

Abstract
Intellectual disability (ID) in Down syndrome (DS) ranges from low normal to severely impaired, and has a significant impact on the quality of life of the individuals affected and their families. Because the incidence of DS remains at approximately one in 700 live births and the life span is now >50 years, development of pharmacotherapies for cognitive deficits is an important goal. DS is due to an extra copy of human chromosome 21 and has often been considered too complex a genetic abnormality to be amenable to intervention. However, recent successes in rescuing learning/memory impairments in a mouse model of DS suggest that this negative outlook may not be justified. In this article, we first review the DS phenotype, chromosome 21 gene content and mouse models, and then discuss recent successes and remaining challenges in the identification of targets for and preclinical evaluation of potential therapeutics.
Introduction
Down syndrome is the most common genetic cause of intellectual disability [1]. It is due to an extra copy of the long arm of human chromosome 21 (HSA21q), and the resulting increase in expression due to gene dosage of the trisomic genes. Brain morphology in DS at birth is largely normal, but postnatal development slows, resulting in reduced volumes of the hippocampus, cerebellum and prefrontal cortex, reduced neuronal densities in specific regions that include the hippocampus and cerebellum, and reduced dendritic branching and spine densities in the hippocampus [2,3]. Cognitive abilities in early infancy are within the lower range of typical development, but again there are decreases over the first decade. Resulting deficits in spatial learning tasks implicate the hippocampus, specific weaknesses in language skills implicate regions of the prefrontal and temporal cortices and the cerebellum, and additional deficits suggest impaired prefrontal cortex function [2-5]. As the brain ages, features similar to those seen in Alzheimer's Disease (AD) develop, including reduced size of, and loss of functional markers in, the basal forebrain cholinergic neurons (BFCNs) that are required for cholinergic input to the hippocampus. Deposition of Aβ has been documented in brains of DS at young ages, AD-like plaques and tangles are universally seen by the age of 30, and an AD-like dementia develops in approximately half of the people with DS over the age of 50 [2, 6]. The postnatal appearance of many abnormalities argues for the potential for therapeutic interventions.
The working hypothesis in DS research is that the phenotypic features are caused by the modest 50% increases in expression of trisomic genes and that these in turn result in modest perturbations of otherwise normal cell functions and pathways. A specific challenge is to identify the subset(s) of HSA21q genes that make major contributions to cognitive deficits and then to identify the most effective targets and timing for controlling the consequences of their overexpression. The complete genomic sequence of HSA21q was completed in 2000 [7], but annotation for gene discovery continues. Our recent review identified >500 genes (Table 1). Only ∼40% are annotated as RefSeqP genes, that is protein coding genes curated in the GenBank and SwissProt databases, and they include a wide diversity of functional classes [8,9]. Based on evolutionary conservation in chimpanzee and mouse orthologous genomic regions, a further ∼30% are unlikely to code for protein (unpublished data). Thus, predicting candidates for causing ID or targets of therapies remains a challenge. Notably, the justification for focusing on the genes within the “Down Syndrome Critical Region” (DSCR) or on the functional synergy between the HSA21q genes DYRK1A and DSCR1 (RCAN1) has been invalidated again (following on from prior work in [10]) by recent analysis of DS due to partial trisomy 21 [11]. This study showed that several cases of DS with significant ID, as measured by an IQ of <50, were not trisomic for these genes. A related work [12] reached similar conclusions.
Table 1.
Overview of HSA21q gene classifications
| Open reading frame characteristics [18; unpublished data] | # genes |
|---|---|
| Protein coding genes in the GenBank RefSeq and SwissProt databases | 164 |
| Keratin-associated protein genes | 43 |
| Novel genes with open reading frames >50 amino acids in length and conserved in chimpanzee | 140 |
| Novel genes lacking open reading frames >50 amino acids in length conserved in chimpanzee | 160 |
| Examples of RefSeqP gene functional classes* [8,9] | |
| Transcription factors/modulators | 18 |
| Protein processing | 10 |
| Cell adhesion/junctions | 8 |
| RNA processing | 9 |
| Mitochondrial function | 7 |
| Reactive oxygen species | 10 |
| MAP kinase and calcineurin pathway interactions | 13 |
| K, Cl, Ca channel subunits | 6 |
, individual genes may belong to more than one class.
In this review, we discuss the identification of targets and testing of potential pharmacoptherapies for cognitive deficits in DS. We describe the phenotypic features and gene content of mouse models, and how these are influencing choices of and responses to drug treatments. We highlight the limitations of mouse as a model system for DS and propose alternatives and additions.
