

Abstract
Shiga toxins (Stx) 1 and 2 are responsible for intestinal and systemic sequelae of infection by enterohemorrhagic Escherichia coli (EHEC). However, the mechanisms through which enterocytes are damaged remain unclear. While secondary damage from ischemia and inflammation are postulated mechanisms for all intestinal effects, little evidence excludes roles for more primary toxin effects on intestinal epithelial cells. We now document direct pathologic effects of Stx on intestinal epithelial cells. We study a well characterized rabbit model of EHEC infection, intestinal tissue and stool samples from EHEC-infected patients, and T84 intestinal epithelial cells treated with Stx1. Toxin uptake by intestinal epithelial cells in vitro and in vivo causes galectin-3 depletion from enterocytes by increasing the apical galectin-3 secretion. This Shiga toxin-mediated galectin-3 depletion impairs trafficking of several brush border structural proteins and transporters, including villin, dipeptidyl peptidase IV, and the sodium-proton exchanger 2, a major colonic sodium absorptive protein. The mistargeting of proteins responsible for the absorptive function might be a key event in Stx1-induced diarrhea. These observations provide new evidence that human enterocytes are directly damaged by Stx1. Conceivably, depletion of galectin-3 from enterocytes and subsequent apical protein mistargeting might even provide a means whereby other pathogens might alter intestinal epithelial absorption and produce diarrhea.
Keywords: Shiga toxin, enterocytes, galectin-3 secretion, apical protein missorting, NHE2
Introduction
Shiga toxin-producing EHEC is one of the major foodborne pathogens by virtue of the frequency in the USA and the severity of the associated illnesses [1]. EHEC infections cause illness in approximately 75 000 people per year in the United States [2]. Recent epidemics such as those related to contaminated leafy green vegetables have increased these totals [2]. While most EHEC-infected individuals develop watery and bloody diarrhea and recover, up to 20% of these patients, mostly children or elderly, develop life-threatening systemic complications, including hemolytic uremic syndrome (HUS) [3,4]. A prospective cohort study [5] provided evidence that antibiotic use significantly increased risks of developing HUS. Improved understanding of Stx-induced intestinal pathophysiology and progression to HUS is necessary to provide the basis for new treatment strategies. Both hemorrhagic colitis and HUS are believed to be thrombotic complications of Stx-induced microvascular injury, since Stx can target globotriaosylceramide (Gb3) receptors on microvascular endothelial cells in gut, kidney, and brain [6].
It has also been assumed that primary damage to enterocytes in EHEC infection results from bacterial attaching and effacing lesions, and that interaction between intestinal epithelial cells (IEC) and Stx are less important. Although there are only a few studies that address the questions of primary damage of IEC by Stx, the majority of them have been done using Gb3-positive IEC models. The absence of detectable amounts of Gb3 in normal human enterocytes [7–9] adds an additional complexity to the problem and prompts the development of new approaches to determine the consequences of the interaction between Stx and enterocytes.
In the current study, we explore direct Stx1 effects on enterocytes. We demonstrate that Stx1 uptake by Gb3-negative IEC causes galectin-3 secretion from their apical domains. This secretion significantly depletes intracellular galectin-3 levels in these cells, studied in vivo and in vitro. Galectin-3 is a β-galactoside-specific lectin implicated in diverse cellular and extracellular signal transduction pathways [10,11]. It is highly expressed in human small intestinal and colonic epithelial cells [12,13]. Decreased galectin-3 levels in human colonic epithelial cells have been previously described in other diarrheal diseases, most notably Crohn’s disease [14,15]. We show here that Stx1 interactions with IEC significantly increase apical galectin-3 secretion in vitro and in vivo. This Stx1-induced galectin-3 depletion from enterocytes leads to the mistargeting of several key structural and absorptive brush border membrane proteins, including dipeptidyl-peptidase IV (DPPIV), villin, and sodium/proton exchanger isoform 2 (NHE2).
Overall, these data demonstrate that apical secretion of galectin-3 by IEC as a result of the direct impact of Stx1 (and also Stx2) on enterocytes might contribute to the development of toxin-induced diarrhea.
Materials and methods
Cell culture
Human colon cancer polarized epithelial T84 cells (A.T.C.C., Manassas, VA) were grown and maintained in DMEM (Dulbecco’s modified Eagle’s medium) F-12 medium (1:1), supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). Monolayers (passage 31–45) were grown on polycarbonate inserts with 0.4-μm pore size (Costar, Cambridge, MA) or on glass coverslips for 7–10 days. Experiments were performed when monolayers achieved a transepithelial electrical resistance TER > 1,500 Ω·cm2.
Materials
Stx1 and recombinant B-subunit of Stx1 (Stx1B) were provided by Dr. A. Kane (Tufts New England Medical Center, Boston, MA). Purified B-subunit of Cholera toxin (CTB) was from List Biological Laboratories, Inc (Campbell, CA). We used the following mouse monoclonal antibodies (mAbs): anti-galectin-3 from BD Pharmingen (for immunoblotting) (San Diego, CA) and from Affinity BioReagents (for immunofluorescence) (Golden, CO), anti-villin (a gift from Dr. M. Arpin, Curie Institute, Paris and BD Transduction Laboratories), anti-DPPIV was a kind gift from Dr. A. Hubbard (Johns Hopkins University, Baltimore, MD), anti-GAPDH from US Biological (Swampscott, MA), anti-EGFR from Upstate (Lake Placid, NY). Secondary fluorescent antibodies and wheat germ agglutinin (WGA) conjugated to the Alexa Fluor 568 were from Invitrogen (Carslbad, CA) or from Rockland Immunochemical Inc. (Gilbertsville, PA). Stx1, Horseradish peroxidase (HRP) and CTB were conjugated with Alexa fluor 680 reactive dye according to the manufacturer’s protocols (Invitrogen). Polyclonal antibodies: anti-NHE2 was a kind gift from Dr. M. Tse (Johns Hopkins University, Baltimore, MD); anti-NHE3 was from Santa Cruz Biotechnology, Inc. (San Diego, CA). All other reagents were purchased from Sigma (St. Louis, MO) unless stated otherwise.
