

Abstract
We used all-atom molecular dynamics simulations to investigate the structure and properties of the actin filament, starting with either the recent Oda model or the older Holmes model. Simulations of monomeric and polymerized actin show that polymerization changes the nucleotide-binding cleft, bringing together the Q137 side chain and bound ATP in a way that may enhance ATP hydrolysis rate in the filament. Simulations with different bound nucleotides and conformations of the DNase I binding loop show that the persistence length of the filament depends only on the loop conformation. The computational modeling reveals how bound phalloidin stiffens actin filaments and inhibits the release of the γ-phosphate from ADP-Pi actin.
Keywords: cytoskeleton, ATP hydrolysis, persistence length, filament, actin
Introduction
The protein actin is highly conserved, abundant in eukaryotic cells, and known to play a role in a wide range of cellular functions. Actin is chiefly found in two forms: monomeric (or globular) actin and its polymerized counterpart, filamentous actin. The dynamic equilibrium between the forms is controlled by a variety of factors within the cell.1 Actin filaments are a central structural feature of all muscle tissue,2 and controlled polymerization of branched networks of actin filaments produce force for cell motility.3
The structural and dynamical properties of actin monomers and filaments depend, at least in part, on the state of the bound nucleotide.4–7 Both forms of actin strongly bind ATP or ADP, but polymerized actin hydrolyzes its bound ATP about 40,000-fold faster8 than monomeric actin,9 but the mechanism is a major unsolved challenge.10 It is assumed that changes in the nucleotide-binding cleft increase the ATPase activity of polymerized actin. Previous molecular dynamics (MD) studies of actin and Arps showed that the conformation of nucleotide-binding cleft depends on the bound nucleotide in monomeric actin and Arps11–13 and actin trimers.12 However these findings do not address the means by which polymerization changes the rate of ATP hydrolysis. Experimental data14,15 showed that residue Q137 is important in ATP hydrolysis, and Oda et al. hypothesized that flattening of the actin molecule moves residue Q137 closer to the bound ATP.16 Crystal structures of monomeric actin5,6 show that Q137 coordinates a water molecule that might attack the bound ATP.
Several dozen crystal structures of actin monomers have been determined since the first crystal structure of ATP-actin bound to DNase I17, but currently no high-resolution crystal structures of filamentous actin are available. The parameters of the actin helix were originally established by x-ray diffraction of whole muscle18,19 and electron microscopy.20–22 The first model of filamentous actin built with a crystal structure of the subunit is now commonly known as the Holmes model.23 Recently Oda et al.16 obtained higher-resolution fiber diffraction data from filamentous actin stabilized by phalloidin and aligned with a superconducting magnet. Their refined model has the same helical arrangement of the actin subunits, but the actin subunit itself is flatter in the filament than is any crystal structure of monomeric actin. In the Oda model filament the two halves of the protein (c.f., Fig. 1) are rotated about 20° so that all four subdomains are close to lying in a plane. Oda et al. and previous workers24 have observed this flattening of the actin subunit in 3D reconstructions of filaments from electron micrographs. These models suggest that flattening of the actin subunit is a main conformational change associated with actin polymerization, and that this change underlies the higher ATPase activity of filamentous actin.16,25
Figure 1.
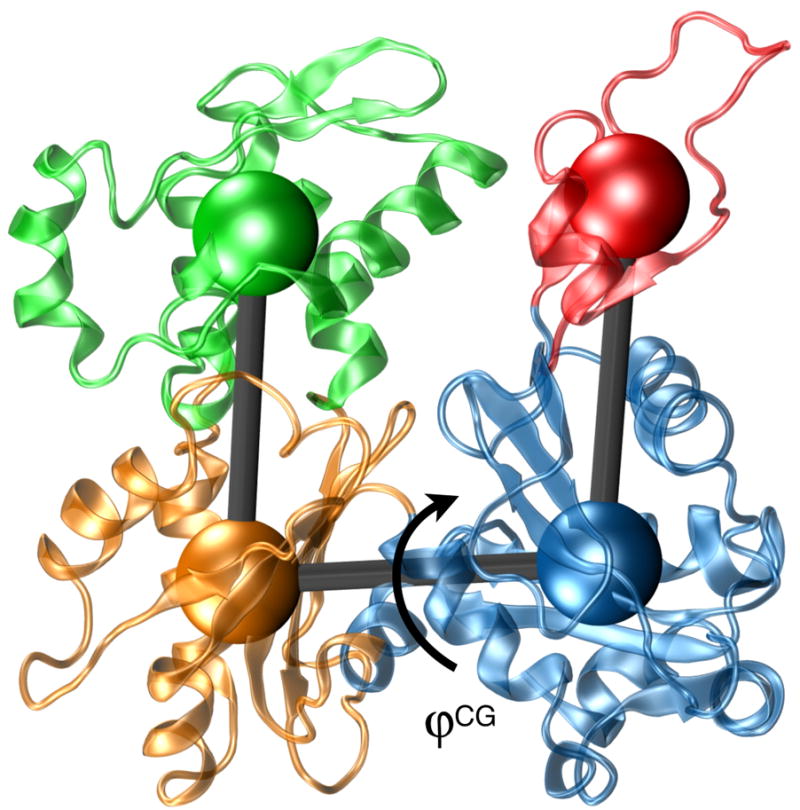
Actin subunit from the Oda et al.16 filament model in atomistic (cartoon) and coarse-grained (CG) representation. The four subdomains shown are: S1 (blue) residues 1-32, 70-144 and 338-375; S2 (red) residues 33-69; S3 (orange) residues 145-180 and 270-337; and S4 (green) residues 181-269. The cartoon representation of the protein is from PDB entry 2ZWH, and the “flatness” order parameter used for analysis is labeled.