Mouse models of DS: genes and phenotypic features
HSA21q RefSeqP genes are conserved in orthologous regions of mouse chromosomes 16, 17 and 10 (MMU16, MMU17 and MMU10) (Figure 1). This multi-chromosomal distribution has so far prevented the development of a mouse model that is trisomic for all orthologs of HSA21q genes. Partial trisomies have, however, been created. They include several for segments of the MMU16 region, one for the MMU17 region, and a mosaic model carrying an almost complete human chromosome 21 [13-17]. Figure 1 and Table 2 describe the genomic regions and gene content of each model. Note that whereas ∼500 genes have been annotated in HSA21q, ∼425 genes have been annotated in the orthologous mouse chromosomal regions, and both lists include many species-specific genes [18; unpublished data]. Thus, for simplicity, we have shown only RefSeqP gene content. Together, these mouse models provide, in varying complements, trisomy of almost all HSA21q RefSeqP orthologs. Table 2 also summarizes the results of behavioral testing which has focused on assessment of hippocampal function (19,20,21; see Box 1). Although it is clear that trisomic gene content influences behavioral abnormalities, genotype-phenotype correlations are not simple. This is illustrated by comparing data from the Ts65Dn, Ts1Cje and Ts1Rhr models, which are trisomic for progressively smaller but completely overlapping sets of genes. Whereas performance in the Morris Water Maze (MWM) is consistent with trisomic gene content, performance in Novel Object Recognition (NOR) appears inconsistent [22,23]. In addition, the Ts1Yah, which is not trisomic for MMU16 genes but rather for twelve genes in the MMU17 region, also displays deficits in NOR; conversely it displays enhanced learning in the MWM [16]. The apparently milder phenotype of the Tc1 model may be due to its mosaicism and/or to internal deletions in HSA21q, leaving significant genes (e.g. ITSN1, RCAN1) disomic [17,24].
Figure 1. HSA21q RefSeqP gene distribution in the major mouse models of DS.
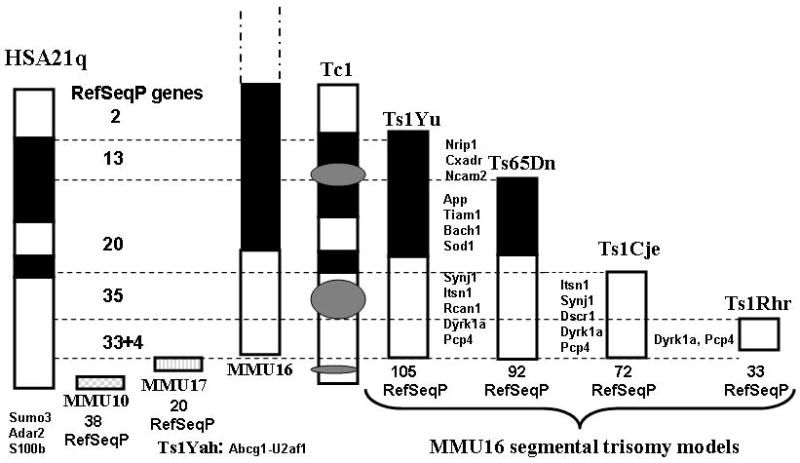
A schematic of HSA21q is shown at the left. Schematics of the orthologous segments of MMU10, 17 and 16, and of the trisomic regions of the major mouse models of DS are shown approximately to scale [13-17]. Dashed horizontal lines indicate regions of HSA21q defined by the differing trisomic segments in MMU16 models. Numbers refer to RefSeqP genes within each region. Locations of representative RefSeqP genes are indicated [8,9,18].
Table 2.