Detection of intracellular Stx
Confluent T84 cells were incubated for varying times with Stx1 conjugated to Alexa 680 fluorescent dye (Stx1-680) in serum free DMEM/F12. Cells were then washed three times with cold PBS to remove extracellular toxin. The fluorescence intensity of Stx1-680, which corresponds to the amount of intracellular Stx1, was measured in total cell lysates (supplemental Material and Methods) by fluorescence plate reader and normalized to the autofluorescence of total cell lysate from untreated cells.
Human tissue specimens
The human tissue samples of EHEC-infected patients collected during the 1993 E. coli O157:H7 epidemic in the Western USA [16] were obtained from the tissue bank of Seattle Children’s Hospital via a Johns Hopkins sponsored material transfer agreement. The samples were coded, so no patient identifiers were linked to the specimens. These studies were approved by the Institutional Review Boards (or equivalent committees) of the Children’s Hospital and Regional Medical Center, Seattle, Washington.
Detection of secreted galectin-3 in cell conditioned media and in stool samples
Following treatments of T84 monolayers with Stx1, B-subunit of cholera toxin (CTB), or Horseradish peroxidase (HRP) for varying times, the media was removed from the upper or lower chambers of culture inserts and the cells were pelleted by gentle centrifugation (1,000 g, 5 min). The supernatants were concentrated using Amicon Ultra Centrifugal Filter Devices MWCO 5,000 (Millipore, Billerica, MA) to a final volume of ~150 μl. The supernatant samples (40 μl) were resuspended in Laemmli sample buffer, boiled for 10 min, subjected to 12% SDS-PAGE, and transferred to nitrocellulose membrane. For galectin-3 detection the membranes were incubated with galectin-3 mAb (1:1,000) in TBS overnight at 4°C. Following three washes in TBS, the blots were incubated with fluorescent dye-conjugated secondary antibody (1:10,000) for 1 h. The blots were washed and resulting bands were detected by infrared imaging scanner. The integrated intensity of fluorescence for each band was quantified using Odyssey software.
To detect galectin-3 in samples from patients infected with EHEC, stool filtrates were collected fresh, immediately diluted in PBS (1:10), centrifuged, filtered through a 0.45 μm filter, as previously described [17,18]. Stools were obtained from infected subjects using a multi-state network of participating microbiology laboratories, and written informed consent was obtained from each subject’s parents. Control stools were obtained in a prospective case control study of the etiology of acute childhood diarrhea, and consisted of community-based healthy controls subjects. These studies were approved by the Institutional Review Boards (or equivalent committees) of the Children’s Hospital and Regional Medical Center, Seattle, Washington University, St. Louis, and the participating institutions from which stools were obtained from infected children.
Rabbit model of EHEC colitis
We used the rabbit cecal model of EHEC colitis [19]. Briefly, in this model the enteroadherrent rabbit E. coli RDEC-1 were transduced with Stx1-converting phage ΦH19A to express Stx1 [19]. To discriminate between Stx1-specific intestinal epithelial pathology and lesions related to the enteric colonization, male New Zealand white rabbits (weighing 2.5–5 kg) were treated with PBS (control animals, n=3), or infected with either 1010 RDEC-1 (control animals, n=3) or 1010 Stx1-producing RDEC-H19A (experimental animals, n=3). Experiments with animals were performed using protocols approved by the Animal Use Committee of the University of New Mexico School of Medicine.
Statistical analysis
Values are presented as mean ± SEM. Statistical significance was determined using Student’s unpaired t- test and p-values <0.05 were considered significant.
Results
Stx1-induced intestinal disease decreases intracellular galectin-3 in IEC
Seven days after infection all three of the rabbits which were challenged with the Stx1-producing RDEC-H19A strain developed bloody diarrhea and severe cecal edema. In contrast, RDEC-1 infected comparison animals developed watery diarrhea with no edema, despite attaching and effacing bacteria in the cecum. PBS-treated control rabbits failed to develop symptoms of disease. Additionally, the RDEC-H19A-infected rabbits lost more weight than either comparison group (data not shown).
We used mass spectroscopy to identify proteins with different abundance in RDEC-H19A vs. RDEC-1 infected animals. Levels of galectin-3 were higher in total cell lysate from cecal tissue of rabbits infected with RDEC-1 than in animals infected with the Stx1-producing RDEC-H19A. Western blots confirmed that galectin-3 was reduced in cecal tissues from RDEC-H19A-infected rabbits compared to either RDEC-1 (Fig. 1A and B) or PBS-treated rabbits (data not shown).
Figure 1. Galectin-3 is downregulated in rabbit cecum during EHEC infection and in T84 intestinal epithelial cells exposed to Stx1.