The conformation of the DB loop in actin filaments is unsettled. The observation that the DB loop of some6 but not all9 crystals of ADP-bound actin form a well-defined α-helix led to the idea that DB loop folds into a helix in conjunction with ATP hydrolysis and phosphate release. The Oda model is based on a form of monomeric actin with an unstructured DNase-I binding loop (DB loop),5,17 but Oda et al. proposed that an extension of the helical DB loop into the neighboring actin subunit in filaments could also explain the observed diffraction patterns.16 This is an intriguing possibility given the evidence that release of the inorganic phosphate subsequent to ATP hydrolysis within filamentous actin softens the filament4,26 as well as increases the susceptibility to attack by severing proteins.27 Additionally, computational studies26 have drawn comparisons between the ATP-bound (DB loop unstructured) and ADP-bound (DB loop folded) forms of filamentous actin. A recent computational study of monomeric and trimeric actin demonstrated that folding of the DB loop in ADP-actin is thermodynamically favorable.12
Motivated by the unanswered questions concerning the structure and properties of actin filaments, we present a large scale all-atom MD study of the Oda and Holmes filament models with variations in both the bound nucleotide and the conformation of the DB loop. Previous MD simulations of actin and actin related proteins (Arps)11–13,26,28,29 explored the structure and dynamics of actin on timescales ranging from a few ns to 100 ns. Actin and Arp simulations based on purely classical MD methods investigated the short timescale properties of the actin monomer or filament, especially the conformation of the DB loop and properties of the nucleotide-binding cleft. Although MD simulations are limited to short time scales, they offer a complementary perspective to experimental techniques, inasmuch as they are fully atomistic in character and therefore permit sampling and observation of all atomic degrees of freedom in the system.
We performed MD simulations in an explicitly solvated aqueous environment of nine different actin filaments based on either the Holmes or Oda models with bound ATP or ADP and either folded or unfolded DB loops, including an additional simulation with bound phalloidin to further replicate the experimental conditions under which the filament models were obtained. We investigated the equilibrium dynamics of each of these filaments with an emphasis on understanding how polymerization stimulates the ATP hydrolysis rate and how the bound nucleotide influences the structure of the DB loop and macroscopic filament properties. The simulations give the persistence length of each filament for comparison with experimental measurements. We also performed a 50 ns simulation of ATP-bound monomeric for comparison with filamentous actin. The total amount of MD simulation performed in this study is over 0.5 μs for the actin filament.
Results
This section summarizes the findings obtained from our MD simulations. Properties reported were calculated from the last 20 ns of each simulation with data collected once the system had equilibrated. The criteria used to establish whether a particular filament simulation had equilibrated are discussed in the methods section. For reference, Table S1 in the Supplementary Material summarizes each of the systems investigated in this study. We systematically varied the filament model, bound nucleotide and the initial conformation of the DB loop. For brevity the results of these eight simulations are presented in shorthand notation to describe each system. For example, H/ATP/unfold refers to the simulation of a Holmes actin filament with bound ATP and the DB loop initially in the unfolded configuration.
Filament and Monomer Structure
The simulations were performed in the isobaric-isothermal (constant NPT) ensemble, and therefore required adjustment of the unit cell dimensions to maintain a constant pressure. As in previous NPT simulations with longitudinal periodicity26 the filament crossover length fluctuates at the beginning but levels out to an approximately constant value, which we report as equilibrium filament lengths in Table 1. The experimentally measured crossover length of the Oda and Holmes filaments is 358 Å, whereas the crossover lengths reported in Table 1 range from 354 to 373 Å. Both of our simulations of ADP Oda filaments have a crossover of 357 Å, in very good agreement with the Oda model with ADP-actin subunits. The equilibrium crossover lengths of all the Oda filaments are smaller than the corresponding Holmes filaments.
Table 1.
Structural properties of actin filaments investigated by molecular dynamics.
| Filament Model | Bound Nucleotide | DNase-I Loop1 | Crossover Length2 (Å) | Filament RMSD3 (Å) |
|---|---|---|---|---|
| Oda | ATP | unfolded | 362(±4) | 5.7(±0.5) |
| ATP | folded | 354(±5) | 6.9(±0.3) | |
| ADP | unfolded | 357(±4) | 5.6(±0.4) | |
| ADP | folded | 357(±3) | 6.4(±0.6) | |
| Holmes | ATP | unfolded | 364(±2) | 8.3(±1.0) |
| ATP | folded | 363(±6) | 10.5(±0.4) | |
| ADP | unfolded | 373(±2) | 7.1(±0.4) | |
| ADP | folded | 366(±5) | 8.2(±0.4) | |
| Oda + phalloidin | ATP | unfolded | 362(±1) | 5.0(±0.2) |
Refers to the initial conformation of the DNase-I binding loop. Further details are provided in the methods section.