Segmental trisomy mouse models of DS
| Mouse model | [Ref] | Chr | # Mb | aRefSeqP | bBehavior | cElectrophysiology; cellular | |||
|---|---|---|---|---|---|---|---|---|---|
| # | Boundaries | Deficits | Normal | Abnormal | Normal | ||||
| Ts1Yu | [14] | MMU16 | 104 | Lip1-Znf295 | ND | ND | |||
| Ts65Dn | [13] | MMU16 | 16 | 94 | Mrpl39-Znf295 | MWM, CFC, NOR, TM | LTP, LTD, BFCN, CB-GC | ||
| Ts1Cje | [13, 22] | MMU16 | 8 | 75 | >Sod1-Znf295 | MWM, CFC, TM | NOR | LTP, CB-GC | BFCN |
| Ts1Rhr | [15, 23] | MMU16 | 3 | 35 | Cbr1-M×2 | NOR, TM | MWM | LTP | LTP-CA1 CB-GC |
| Ts1Yah | [16] | MMU17 | 0.5 | 12 | Abcg1-U2af1 | NOR | MWM* | LTP* | |
| Tc1 | [17, 24] | HSA21 | ∼30 | DEL: CXADR; IFNGR1, ITSN1, KCNE1, RCAN1 | NOR-stm, MWM-stm | NOR-Ltm, MWM-Ltm; TM | LTP-1; CB-GC | LTP-24 | |
RefSeqP: # of curated protein coding genes in the orthologous region of HSA21q. Boundaries: genes at the centromeric and telomeric ends of the trisomic segment.
Behavior: see Box 1. Stm, short term memory; Ltm, long term memory.
LTP, long term potentiation; LTP-1, LTP-24, LTP measured at one and 24 hours after initial stimulation. LTD, long term depression; BFCN, basal forebrain cholinergic neurons; CB-GC, cerebellar granule cells.
, enhanced compared with euploid controls.
Box 1.
Behavioral tests for hippocampal function
Morris Water Maze (MWM)
During training sessions over several days, mice are repeatedly placed in a large pool of water and swim until they locate a submerged platform which allows them to escape the water. Mice cannot see the platform because the water is made opaque using a dye, and they must learn the location using visual cues on the walls of the pool or room. One measure of learning is the decrease in time and path length required to reach the platform with increased numbers of trials. A second measure of learning is the “probe trial”, in which the platform is removed from the pool and the proportion of time the animal spends searching in the correct quadrant of the pool is recorded.
Novel Object Recognition (NOR)
During training sessions, mice are allowed to explore two objects in an open field. For the test session, one of the two objects is replaced with a novel object. Memory is measured as the proportion of time the animal spends investigating the novel object versus the familiar object.
Context Fear Conditioning (CFC)
The mouse is placed in a novel context (chamber) and allowed to explore for a few minutes. The mouse then receives an electric shock and is returned to the home cage. Learning and memory of the context are measured by returning the mouse to the shock chamber for a short time, and recording the proportion of the time the mouse “freezes” (cessation of all movement except breathing).
T-Maze (TM) and Y-Maze (YM)
T- and Y-mazes are composed of three arms and measure the innate preference of an animal to spontaneously alternate arm entries in order to explore an arm that has not been explored in the previous trial. Memory is measured as the proportion of successive entries into a different arm relative to the total number of opportunities for choice.
Water Radial Arm Maze (water-RAM)
The apparatus is composed of an eight-armed radial maze submerged in water. Four of the eight arms have submerged escape platforms. During training the mouse is placed in a starting arm and swims to find an arm that contains a platform. With repeated training, the mouse uses visual cues within the room to learn the arm-locations of escape platforms. The number of correct arm entries to total arm entries is used as a measure of learning.
T65Dn is the best-studied model of DS. Additional phenotypic features of this model with direct correlates to observations in DS include abnormalities in dendritic spine number and morphology, abnormalities in neuron number in hippocampal regions, degeneration of BFCN and decreased cerebellar volume due to decreased granule and molecular cell numbers [reviewed in 3,13; 25,26]. Electrophysiological studies, as measures of synaptic plasticity related to learning and memory, have demonstrated regional abnormalities in hippocampal long term potentiation (LTP) [13]. Thus, although trisomic for only about half of the HSA21q genes, these mice are nevertheless a useful model of many neurological features of DS. Table 2 also shows that, as with behavior, there are similarities and differences among the mouse models in BFCNs, cerebellar and electrophysiological abnormalities.
In summary, there is no perfect mouse model of DS. Compromises must be made regarding species specificities (human vs. mouse gene content), technical challenges of generating trisomic gene complements, oversimplification if too few genes are trisomic, and the true complexity of assaying effects of 500 genes, many of which interact. The different trisomic gene content of mouse models, incomplete in each case, undoubtedly affects aspects of the phenotypes that can be observed. Over-interpretation of the effects of pharmacotherapies must be avoided.
Approaches to pharmacotherapeutic target identification
Rational targets for potential pharmacotherapies include (1) overexpression of individual HSA21q genes; (2) neuroanatomic or electrophysiological abnormalities seen or predicted in DS, even though hypotheses on causative HSA21q genes are lacking; and (3) perturbations in molecular pathways relevant to DS cognitive deficits predicted from the integrated functions of HSA21q genes. Table 3 lists recent attempts, with the Ts65Dn model, using each type of target. Notably, these treatments were largely delivered postnatally, an advantage when considering the potential extension to human intervention.