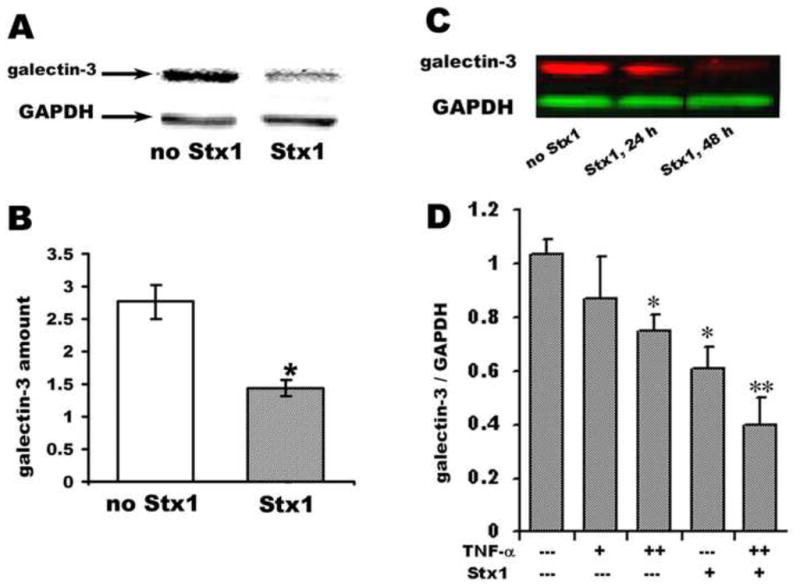
(A) Representative immunoblot of galectin-3 in total cell lysates from cecum of a rabbit infected either with RDEC-1 (no Stx1), or RDEC-H19A (Stx1).
(B) Quantification of galectin-3 immunoblot data normalized to GAPDH expression show significant (*p<0.05) decrease in rabbit cecal galectin-3 in animals infected with Stx-producing strain compared with rabbit infected with no Stx1-producing strain.
(C) Representative immunoblot of galectin-3 in total cell lysates from control T84 cells or cells incubated with 200 ng/ml of Stx1 (lowest concentration at which significant and reproducible downregulation of galectin-3 was detected after 24h of exposure to Stx1) over time.
(D) Quantification of the galectin-3 protein amount in T84 cells treated for 24 h either with TNF-α: (+ − 10 ng/ml; ++ − 50 ng/ml) or Stx1 (200 ng/ml) or their combination. (*) Significant compared to the control (p<0.05); (**) significant compared to either Stx1, or TNF-α alone (p<0.05).
Galectin-3 is a β-galactose binding protein [10] that is expressed in a variety of human cells, including small intestinal and colonic enterocytes. Galectin-3 is localized predominantly to the cytoplasm and sub-apical region of these polarized IEC [12,13]. Immunofluorescence microscopy confirmed that the significant decrease in the galectin-3 expression in cecum was mostly attributed to absence from epithelial cells in rabbits infected with Stx1-producing bacteria. There was little change in galectin-3 expression by immune cells present in lamina propria (Supplemental Fig. 2). The average fluorescence intensities of galectin-3 staining in cecal epithelial cells in PBS treated (negative controls) or REDC-1-infected rabbits (no Stx1) vs. RDEC-H19A (Stx1-producing) infected animals were 192.3± 6.5 per cell, 181.7 ±7.6 per cell and 117.6±4.7 per cell, respectively, (p<0.05 compared with either PBS or RDEC-1; 120 cells measured for each condition).
Stx1 uptake by IEC decreases the intracellular amount of galectin-3
We tested if the decreased amounts of intracellular galectin-3 in EHEC-induced intestinal disease were due to direct interaction between Stx1 and IEC rather than to secondary effects mediated by ischemia or inflammation. We examined the effects of Stx1 on galectin-3 levels using in vitro T84 cell model. Incubation of T84 cells with Stx1 alone significantly decreased the amount of galectin-3 in T84 cells (Fig. 1C), indicating a direct effect of Stx1 on IEC.
We then explored possible mechanisms for Stx1effects on galectin-3 levels. It has been shown that Stx1 does not inhibit protein synthesis in T84 cells [9,25]. We thus tested Stx1 effects on galectin-3 mRNA expression. T84 cells were incubated apically for 24 h with 200 ng/ml or 400 ng/ml of Stx1. The non-catalytic B-subunit of Stx1 (Stx1B, 1 μg/ml) was used as a negative control. We used TNF-α as a positive control for galectin-3 mRNA downregulation in T84 cells, since in Crohn’s disease galectin-3 expression has been shown to be markedly reduced in enterocytes due to inhibition of galectin-3 mRNA synthesis by TNF-α [14]. Neither Stx1 nor Stx1B affected galectin-3 mRNA expression in T84 cells. By comparison, basolateral treatment of T84 monolayers for 24 h with TNF-α (10 ng/ml or 50 ng/ml) decreased the galectin-3 mRNA levels by 7± 3% (p=0.07, n=3) and 22 ±5 % respectively (p<0.05, n =3) compared to non-treated cells. Neither Stx1 nor TNF-α affected GAPDH mRNA levels. We conclude that the Stx1-induced decrease in galectin-3 does not occur due to a decrease in galectin-3 mRNA.