The crossover length is calculated by averaging the unit cell length over the last 20 ns of each simulation. Simulations were performed of one filament crossover distance (13 actin subunits) with periodicity along the longitudinal axis of the filament.
The RMSD (of 4875 Cα atoms) is calculated with respect to the initial structure and using the last 20 ns of each data set as described in the methods section.
Our simulations of Oda filaments were more stable than simulations of Holmes models based on the root-mean-square deviations (RMSD), reported in Table 1 as ensemble average RMSD of all 4875 Cα atoms in an actin 13-mer calculated with respect to the starting structure. This suggests that the higher-resolution Oda model more accurately captures the subunit interactions that ultimately give rise to stable filament dynamics. The significantly larger filament RMSD values for the Holmes model simulations also indicate that the individual subunits must undergo some conformational shifts within the respect to the filament superstructure prior to equilibration. The RMSD of filaments were significantly higher than individual actin monomers or actin related proteins Arp2 and Arp3. The RMSD determined from MD simulations of actin or Arp2/Arp3 were reported to range from 2–2.5 Å.11–13,26,28 However, because of the supramolecular nature of the actin filament, i.e., the actin subunits are only constrained to each other by non-covalent bonds, it is not surprising that the RMSD of the entire actin 13-mer was higher than actin monomers, especially considering the fluctuations in filament crossover length.
For control purposes we performed simulations of actin filaments with bound ATP and a folded DB loop region. As detailed below in the discussion of the filament stiffness, the purpose of this control simulation was not to simulate a known experimental state of actin, but to test a hypothesis regarding the connection between the state of the nucleotide, the DB loop conformation, and the filament stiffness. In the ATP/(DB fold) Oda filament model simulation we found that the DB loop unfolded in the individual actin subunits during simulations of the state. In contrast, the folded DB loop in the ADP filament simulations was much more stable, in agreement with recent folding simulations that showed a thermodynamic driving force for a folded DB loop within actin trimers based on the Oda filament model.12 The observed unfolding in the ATP/fold filament occurred slowly with respect to the simulation timescale and by the end of the simulation most of the DB loops were partially or fully unfolded. This is consistent with previous work that showed unfolding of the DB loop during simulation of ATP-actin monomers28 and that the ATP-actin trimers are thermodynamically unfavorable for folding of the DB loop region.12 Additionally, these studies were performed with the CHARMM22/27 and AMBER99SB (respectively) force fields. Consistent results for the ATP-actin (DB fold state) have now been obtained from three separate studies and two different force fields. Finally, these observations are consistent with the absence of experimental evidence folded DB loops in ATP-actin.
We characterized the flatness actin molecules with a four subunit coarse-grained (CG) representation of actin (Fig. 1).13,17,30 The model consists of four actin subdomains, which provide an intuitive means to understand actin’s global shape and structure. The 4-site CG model is completely specified with six internal coordinates, typically chosen to be three bonds, two bending angles and a single dihedral. The flatness of subunits within a filament depends on the dihedral angle, which we chose as an order parameter to monitor in the atomistic MD simulations. The 4-site CG model accurately captures the flattening reported in the Oda model: the dihedral angles of crystallized monomeric actin range from 156 to 159° (obtained from PDB entries 1J6Z6 and 1NWK5, respectively; whereas the dihedral angle for the Oda actin filament model is 176° (PDB 2ZWH16). An angle of 180° corresponds to a completely planar (flat) monomer in the CG model.
We used the CG structures produced by our atomistic MD simulations to calculate probability densities for dihedral angles of the actin subunits in filaments based on the Oda and Holmes models with bound ATP or ADP and unfolded or folded DB loops (Fig. 2). Each panel of the figure also shows the starting angles and the sample mean and skewness (γ), or third moment of the distribution defined as:
| (1) |
where N is the total number of dihedral angles (ϕ), e.g. the number of trajectory snapshots times 13. The skewness measures the asymmetry of a distribution – a distribution that is positively skewed appears to lean to the left and have a longer tail to the right, and vice versa.
Figure 2.

Probability distributions for dihedral angle in the four-site CG model. Each panel contains the distributions for the Oda (solid) and Holmes (dashed) models for the four different nucleotide/DB loop pairs. The width of the bars is 1 deg, and the initial value for each model is labeled with a vertical line. The sample mean and skewness are listed in each panel.