Table 3.
Pharmacological and genetic therapeutic interventions using the Ts65Dn mouse model
| Phenotypic target | Treatment | Mechanism | Outcome | |
|---|---|---|---|---|
| Targeting HSA21q genes | ||||
| Abnormal endosomes; failed NGF transport; neurodegeneration | Ts65Dn × App KO | Reduce APP to disomy | Rescued; improved; rescued | [28,29] |
| LTP; L/M deficit (NOR) |
EGCG | Inhibitor of DYRK1A | Rescued | [31,32] |
| Targeting phenotypic features | ||||
| Neurodegeneration | Nerve growth factor | BFCN-HP signaling | Improved | [35] |
| L/M deficit (MWM) | Donepezil | Inhibit AchE | Failed | [47] |
| Neurodegeneration; L/M deficit (water-RAM); neuroinflammation |
Minocycline | Anti-inflammatory | Prevented; improved; reduced | [39] |
| Cerebellar granule cell deficiency | SAG 1.1 | Synthetic activator of Hedgehog pathway | Rescue | [25] |
| L/M deficit (MWM) | Piracetam | Nootropic | Failed | [33] |
| Excess inhibition; LTP; L/M deficits (NOR, TM: MWM) |
Pentylenetetrazole | Antagonist of GABAA receptor | Rescued | [47;37] |
| Targeting pathway perturbations | ||||
| Impaired adult neurogenesis | Fluoxetine | Inhibit serotonin reuptake | Rescued | [49] |
| L/M (CFC) | Memantine | Antagonist of NMDA receptor | Rescued | [55] |
| Developmental delay | NAPVSIPQ+SALLRSIPA | Neuroprotective peptides | Rescued | [59,60] |
| Oxidative stress; L/M (water-RAM); neurodegeneration |
Vitamin E | Antioxidant | Reduced; improved; prevented | [61] |
| L/M deficit (MWM) | SGS-111 (piracetam analogue) | Inhibitor of oxidative damage & apoptosis | Failed | [65] |
Targeting individual HSA21q genes
Overexpressing transgenic lines created from genomic constructs are available for the HSA21q or orthologous mouse genes encoding APP, SOD1, SIM2, SYNJ1, S100B and DYRK1A (see Table 4 for gene function descriptions) [reviewed in 13,27]. Each model displays learning and memory deficits in one or more hippocampal tasks, suggesting that amelioration of ID in DS could require regulation of expression of many HSA21q genes. To do this directly would probably require multiple drugs with attendant complications of adverse interactions. We mention two additional single gene manipulations. First, when Ts65Dn mice were crossed with a knockout of the App gene, thus reducing App to disomy, the development of some cellular abnormalities, including degeneration of BFCN, was prevented [28,29]. This genetic manipulation also rescued deficits in axonal transport of nerve growth factor (NGF), but only to levels 60% of normal, thus showing that additional genes contribute to this feature [29]. Second, activity of the DYRK1A protein kinase is inhibited by the chemical epigallocatechin (EGCG), a component of green tea [30]. Treating Ts65Dn with EGCG corrected deficits in LTP [31], and feeding green tea to a Dyrk1a single gene transgenic improved performance in NOR [32]. However, Dyrk1a is trisomic in each model listed in Table 2, yet these models vary in MWM and NOR deficits. Therefore, functional interactions exist between DYRK1A and other HSA21q proteins, indicating that reducing levels of Dyrk1a alone may not affect all hippocampus-based learning/memory tasks and is unlikely to have the same consequences in the context of a complete trisomy of HSA21q. Whether this is a serious drawback needs to be determined by analyzing additional models.
Table 4.