To confirm that Stx1 and TNF-α decrease intracellular galectin-3 protein levels by different mechanisms, we studied the effect of the combination of Stx1 and TNF-α vs. TNF-α alone (Fig. 1D). As expected, 50 ng/ml TNF-α decreased the amount of intracellular galectin-3 (compared to non-treated cells) by ~ 25%, resembling the decreases in galectin-3 mRNA. The combination of Stx1 and TNF-α resulted in even larger decreases in the levels of galectin-3 protein. These data indicate that besides a decrease in galectin-3 synthesis by the TNF-α-mediated effect on its mRNA, Stx1 interaction with T84 cells has additional effects on galectin-3 expression that cannot be attributed to inhibition of protein synthesis per se.
Stx1 uptake by IEC stimulates apical galectin-3 secretion
Since galectin-3 is secreted from many types of cells [26,27,11] we next tested the possibility that Stx1 uptake by IEC might increase galectin-3 secretion and thereby diminish its intracellular content. We incubated T84 monolayers with Stx1-680 (200 ng/ml apically) and measured both Stx1 uptake and the relative amount of galectin-3 over time in conditioned medium (both apical and basolateral) by immunoblotting.
The intracellular content of Stx1 in T84 cells, which reflects endocytosed toxin and is quantified by Stx1-680 fluorescence intensity, increased over 48 h of incubation (Fig. 2A and B). This Stx1 uptake was accompanied by significantly increased galectin-3 levels in apical conditioned medium (Fig. 2C and D). This latter increase was not due to cell death mediated by Stx1, because the cytosolic protein GAPDH was undetectable in conditioned media from both control cells and cells incubated with Stx1. In contrast, apical Stx1 at levels of up to 0.4 μg/ml Stx1 for 48 h did not alter basolateral galectin-3 secretion. In basolateral conditioned medium, galectin-3 levels were undetectable in both control and Stx1-treated cells. The relationship between apical Stx1 concentration and amount of galectin-3 secreted into the apical medium was dose dependent (Supplemental Fig. 3A and B).
Figure 2. Galectin-3 is apically secreted from enterocytes in response to Stx1 uptake in both in vitro cell model and in EHEC-infected patients.

(A) Image of Stx1 uptake by the T84 cells measured as fluorescence intensity of Stx1-680 in 100 μg of protein from total cell lysates over incubation time.
(B) Quantification of time dependency of Stx1 uptake by T84 cells (Stx1 apical concentration 0.2 μg/ml). *Statistically significant difference (p<0.05) compared to Stx1 uptake at 6 h.
(C) Immunoblot of apically secreted galectin-3 over time in 100 μl conditioned media.
(D) Time dependency of galectin-3 secretion in response to apical 0.2 μg/ml Stx1calculated from immunoblots and normalized per amount of galectin-3 secreted by non-treated T84 cells at 24 h (n= 4 experiments for each time point). * Statistically significant difference (p<0.05) compared to galectin-3 secretion at 48 h in control cells.
(E) Representative immunoblot of galectin-3 and DPPIV recovered from proteins in stool filtrates from healthy controls and EHEC-infected individuals.
Stx1 represents a family of AB5 toxins [1,3]. An unrelated AB5 toxin, cholera toxin, was tested in the same model to determine the specificity of Stx1 effect on stimulation of galectin-3 secretion. Incubation of T84 cells with CTB (1 μg/ml) over 48 h affected neither apical nor basolateral secretion of galectin-3. Thus, apical level of galectin-3 in control cells was 1.3±0.3 and 1.0±0.4 in CTB-treated cells. Stx1 uptake by T84 cells is receptor-independent [25,22,9,8]. To further prove the specificity of Stx1-induced galectin-3 secretion, we tested whether uptake of HRP, a large molecular weight fluid phase marker also stimulates galectin-3 secretion. Incubation of T84 cells with 5 μg/ml HRP up to 48 h influenced neither apical nor basolateral secretion of galectin-3 from T84 cells. Apical galectin-3 level was 1.4±0.2 in HRP-treated cells. Galectin-3 was below detection level in basolateral medium in control cells and in cells treated with CTB or HRP.
Taken together, these data demonstrate that Stx1 specifically decreases the amount of galectin-3 in IEC by increasing apical galectin-3 secretion.
Secreted galectin-3 is detected in stool filtrates from patients infected with Stx1 and Stx2-producing bacteria
To determine whether galectin-3 secretion occurs in human infections with Stx-producing bacteria, the stool concentrations of galectin-3 were examined in patients with EHEC. For these experiments, stool filtrates (n=17) from a cohort of patients with documented infection with Stx-producing E. coli O157:H7 were analyzed by galectin-3 immunoblots. In parallel, 17 samples of stool filtrates from healthy individuals (matched by age) were also examined. Galectin-3 was detected in all seven stool filtrates collected on or before day 5 of illness with E. coli O157:H7 (Fig. 2E, Supplemental Table 1), and less consistently later in illness (n=2). Galectin-3 was not detected in any of seventeen normal samples (Fig. 2E). Presence of galectin-3 in stools from EHEC infected patients was not related simply to cell death, but represented secretion, because the apical protease DPPIV [28] was readily detectable in filtrates from healthy uninfected controls, but was below detection level in stools from EHEC-infected patients.
Galectin-3 depletion in Stx1-induced injury of enterocytes leads to the mistargeting of brush border proteins
Lack of galectin-3 has recently been shown [29] to impair the trafficking of several brush border membrane proteins. DPPIV and actin binding proteins ezrin and villin were missorted to the basolateral membranes in ileal villus cells of galectin-3 null mice. These results, taken together with our observation that DPPIV is absent from the stools of EHEC-infected patients, led us to hypothesize that the acute loss of intracellular galectin-3 by enterocytes due to Stx1 uptake might also lead to missorting of apical proteins and thus contribute to Stx1-induced intestinal epithelial pathology. To test this hypothesis we compared the expression and intracellular distribution of the apical membrane proteins DPPIV, villin, and NHE2 in control T84 cells vs. T84 cells treated with Stx1. Exposure of T84 cells to Stx1 for 24 h caused substantial re-distribution of all three tested proteins from the apical to the basolateral region (Fig. 3).
