, Han KH. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood. 2005;105:1405–1407. doi: 10.1182/blood-2004-08-3178. [DOI] [PubMed] [Google Scholar]