For both ATP and ADP filaments with folded or unfolded DB loops the means of the distributions of dihedral angles are similar to the starting filament model, 169° for the Oda model and 157° for the Holmes model. Given enough simulation time the two models presumably would converge to the same dihedral angle, however such large-scale rearrangement is not generally observed in MD simulations. However, it is illustrative to describe the overall tendencies of the “flatness” order parameters with respect to the initial and mean values. The Oda ADP models had the narrowest distribution of dihedral angles from 156°–180° with a skew away from the mean and initial dihedral values toward a less flat conformation. All of the other systems had larger ranges of dihedral angles. Under all four conditions the simulations of Oda filaments were less flat than the initial model. While the Holmes filaments maintained an average dihedral angle approximately equal to the starting value, the simulations skewed the distributions toward flatter dihedral angles in both Holmes filaments with unfolded DB loops and in the other direction with folded DB loops. Both of the ATP/fold simulations show dihedral angle distributions that are bimodal. The heights of the two peaks in each distribution are constant for the entire 20 ns period over which the data were collected indicating that these data are equilibrated on the timescale of the simulations performed in this paper. Additionally we performed the same dihedral angle analysis of the ATP/unfold+phalloidin simulation. A comparison between the two filaments (with and without phalloidin) is shown in Supplementary Fig. S1. The phalloidin angle distribution is narrower, symmetric, and also more closely centered on the starting value from the Oda et al. structure. This is not surprising given the well-known stabilizing effect that phalloidin has on actin filaments. Additionally, since the Oda model was derived using actin filaments with bound phalloidin, it is possible that the phalloidin-free filament systems simulated in this work may eventually undergo conformational changes on significantly longer timescales (e.g. microsecond or longer) as they approach their equilibrium states.
We investigated the dynamical properties of the backbone dihedral angles between residues 141-142 and 336-337 (the so-called “hinge-region” of actin) that Oda et al. used to explain the domain rotation that flattened the subunit in their model.16 We examined the distributions of these backbone dihedral angles for each subunit in the ADP/fold and ATP/unfold filament systems and used Ramachandran plots of these angles to display the distributions for the ATP/unfold and ADP/fold systems (Fig. 3). For residues 141-2, the data are very similar for all systems. The Oda and Holmes models differ in their initial φ values, but the ensemble data overlap nearly completely on the 50–100 ns timescale. The backbone angles for residues 336-337 show the few observed differences between the two systems more clearly. In both residues 336 and 337 the observed range of φ angles has nearly complete overlap for both filament models. In contrast the ψ angles have broader distributions and regions that do not overlap. It is interesting to see, especially in the case of residue 337 that the Oda and Holmes φ values almost form boundaries of the range of observed values. The ensemble of backbone angles for these residues has a very high standard deviation, and it was therefore not possible to obtain a meaningful correlation between the flatness order parameter we developed and the configuration of residue 141-142 / 336-337. However, qualitatively, there is a link between the observation that the subunits within the Oda filaments generally became “less flat” and the backbone dihedral distributions for the residues that are linked to the subunit flattening overlap significantly with those from the classical Holmes model.
Figure 3.

Ramachandran plots for residues 141-142 and 336-337 in actin filaments derived from NPT MD simulations. The initial values for the Oda and Holmes filaments are represented by circle and triangles, respectively. The regions encompassing the values observed during MD simulation are marked with a dashed line (Oda) or solid line (Holmes). The lines are drawn with a contour level that encloses 100% of the observed backbone angles for the specific residues that are indicated. The Oda actin subunit is shown at the top with the residues of interest labeled in red.
To better understand how the different filament models and bound nucleotide influence the actin-actin contacts we calculated the average actin trimer structure from each filament simulation using the last 20 ns of each simulation. Trimers contain the fundamental contacts common to all actin filaments, designated as the (i − i+1) contacts along the short-pitch helix, and the (i − i+2) contacts along the long-pitch helix. To calculate the average actin trimer from a given simulation each actin 13-mer was decomposed into eight separate trimers, e.g., A1-A2-A3, A2-A3-A4, and the average Cα position for each trimer was then calculated (Fig. 4). Residues in adjacent subunits were designated as “in contact” if their C atoms were closer than 10 Å (Table S2).
Figure 4.

Inter-subunit contact architecture of Oda and Holmes actin filaments derived from MD simulations. The average Cα position is depicted as a tube model to illustrate the cross-strand (i −i+1) and intra-strand (i −i+2) contacts. The initial structure is compared to the ATP/unfold and ADP/fold simulations. Contacts are shown in blue (subunit i) or black (subunits i+1 and i+2), and a contact is defined as two Cα atoms being less than 10 Å apart. Table S2 details the specific residues in contact for all of the models investigated.
The MD simulations reduced the contacts between subunits along both the short-pitch and long-pitch helices compared with the initial Oda model. It is interesting to note that the folded and unfolded DB loops had roughly the same number of contacts, consistent with the work of Oda et al., who noted that both DB loop conformations fit the observed fiber diffraction data.16 It is also noteworthy that the MD simulations shifted the DB loop from an out of contact position in the Holmes model into contact with the next subunit along the short-pitch helix, but these contacts are not as extensive as those created by “flattening” in the original Oda model or after the simulations.
Properties of Nucleotide-Binding Cleft
Our MD simulations show the time evolution of all the atomic degrees of freedom in the system and allow observations of the dynamics of the nucleotide-binding cleft (although limited to the timescales accessible by MD). Table 2 lists structural parameters that describe the behavior of the gamma phosphate group in simulations with bound ATP and the whole filaments for comparison with monomeric actin. The table lists average distances or bond lengths obtained over the last 20 ns of each simulation.
Table 2.