Figure 3. Stx1 interactions with T84 cells cause mistargeting of apical membrane proteins DPPIV, villin and NHE2 into the basolateral compartments through the loss of intracellular galectin-3.

Single XY projections from 3D reconstruction and optical XZ projections of intracellular distribution of protein of interest (in green by immunostaining with corresponding primary antibody and Alexa Fluor 488 secondary antibody). Cell nuclei (blue) labeled by Hoechst.
To test whether this shift in distribution of apical membrane proteins reflects the loss of intracellular galectin-3, we depleted galectin-3 from T84 cells using three different galectin-3 specific shRNA (shGal-3) sequences that we delivered to the cells using lentiviruses (Supplemental Methods). All three shRNA constructs significantly decreased galectin-3 expression in T84 cells (Supplemental Fig. 4). The intracellular distribution of DPPIV, NHE2, and villin was altered in galectin-3 depleted cells compared to control T84 cells. All three proteins were present basolaterally similarly to these in T84 cells exposed to Stx1 (Fig. 3). In contrast to these brush border proteins, the basolateral EGF receptor (EGFR, Supplemental Fig. 5A and B) and the tight junctional protein ZO-1 (data not shown) displayed its normal distribution patterns in T84 cells that were treated with Stx1 or depleted of galectin-3. The expression level of these proteins as examined by immunoblotting did not change either in Stx1-exposed cells or in galectin-3-depleted cells (Supplemental Fig. 5C–F).
To further confirm that impaired sorting of these brush border proteins due to Stx1-induced loss of galectin-3 is a characteristic feature of human enterocytes in EHEC-induced disease, colonic tissue samples from patients with EHEC infection were examined by immunofluorescence for expression of DPPIV, NHE2, and villin. The distribution of all three proteins in tissue from EHEC-infected patients was greatly altered in comparison to distribution identified in normal control colonic samples (Fig. 4). NHE2 was absent from the brush border of colonocytes in EHEC-infected patients in both surface and crypt cells. NHE2 was present in intracellular vesicles in surface cells and was laterally distributed in crypt cells (Fig. 4A–D). DPPIV, which is apically expressed in normal human enterocytes (Fig. 4E), was distributed all through the cytoplasm and basolateral part of these cells in patients with EHEC infection (Fig. 4,F). Villin was largely present intracellularly in EHEC infection, and not on the apical surface where it is found in normal colonic tissue (Fig. 4G,H).
Figure 4. NHE3, DPPIV and villin are mistargeted from apical surface to the basolateral compartments in IEC from patients infected with Stx1 producing EHEC bacteria.

Representative immunofluorescence images of NHE2 distribution (green) in (A) normal surface colonic tissue; (B) in colonic surface in EHEC-infection, WGA (red) to outline the apical surface of enterocytes; (C) NHE2 (green) apical distribution in normal crypt epithelial cells and (D) in crypt colonocytes from EHEC-infected patient, WGA (red) to outline the apical surface of enterocytes; (E) apical distribution of DPPIV (green) in normal human tissue and (F) intracellular pattern of DPPIV in IEC from EHEC-infected patient; (G) apical distribution of villin (red) in normal human tissue and (H) villin intracellular pattern in IEC from EHEC-infected patient. Nuclei (blue) by Hoechst in all panels.
We additionally examined cecal tissue from rabbits infected with RDEC-1 (control) vs. Stx1-producing RDEC-H19A bacteria for changes in the distribution of DPPIV and NHE2. Stx1-induced loss of galectin-3 from cecal enterocytes was accompanied by loss of DPPIV from the apical membranes, similar to this in human enterocytes and T84 cells (Supplemental Fig. 6). NHE2 was detected neither in control nor in Stx1-exposed cecal tissue samples. We believe that this may have been due to the absence of NHE2 from this part of rabbit intestine or to the features of antibodies that we used. However, NHE3, another member of sodium/proton exchanger family [30], was targeted to cytosol in cecal tissue from rabbits infected with Stx1-producing RDEC-H19A bacteria compared with its predominantly apical pattern in tissue from RDEC-1 infected animals (Supplemental Fig. 6).
NHE2-mediated sodium absorption is significantly inhibited due to the loss of galectin-3 from IEC
NHE2 is the major colonic apical sodium absorptive protein. Its loss from the apical surface of colonocytes in EHEC infection might contribute to the Stx1-induced watery diarrhea. To test whether NHE2 missorting led to the decrease in the sodium absorption by colonocytes, we compared NHE2 activity in T84 cells exposed to Stx1 to that in cells in which shRNA treatment depleted galectin-3 and to that in control cells with apically expressed NHE2 (Supplemental Methods). Mistargeting of NHE2 due to Stx1 uptake significantly inhibited the rate of Na+/H+ exchange mediated by NHE2 (Fig. 5). Of note, NHE3 is absent from T84 cells [31]. Depletion of galectin-3 by shRNA which mislocates NHE2 was also sufficient to inhibit NHE2 activity (Fig. 5).
Figure 5. Loss of galectin-3 due to Stx1 uptake or by shRNA significantly inhibits NHE2 activity in T84 cells.

(A) Patterns of Na-dependent intracellular alkalinization in control T84 cells, or cells treated for 24 h with 200 ng/ml of Stx1, or with depleted galectin-3.
(B) Rate of Na/H exchange activity (DpH/min) was significantly (*p<0.05) higher in control conditions compared with this in cells treated either with 200 ng/ml of Stx1 or with depleted galectin-3; n=3 experiments per each condition.
We conclude that the Stx1-mediated loss of galectin-3 from the enterocytes is likely to play a substantial role in EHEC-induced intestinal disease. The loss of galectin-3 causes the mistargeting of some brush border proteins and significantly inhibits sodium absorption in ways that are likely to contribute to Stx-induced diarrhea.
Discussion
Here we demonstrate that Shiga toxins produced in EHEC infection cause the depletion of galectin-3 from IEC. We used both in vivo and in vitro models to determine the mechanisms of galectin-3 depletion from the enterocyes due to EHEC infection and found that Stx-induced apical secretion of galectin-3 is mainly responsible. This is in contrast to chronic inflammatory intestinal disease where loss of galectin-3 from enterocytes has been mostly associated with TNF-α induced downregulation of galectin-3 mRNA [14,15].
Several galectins, including galectin-3, are secreted by ectocytosis, a mechanism that differs from the classical secretory vesicular trafficking through the endoplasmic reticulum and Golgi and is independent of a recycling pathway [26,27,32]. Galectin-3 is usually secreted in small amounts towards the apical (lumenal) rather than the basal surface [26]. The mechanism of Stx1 stimulation of galectin-3 secretion has yet to be elucidated. We have recently shown that macropinocytosis, an actin-driven endocytic process, is likely to be responsible for Stx uptake by enterocytes in the absence of Gb3 receptors [22]. Thus, we speculate that Stx1 macropinocytosis might stimulate apical ectocytosis and an ensemble of proteins, including galectin-3, might then be released.
Secretion of galectin-3 is not unique to EHEC infection. Adhesion of Helicobacter pylori to gastric lining stimulates galectin-3 secretion by gastric epithelial cells [33], indicating that loss of galectin-3 from enterocytes might represent a common step of damage of IEC by bacterial infections and bacterial products in acute and chronic conditions.
Galectin-3 is a lactose-binding multifunctional protein [11] and its intracellular depletion might compromise IEC homeostasis in several ways. A study in galectin-3 null mice demonstrated that galectin-3 plays an important role in trafficking of many brush border proteins and also in development of the brush border cytoskeletal architecture [34,29]. Our data presented here are in good agreement with these findings and further show the pathophysiologic consequences of the loss of galectin-3 from IEC. Namely Stx1-induced loss of galectin-3 from the enterocytes leads to the missorting of several apical membrane proteins, including DPPIV, villin and NHE2 to the basolateral and cytoplasmic compartments in vitro and in vivo. It has been previously shown that purified Stx2 was able to inhibit water absorption across the human colonic mucosa without affecting the electrical parameters [35]. Inhibition of electroneutral colonic Na+/H+ exchange by mistargeting NHE2 might be responsible for such changes in water absorption and consequent diarrhea. Our finding that secreted galectin-3 appears in human stool samples early in the infection which corresponds to the stage of watery diarrhea strongly support our data that depletion of galection-3 from enterocytes by Stx uptake contributes to the watery diarrhea. Mislocalization of DPPIV, an important hydrolase in the digestion of dietary proline-rich proteins, might significantly compromise the digestive functions of enterocytes. Thus, mistargeting of both NHE2 and DPPIV might contribute to the EHEC-induced diarrheal disease. Intracellular and basolateral distribution of DPPIV might explain the absence of this usually lumenaly secreted protein in stools from EHEC-infected patients. Interestingly, the missorting of DPPIV to the basolateral membrane has also been found in rotavirus infected polarized intestinal epithelial Caco-2 cells [36]. Similarly to Stx1 uptake, rotaviral infection causes significant changes in the distribution of brush border-associated cytoskeletal proteins in Caco-2 cells, including villin, without changes in their mRNA level [36,37]. Although it is not known, whether the misplacement of apical proteins in rotaviral infection is a result of loss of galectin-3, this similarity in apical protein missorting due to different enteric infections further support its role in the development of absorptive diarrhea.
In conclusion, data presented above demonstrate that Stx1 upon its release by EHEC bacteria into the gut lumen has direct impact on IEC independent of later development of ischemia and inflammation from endothelial effects. Stx-stimulated depletion of galectin-3 at the early stages of Stx-induced illness that leads to missorting of apical structural and absorptive proteins might contribute to the EHEC-induced watery diarrhea. Loss of galectin-3 from the enterocytes might be a common step in infectious absorptive diarrheal diseases.
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1DK58928, R24DK064388, The Johns Hopkins Basic Research Digestive Disease Development Core Center, DK52081, and P30DK 034928. We acknowledge the Ross Confocal Facility and assistance of Mr. John Gibas, Mouse Physiology Core and the assistance of Mrs. Jennifer Sipes, human tissue and stool sample handling of Jody L. Mooney, Sandra L. Watkins, Eileen J. Klein, Donna M. Denno, and Kathleen Patterson.