Geometric parameters that characterize the nucleotide-binding pocket in actin subunits from MD simulations.
| Filament Model | Bound Nucleotide | DNase-I Loop1 | B12 (Å) | B22 (Å) | B32 (Å) | B42 (Å) |
|---|---|---|---|---|---|---|
| Oda | ATP | unfolded | 4.6(±0.2) | 5.7(±0.4) | 4.5(±0.4) | 4.0(±0.5) |
| ADP | unfolded | 3.6(±0.5) | ||||
| ADP | folded | 5.8(±1.5) | ||||
| Holmes | ATP | unfolded | 5.8(±0.3) | 4.9(±0.3) | 3.8(±0.4) | 3.3(±0.8) |
| ADP | unfolded | 2.6(±0.6) | ||||
| ADP | folded | 3.3(±0.8) | ||||
| G-actin | ATP | unfolded | 6.1(±.4) | 3.4(±.1) | 5.2(±.2) | 3.0(±1.0) |
| Oda3 (initial structure) | ADP | unfolded | 5.1 | 4.3 | ||
| Monomeric actin3 (initial structure) | ATP | unfolded | 7.1 | 3.9 | ||
| Oda | ATP phalloidin | unfolded | 4.7(±0.2) | 4.2(±0.7) | 5.7(± 0.4) | 4.0(±0.8) |
Refers to the initial conformation of the DNase-I binding loop. Further details are provided in the methods section.
Definition of geometrical parameters: B1: distance between Q137 Cδ side chain atom and Pγ atom of bound ATP, B2: Distance between Pγ atom of bound ATP and O atom of closest coordinating water molecule, B3: Distance between Pγ atom of bound ATP and Cα atom of G156, B4: Distance between O atom in Q137 side chain and H atom of closest coordinating water molecule.
The initial Oda structure properties were taken from PDB entry 2ZWH. The initial G-actin structure was taken from PDB entry 1NWK and prepared as described in the text.
The average distance between the Cδ atom of Q137 and the Pγ of the bound ATP is 1.2 Å shorter in simulations of the Oda model than in simulations of the Holmes model. The actual coordination occurs through a non-bonded interaction between an O atom of the γ-PO3 of ATP and one of the hydrogen atoms on the NH2 group bound to the Cδ atom of Q137 side chain (Fig. 5). The slight reorientation of the “hinge region” including residues 137 – 146 flattens the actin subunit in the Oda model and allows orientation of the Q137 side chain anchored to the Pγ group, whereas the Holmes model does not. We also observed that γ-PO3 group moves somewhat further away from the protein backbone in simulations of Oda models than Holmes models (item B3 in Table 2). The observed variances for B3 are higher than those for B1, making the magnitude of the difference difficult to discern.
Figure 5.

Coordination of Q137 side chain with gamma phosphate group of bound ATP in actin filament simulations shown in stereo view. The protein is drawn with a cartoon representation, and the Q137 side chain and bound ATP molecule are shown in licorice format. Panels A and B show the initial configuration for the Oda and Holmes filament models, respectively. Panels C and D show a representative structure for the Oda and Holmes models, respectively. The distance between the NH2 group and an O atom on the terminal PO3 group of ATP is illustrated with a dashed line. Water molecules within 2.5 Å of the ATP molecule or Q137 side chain are shown.
Water coordinated differently with the γ-phosphate group of ATP and the side chain of Q137 in simulations based on Oda and Holmes models (items B2 and B4 in Table 2). Water shows a stronger tendency (shorter distance) to coordinate with one of the oxygen atoms of the γ-phosphate group of ATP (item B2) in the Holmes model compared to the Oda model. Water also coordinated more strongly (with a shorter distance) to the Q137 side chain in simulations of the Holmes model than the Oda model (distance B4 in Table 2).
Simulation of ATP-bound monomeric actin for 50 ns showed that the γ-phosphate of ATP is separated further from both the protein backbone and the Q137 side chain than in filaments. The γ-phosphate is less constrained by the protein but more likely to coordinate with water in the nucleotide-binding cleft of the monomer than in any of the filament models.
Persistence Length of Actin Filaments
We used the greatly increased computing power available since the original estimates persistence lengths of actin filaments from MD simulations26 to re-examine the hypothesis that the stiffness of filaments depends on the DB loop conformation (Table 3). As in the previous work, the values obtained from MD simulations based both the Holmes and Oda models (in the ADP/folded and ATP/unfolded conformations) agreed well with experimentally measured4,31 persistence lengths (using ADP-BeFe3− actin to stand in for short lived ATP-actin). Persistence lengths were almost two-fold higher with an unfolded DB loop than with a helical DB loop. The control studies with opposite DB loop conformations (ADP/unfold and ATP/fold) supported the hypothesis the conformation of the DB loop has a stronger influence on persistence length than the bound nucleotide. The table includes two values for the simulations of the Oda model with ATP and folded DB loop. During the simulation the DB helix unfolded and the stiffness increased. Simulation of the Oda model of ATP actin filaments and including the bound phalloidin ligands gave a persistence length very similar to experiment4,31 and somewhat larger than obtained experimentally with ADP-BeF3−. We note that phalloidin was included in the experimental preparation of the actin gels used in fiber diffraction experiments but not explicitly included in the model calculations of the Holmes and Oda models as discussed23.
Table 3.