Abbreviations
- Stx
Shiga toxin
- Stx1B
B-subunit of Shiga toxin 1
- EHEC
enterohemorrhagic Escherichia coli
- Gb3
globotriaosylceramide
- TNF-α
tumor necrosis factor α
- CTB
B-subunit of cholera toxin
- HRP
horse radish peroxidase
- DPPIV
dipeptidylpeptidase IV
- RT-PCR
reverse-transcription polymerase chain reaction
- IEC
intestinal epithelial cells
Footnotes
Conflict of interests: The authors disclose that Dr. Mark Donowitz is owner of Tranzmembrane Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Serna A, 4th, Boedeker EC. Pathogenesis and treatment of Shiga toxin-producing Escherichia coli infections. Curr Opin Gastroenterol. 2008;24:38–47. doi: 10.1097/MOG.0b013e3282f2dfb8. [DOI] [PubMed] [Google Scholar]
- 2.Center for Disease Control and Prevention. Ongoing multistate outbreak of Escherichia coli serotype O157:H7 infections associated with consumption of fresh spinach--United States, September 2006. Morb Mortal Wkly Rep. 2006;55:1045–1046. [PubMed] [Google Scholar]
- 3.Sandvig K, Van Deurs B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol Review. 1996;76:949–966. doi: 10.1152/physrev.1996.76.4.949. [DOI] [PubMed] [Google Scholar]
- 4.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 5.Tarr PI, Watkins SL, Neill MA. Risk of hemolytic uremic syndrome from antibiotic treatment of Escherichia coli O157:H7 colitis. JAMA. 2002;288:3111–3112. doi: 10.1001/jama.288.24.3111-jlt1225-2-3. [DOI] [PubMed] [Google Scholar]
- 6.Bielaszewska M, Karch H. Consequences of enterohaemorrhagic Escherichia coli infection for the vascular endothelium. Thromb Haemost. 2005;94:312–318. doi: 10.1160/TH05-04-0265. [DOI] [PubMed] [Google Scholar]
- 7.Ergonul Z, Clayton F, Fogo AB, Kohan DE. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr Nephrol. 2003;18:246–253. doi: 10.1007/s00467-002-1025-9. [DOI] [PubMed] [Google Scholar]
- 8.Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc Natl Acad Sci USA. 2005;102:19087–19092. doi: 10.1073/pnas.0506474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schüller S, Frankel G, Phillips AD. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004;6:289–301. doi: 10.1046/j.1462-5822.2004.00370.x. [DOI] [PubMed] [Google Scholar]
- 10.Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Atca. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Vasta GR. Role of galectins in infection. Nature Rev Microbiology. 2009;7:424–438. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huflejt MI, Jordan ET, Gitt MA, Barondes SH, Leffer H. Strikingly different localization of galectin-3 and galectin-4 in human colon adenocarcinoma T84 cells. Galectin-4 is localized at sites of cell adhesion. J Biol Chem. 1997;272:14294–14303. doi: 10.1074/jbc.272.22.14294. [DOI] [PubMed] [Google Scholar]
- 13.Nio J, Kon Y, Iwanaga TJ. Differential cellular expression of galectin family mRNAs in the epithelial cells of the mouse digestive tract. J Histochem Cytochem. 2005;53:1323–1334. doi: 10.1369/jhc.5A6685.2005. [DOI] [PubMed] [Google Scholar]
- 14.Jensen-Jarolim E, Gscheidlinger R, Oberhuber G, Neuchrist C, Lucas T, Bises G, Radauer C, Willheim M, Scheiner O, Liu FT, Boltz-Nitulescu G. The constitutive expression of galectin-3 is downregulated in the intestinal epithelia of Crohn’s disease patients, and tumour necrosis factor alpha decreases the level of galectin-3-specific mRNA in HCT-8 cells. Eur J Gastroenterol Hepatol. 2002;14:145–152. doi: 10.1097/00042737-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Muller S, Schaffer T, Flogerzi B, Fleetwood A, Weimann R, Schoepfer AM, Seibold F. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm Bowel Dis. 2006;12:588–597. doi: 10.1097/01.MIB.0000225341.37226.7c. [DOI] [PubMed] [Google Scholar]
- 16.Murray KF, Patterson K. Escherichia coli 0157:H7-induced hemolytic-uremic syndrome: histopathologic changes in the colon over time. Pediatr Dev Pathol. 2000;3:232–239. doi: 10.1007/s100249910030. [DOI] [PubMed] [Google Scholar]
- 17.Cornick NA, Jelacic S, Ciol MA, Tarr PI. Escherichia coli O157:H7 infections: discordance between filterable fecal shiga toxin and disease outcome. J Infect Dis. 2002;186:57–63. doi: 10.1086/341295. [DOI] [PubMed] [Google Scholar]
- 18.Lathem WW, Grays TT, Witowski SE, Torres AG, Kaper JB, Tarr PI, Welch RA. StcE, a metalloprotease secreted by Escherichia coli O157:H7, specifically cleaves C1 esterase inhibitor. Mol Microbiol. 2002;45:277–288. doi: 10.1046/j.1365-2958.2002.02997.x. [DOI] [PubMed] [Google Scholar]
- 19.Sjogren R, Neill R, Rachmilewitz D, Fritz D, Newland J, Sharpnack D, Colleton C, Fondacaro J, Gemski P, Boedeker E. Role of Shiga-like toxin I in bacterial enteritis: comparison between isogenic Escherichia coli strains induced in rabbits. Gastroenterology. 1994;106:306–317. doi: 10.1016/0016-5085(94)90587-8. [DOI] [PubMed] [Google Scholar]