Comparison of experimental and calculated persistence lengths for actin filaments. All persistence lengths are given in units of μm.
| Filament1 | Lp (μm) |
|---|---|
| ATP unfolded | |
| Experiment2 | 13.5 |
| Oda | 15.9(±0.3) |
| Holmes | 14(±0.3) |
| ADP folded | |
| Experiment3 | 9.1(±0.3) |
| Oda | 8.7(±0.1) |
| Holmes | 8.2(±0.1) |
| ADP unfolded | |
| Holmes | 14.8(±0.3) |
| Oda | 17.2(±0.4) |
| ATP folded | |
| Oda4 | 13.6(±0.1) |
| Oda4 | 8.6(±0.1) |
| Holmes | 7(±0.05) |
| ATP unfold + phalloidin | |
| Experiment2 | 18(±1) |
| Oda | 20.5(±0.4) |
The filament model is described by whether it is a model or experimental determination of the persistence length, the state of the bound nucleotide and the initial conformation of the DNase-I loop. Further details on model preparation are provided in the methods section. The configuration of the DNase-I loop was not determined via experiment, but inferred based on previous experiments and simulations as well as the present work.
Experimental values from Isambert et al.4 with ADP-BeFe3− or ATP + phalloidin.
Experimental value and error obtained via averaging results from Isambert et al.4 and McCullough et al.31
The larger Oda value is obtained from the last 20 ns of NPT simulation when the DB loop regions became unfolded. The smaller value is from an earlier point in the simulation when equilibration had not yet been achieved (see Methods section), but the DB loop region was still folded.
Simulations of Filaments With Bound Phalloidin
Simulation an Oda model of an ATP-actin filament with bound phalloidin showed that the orientation of the phalloidin shifts from the initial position proposed by Lorenz et al.32 to a new equilibrium site with a large increase contacts between phalloidin and the (i − i+1) subunit along the short-pitch helix in the region of residues 69—78 on the adjacent subunit (Fig. 6). This region contains the evolutionary conserved residue 73 (3-methylhistidine)5 that is close to the nucleotide binding pocket and was previously shown to coordinate the Pi group upon ATP hydrolysis.12 The number of cross-strand contacts between phalloidin and the (i − i+1) actin subunits increases from an initial value of 2 to 6.9(±0.4) for the entire subunit and from 2 to 4.4(±0.4) when only considering the important range of residues 69 – 78. A phalloidin – actin contact is defined when the two Cα atoms are located within 10.0 Å of each other. This increase in contacts between the helix strands points to a quantitative mechanism by which phalloidin stiffens the actin filament. In principle this observed correlation between the cross-strand contact architecture and the phalloidin binding site could be further explored using a non-equilibrium technique such as umbrella sampling, metadynamics, or steered MD; however, this would be a very computationally challenging calculation and is beyond the scope of the present work.
Figure 6.

Effect of bound phalloidin on contact architecture in actin filament simulations. The ribbon diagrams show subunits i (blue), i+1 (brown) and i+2 (green) and of phalloidin (licorice representation). Red spheres show the positions of all Cα atoms within 10.0 Å of the bound phalloidin. The two panels show stereo views for initial (top) and representative configuration (bottom) that illustrate how bound phalloidin increases the inter-strand (i − i+1) contacts in actin filaments.
Simulation of the Oda model with bound ATP, bound phalloidin and an unfolded DB loop gave persistence length close to the experimental value.4 It is interesting that the RMSD for the filament with phalloidin is the lowest of all the filaments investigated, so phalloidin even stabilizes filaments on the 50 ns time scale. The nucleotide-binding cleft of subunits in filaments with bound phalloidin was very similar to ATP-actin filaments without phalloidin.
Discussion
Our extensive MD simulations of actin filaments based on both the Holmes23 and Oda16 filament models with ADP or ATP and two conformations of the DNase I binding loop have provided valuable insights into the means by which actin polymerization enhances ATP hydrolysis, and ATP hydrolysis in the actin subunit influences the structure and dynamics of filaments. Our simulations of actin with bound phalloidin address the means by which phalloidin stiffens actin filaments and suppresses ATP hydrolysis and/or release of the inorganic phosphate (Pi) after hydrolysis. The simulations performed in this work represent the most extensive sampling via MD simulation to date of the actin filament. Simulations on the 50 to 100 ns timescale such as those presented herein are useful for characterizing the local equilibrium of biomolecular assemblies, but cannot provide quantitative information regarding the equilibrium properties of rare events (e.g., certain protein conformational changes) that occur on significantly longer timescales (i.e., microsecond or longer).
The Oda filament model offered a potential explanation for how flattening of the actin subunit might enhance the rate of ATP hydrolysis by four orders of magnitude by locating the Q137 side chain closer to the γ-phosphate group of ATP. Actin residue Q137 is critical for ATP hydrolysis15, fiber diffraction methods do not currently have sufficient resolution to locate side chain atoms. Our simulations confirmed that flattening of the actin subunits during polymerization moves the Q137 side chain close to the γ-phosphate group of ATP. Residue Q137 is part of the hinge region that participates in flattening, which moves its side chain upward toward the nucleotide-binding pocket. This repositioning results in a pronounced increase in the coordination between the side chain and the γ-phosphate group, offering improved stabilization (and susceptibility to hydrolysis) of the triphosphate group compared with its position in actin monomers and in subunits in the Holmes filament model. Our simulations of ATP-bound monomeric actin confirmed this difference in the nucleotide-binding pockets of monomeric and filamentous actin.
Our simulations also address the relationship between the conformation of the DNase I loop and the stiffness of the actin filament. Previous simulations26 showed that Holmes filaments with ADP/folded and ATP/unfolded subunits have persistence lengths in good agreement with those measured by experiment, leading to the hypothesis that that the conformation of the DB loop is a dominant factor in defining rigidity and filament related properties. Our recalculation of the Chu and Voth data and additional control studies of both the Oda and Holmes filament models in the ATP/folded and ADP/unfolded configurations support this hypothesis. Additionally, recent simulations12 show that folding of the DNase I loop into an alpha helical conformation is thermodynamically favorable in actin trimers, especially within the Oda filament model. Taken together, this data points to a mechanism where i) polymerization flattens the actin subunits, promoting ATP hydrolysis, ii) dissociation of γ-phosphate induces changes in the DNase I loop, and iii) changes in the DNase I loop cause softening of the filament, i.e., reduction of its persistence length.
Our simulations of actin with bound phalloidin32 agree with experimentally measured effects of phalloidin on actin filaments. Phalloidin reoriented into a more favorable position and increased the (i − i+1) inter-strand contacts between subunits along the short-pitch helix, which explains how phalloidin stiffens and stabilizes filaments.4 Future studies from our group will use MD simulations to investigate the mechanism of Pi release and explore the atomistic deals of how phalloidin interferes with this process.
Methods
The coordinates for filamentous actin were based on the Oda16 or Holmes23 models. Oda filaments were generated with either the DB loop in a folded or unfolded conformation. The initial unfolded conformation used was published by Oda et al. in PDB entry 2ZWH.16 The initial folded conformation was built by taking the folded DB loop from PDB entry 1J6Z6, splicing it into the 2ZWH structure, and relaxing the structure with an energy minimization routine. The initial coordinates for the filaments were obtained in an identical manner to a previously published study26 from our group. Phalloidin binding site coordinates were obtained from the published data by Lorenz et al.32 The filament was placed in a box of explicit TIP3P water molecules33 with a minimum distance of 15 Å between the protein and the border of the periodic boundary conditions. The charge of the system was neutralized via random placement of counter-ions (KCl) using the VMD autoionize plugin34 The size of each system is detailed in Table S1 of the Supplementary Material.
All MD calculations were performed using NAMD.35 The CHARM22/2736 force field was used in conjunction with the particle mesh Ewald sum method37 for calculating electrostatic interactions. All intramolecular hydrogen bonds were constrained using the SHAKE38 algorithm allowing for an integration timestep of 2 fs. After heating the system to 310 K, a 100 ps equilibration was performed in the canonical ensemble by velocity rescaling. Following this, the simulations were continued in the isobaric-isothermal (constant NPT) ensemble (310 K, 1.01325 bar) through the use of Langevin dynamics and the Langevin piston method via its implementation in NAMD.39,40 The damping coefficient used for Langevin dynamics was 0.5 ps−1 and the Langevin piston was controlled using a piston period decay of 2 ps. Each simulation was continued for at least 50 ns, or until the unit cell length had equilibrated and sufficient equilibrated sampling of the NPT ensemble was achieved. The strategy we employed was to run the NPT simulation until the box length along the filament axis stabilized (normally 30 ns), and then continued for an additional 20 ns “production phase.” The exact length of each simulation is provided in Table S1. Unless otherwise noted, results reported from MD simulations are based on the last 20 ns of each trajectory. The unit cell length was the slowest property to equilibrate in the simulations performed in this work. By comparison, the filament RMSD (< 10 ns), system pressure (< 1 ns), and total energy (< 10 ns) equilibrated far faster. Analysis of MD calculations and creation of all protein figures was performed using Visual Molecular Dynamics (VMD).34 Error estimation was performed with a standard block-averaging technique.41,42 For average values of a property of a single actin subunit, that were derived from filament simulations, e.g., water coordination, the error for that property for each subunit was estimated via block-averaging, and the subsequent 13 error estimates were averaged to obtain an overall average error estimate.