- 20.Schwaderer AL, Vijayakumar S, Al-Awqati Q, Schwartz GJ. Galectin-3 expression is induced in renal beta-intercalated cells during metabolic acidosis. Am J Physiol Renal Physiol. 2006;290:F148–158. doi: 10.1152/ajprenal.00244.2005. [DOI] [PubMed] [Google Scholar]
- 21.Kovbasnjuk O, Edidin M, Donowitz M. Role of lipid rafts in Shiga toxin 1 interaction with apical surface of CaCo-2 cells. J Cell Sci. 2001;114:114, 4025–4031. doi: 10.1242/jcs.114.22.4025. [DOI] [PubMed] [Google Scholar]
- 22.Malyukova I, Murray KF, Zhu C, Boedeker E, Kane A, Patterson K, Peterson JR, Donowitz M, Kovbasnjuk O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am J Physiol Gastrointest and Liver physiology. 2009;296:G78–92. doi: 10.1152/ajpgi.90347.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murtazina R, Kovbasnjuk O, Zachos N, Li X, Chen Y, Hubbard A, Hogema BM, Steplock D, Seidler U, Hoque KM, Tse CM, De Jonge HR, Weinman EJ, Donowitz M. Tissue specific regulation of NHE3 activity in NHERF1 null mice: cAMP inhibition is differentially dependent on NHERF1 and EPAC in ileum versus proximal tubule. J Biol Chem. 2007;282:25141–25151. doi: 10.1074/jbc.M701910200. [DOI] [PubMed] [Google Scholar]
- 24.Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na+/H+ exchanger. Eur J Med Chem. 2003;38:547–554. doi: 10.1016/s0223-5234(03)00100-4. [DOI] [PubMed] [Google Scholar]
- 25.Jones NL, Islur A, Haq R, Mascarenhas M, Karmali MA, Perdue MH, Zanke BW, Sherman PM. Escherichia coli Shiga toxins induce apoptosis in epithelial cells that is regulated by the Bcl-2 family. Am J Physiol Gastrointest Liver Physiol. 2000;278:G811–819. doi: 10.1152/ajpgi.2000.278.5.G811. [DOI] [PubMed] [Google Scholar]
- 26.Lindstedt R, Apodaca G, Barondes SH, Mostov KE, Leffler H. Apical secretion of a cytosolic protein by Madin-Darby canine kidney cells. Evidence for polarized release of an endogenous lectin by a nonclassical secretory pathway. J Biol Chem. 1993;268:211750–11757. [PubMed] [Google Scholar]
- 27.Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005;27:375–387. doi: 10.1007/s00281-005-0004-1. [DOI] [PubMed] [Google Scholar]
- 28.Slimane TA, Lenoir C, Sapin C, Maurice M, Trugnan G. Apical secretion and sialylation of soluble dipeptidyl peptidase IV are two related events. Exp Cell Res. 2000;258:184–194. doi: 10.1006/excr.2000.4894. [DOI] [PubMed] [Google Scholar]
- 29.Delacour D, Cramm-Behrens CI, Drobecq H, Le Bivic A, Naim HY, Jacob R. Loss of galectin-3 impairs membrane polarisation of mouse enterocytes in vivo. J Cell Sci. 2008;121:458–465. doi: 10.1242/jcs.020800. [DOI] [PubMed] [Google Scholar]
- 30.Donowitz M, Mohan S, Zhu CX, Chen TE, Lin R, Cha B, Zachos NC, Murtazina R, Sarker R, Li X. NHE3 regulatory complexes. J Exp Biol. 2009;212:1638–1646. doi: 10.1242/jeb.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beltran AR, Ramirez MA, Carraro-Lacroix LR, Hiraki Y, Reboucas NA, Malnic G. NHE1, NHE2, and NHE4 contribute to regulation of cell pH in T84 colon cancer cells. Pflugers Arch. 2008;455:799–810. doi: 10.1007/s00424-007-0333-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhu WQ, Ochieng J. Rapid release of intracellular galectin-3 from breast carcinoma cells by fetuin. Cancer Res. 2001;61:1869–1873. [PubMed] [Google Scholar]
- 33.Fowler M, Thomas RJ, Atherton J, Roberts IS, High NJ. Galectin-3 binds to Helicobacter pylori O-antigen: it is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell Microbiol. 2006;8:44–54. doi: 10.1111/j.1462-5822.2005.00599.x. [DOI] [PubMed] [Google Scholar]
- 34.Delacour D, Cramm-Behrens CI, Drobecq H, Le Bivic A, Naim HY, Jacob R. Requirement for galectin-3 in apical protein sorting. Curr Biol. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 35.Fiorito P, Burgos JM, Miyakawa MF, Rivas M, Chillemi G, Berkowski D, Zotta E, Silberstein C, Ibarra C. Effect of Shiga toxin 2 on water and ion transport in human colon in vitro. Dig Dis Sci. 2000;45:480–486. doi: 10.1023/a:1005480720832. [DOI] [PubMed] [Google Scholar]
- 36.Jourdan N, Brunet JP, Sapin C, Blais A, Cotte-Laffitte J, Forestier F, Quero AM, Trugnan G, Servin AL. Rotavirus infection reduces sucrase-isomaltase expression in human intestinal epithelial cells by perturbing protein targeting and organization of microvillar cytoskeleton. J Virol. 1998;72:7228–7236. doi: 10.1128/jvi.72.9.7228-7236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorrot M, Vasseur M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol J. 2007;21:4, 31. doi: 10.1186/1743-422X-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.