Supplementary Material
Acknowledgments
This work was supported in part by National Science Foundation International Research Fellows Program (OISE-0700080) (to JP) and NIH research grant GM066311 (to TDP). Computational support was provided by NSF Teragrid resources at the Pittsburgh Supercomputing Center and the National Institute for Computational Sciences. The authors wish to thank Niels Volkmann for directing them to the bound phalloidin coordinates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 2.Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 3.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 4.Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, Carlier MF. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J Biol Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 5.Graceffa P, Dominguez R. Crystal structure of monomeric actin in the ATP state. J Biol Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- 6.Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 7.Belmont LD, Orlova A, Drubin DG, Egelman EH. A change in actin conformation associated with filament instability after Pi release. Proc Natl Acad Sci USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanchoin L, Pollard TD. Hydrolysis of ATP by polymerized actin depends on the bound divalent cation but not profilin. Biochemistry. 2002;41:597–602. doi: 10.1021/bi011214b. [DOI] [PubMed] [Google Scholar]
- 9.Rould MA, Wan Q, Joel PB, Lowey S, Trybus KM. Crystal structures of expressed non-polymerizable Monomeric actin in the ADP and ATP states. J Biol Chem. 2006;281:31909–31919. doi: 10.1074/jbc.M601973200. [DOI] [PubMed] [Google Scholar]
- 10.Reisler E, Egelman EH. Actin’s structure and function: What we still do not understand. J Biol Chem. 2007;282:36133–36137. doi: 10.1074/jbc.R700030200. [DOI] [PubMed] [Google Scholar]
- 11.Dalhaimer P, Pollard TD, Nolen BJ. Nucleotide-mediated conformational changes of monomeric actin and Arp3 studied by molecular dynamics simulations. J Mol Biol. 2007;376:166–183. doi: 10.1016/j.jmb.2007.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfaendtner J, Branduardi D, Parrinello M, Pollard TD, Voth GA. Nucleotide-dependent conformational states of actin. Proc Natl Acad Sci. 2009;106:12723–12728. doi: 10.1073/pnas.0902092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfaendtner J, Voth GA. Molecular dynamics simulation and coarse-grained analysis of the Arp2/3 complex. Biophys J. 2008;95:5324–5333. doi: 10.1529/biophysj.108.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vorobiev S, Strokopytov B, Drubin DG, Frieden C, Ono S, Condeelis J, Rubenstein PA, Almo SC. The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc Natl Acad Sci. 2003;100:5760–5765. doi: 10.1073/pnas.0832273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasa M, Maeda K, Narita A, Maéda Y, Oda T. Dual roles of Gln137 of actin revealed by recombinant human cardiac muscle alpha-actin mutants. J Biol Chem. 2008;283:21045–22103. doi: 10.1074/jbc.M800570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oda T, Iwasa M, Aihara T, Maeda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin: DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 18.Huxley HE. X-ray analysis and the problem of muscle. Proc R Soc Lond, Ser B: Biol Sci. 1953;141:59–62. doi: 10.1098/rspb.1953.0017. [DOI] [PubMed] [Google Scholar]
- 19.Huxley HE. Electron microscope studies of the organisation of the filaments in striated muscle. Biochim Biophys Acta. 1953;12:387–394. doi: 10.1016/0006-3002(53)90156-5. [DOI] [PubMed] [Google Scholar]
- 20.Hanson J, Lowy J. The structure of F-actin and of actin filaments isolated from muscle. J Mol Biol. 1963;6:46–60. [Google Scholar]
- 21.Moore PB, Huxley HE, DeRosier DJ. Three-dimensional reconstruction of F-actin, thin filaments and decorated thin filaments. J Mol Biol. 1970;50:279–295. doi: 10.1016/0022-2836(70)90192-0. [DOI] [PubMed] [Google Scholar]
- 22.DeRosier DJ, Moore PB. Reconstruction of three-dimensional images from electron micrographs of structures with helical symmetry. J Mol Biol. 1970;52:355–369. doi: 10.1016/0022-2836(70)90036-7. [DOI] [PubMed] [Google Scholar]
- 23.Holmes KC, Popp D, Gebhard W, Kabsch W. Atomic model of the actin filament. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 24.Volkmann N, Liu H, Hazelwood L, Krementsova EB, Lowey S, Trybus KM, Hanein D. The structural basis of myosin V processive movement as revealed by electron cryomicroscopy. Mol Cell. 2005;19:595–605. doi: 10.1016/j.molcel.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Holmes KC. Structural biology: actin in a twist. Nature. 2009;457:389–390. doi: 10.1038/457389a. [DOI] [PubMed] [Google Scholar]
- 26.Chu JW, Voth GA. Allostery of actin filaments: Molecular dynamics simulations and coarse-grained analysis. Proc Natl Acad Sci USA. 2005;102:13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De La Cruz EM. How cofilin severs an actin filament. Biophysical Reviews. 2009;1:51–59. doi: 10.1007/s12551-009-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng X, Diraviyam K, Sept D. Nucleotide effects on the structure and dynamics of actin. Biophys J. 2007;93:1277–1283. doi: 10.1529/biophysj.107.109215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wriggers W, Schulten K. Stability and dynamics of G-actin: Back-door water diffusion and behavior of a subdomain 3/4 loop. Biophys J. 1997;73:624–639. doi: 10.1016/S0006-3495(97)78098-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu JW, Voth GA. Coarse-grained modeling of the actin filament derived from atomistic-scale simulations. Biophys J. 2006;90:1572–1582. doi: 10.1529/biophysj.105.073924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCullough BR, Blanchoin L, Martiel JL, De la Cruz EM. Cofilin increases the bending flexibility of actin filaments: implications for severing and cell mechanics. J Mol Biol. 2008;381:550–558. doi: 10.1016/j.jmb.2008.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorenz M, Popp D, Holmes KC. Refinement of the F-actin model against X-ray fiber diffraction data by the use of a directed mutation algorithm. J Mol Biol. 1993;234:826–836. doi: 10.1006/jmbi.1993.1628. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, Gumbart J. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 38.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of cartesian equations of motion of a system with constraints: molecular-dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 39.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-pressure molecular-dynamics simulation - the Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 40.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 41.Flyvbjerg H, Petersen HG. Error-estimates on averages of correlated data. J Chem Phys. 1989;91:461–466. [Google Scholar]
- 42.Frenkel D, Smit B. Understanding molecular simulation: From algorithms to applications. MPG Books; Bodman: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.