HSA21q-encoded protein functional interactions
| HSA21q gene | Function [8,9,45,67] | HSA21q interactors |
|---|---|---|
| MM16 | ||
| NRIP1 | Nuclear Receptor Interacting Protein; transcription modulator; represses activity of estrogen and glucocorticoid receptors; repression increased by SUMO3 modification | SUMO3 |
| APP | Amyloid Precursor Protein; mutated or duplicated in some familial AD; source of neurotoxic amyloid β (Aβ) peptide which accumulates in AD plaques, inhibits NMDA receptor activity and contributes to ROS; APP processing is altered by overexpression of SUMO3; L/M | SUMO3; SOD1 |
| BACH1 | Transcription factor; inhibits expression of oxidative responses genes, increases ROS | |
| TIAM1 | Guanine nucleotide exchange factor specific for RAC; requires phosphoinositols for membrane localization; interacts with NMDA receptor; overexpression results in abnormalities of dendritic spines | SYNJ1 |
| SOD1 | Superoxide dismutase; produces ROS; L/M | APP |
| SYNJ1 | Synaptojanin; phosphoinositol-phosphatase; synaptic vesicle endocytosis; L/M | DYRK1A, ITSN1, TIAM1 |
| ITSN1 | Intersectin; multi-domain adaptor protein; synaptic vesicle endocytosis; cytoskeletal regulation; guanine nucleotide exchange activity for cdc42; interacts with NMDA receptors; overexpression causes abnormalities of dendritic spines | SYNJ1 |
| KCNJ6 | Inwardly rectifying potassium channel; mediates activity of serotonin receptor 5HT1A and neurogenesis | |
| RCAN1 | Regulator of Calcineurin; inhibits phosphatase activity of the Ca-calmodulin dependent protein phosphatase calcineurin; activated by ROS | PCP4 |
| SIM2 | Singleminded; transcription factor; L/M | |
| DYRK1A | Protein kinase; substrates include Tau, transcription factors, pre-mRNA processing factors | SYNJ1 |
| PCP4 | Purkinje Cell Protein; inhibitor of calmodulin which is required for calcineurin activity | RCAN1 |
| MMU10 | ||
| SUMO3 | Small ubiquitin-like modifier protein; modifies target protein activity; represses transcriptional activity of ELK; increases activity of NRIP1 | NRIP1 |
| ADAR2 | Adenosine deaminase that acts of pre-mRNA; targets include the serotonin receptor 2C pre-mRNA; alters the amino acid sequence and functional properties | |
| S100B | Ca-binding protein; promotes neuronal survival when oxidized and apoptosis when reduced; L/M |
ROS, reactive oxygen species; L/M, learning and memory deficits in overexpressing transgenic model. MMU16, MMU10, orthologs of HSA21q proteins map to mouse chromosome 16 and 10 respectively.
These examples illustrate the primary difficulties of targeting therapeutics to HSA21q genes – how many genes need to be regulated simultaneously? How many drugs would this require and what gene and/or drug interactions might occur in full trisomies to produce adverse effects? How can these be assayed in incomplete mouse models?
Targeting phenotypic features
The difficulties in identifying and targeting multiple HSA21q genes can be avoided by instead directly targeting the phenotypic consequences of trisomy. For the examples in Table 3, information on the underlying HSA21q causative genes and/or HSA21q gene-drug interactions is lacking, unconfirmed or incomplete.
Two studies documented degeneration of BFCNs in older Ts65Dn mice, by measuring a progressive decrease in choline acetyl transferase and NGF receptor p75 protein markers [33,34]. This degeneration was associated with a deficit in axonal retrograde transport of NGF, which was reduced to <10% of normal levels. Intracerebroventricular injection of NGF into 18-month-old Ts65Dn mice reversed the abnormalities in both size and marker levels of the BFCNs [35].
One method to increase levels of choline acetyl transferase in BFCNs is to inhibit its degradation by inhibiting acetylcholine esterase (AChE) activity. Donepezil, an FDA-approved drug for AD, is an AChE inhibitor and a clinical trial on young adults with DS has reported some positive results [36]. However, when Ts65Dn mice were treated with Donepezil continuously from the age of seven months through training and testing in the MWM at ten months, no improvement in learning and memory was observed [37]. This is in contrast to positive results with mouse models for some other neurological abnormalities [38].
Neuro-inflammation is a possible contributing factor to degeneration in DS and AD, and elevated levels of neuro-inflammatory markers have also been observed in Ts65Dn [39]. Minocycline, a member of the tetracycline family, has anti-inflammatory properties and has been used with some success in clinical trials on individuals with Parkinson's Disease and Huntington's Disease [40]. Treatment of Ts65Dn mice with minocycline for 75 days starting at seven months of age resulted in improved performance in the water version of the radial arm maze (water-RAM), although it did not reach levels of euploid controls [39]. Treatment also prevented the loss of functional markers in BFCNs, suggesting further that minocycline at the very least delays degeneration.
The reduced densities of granule cell neurons in the cerebellum of Ts65Dn, a deficiency also seen in brains of individuals with DS [26], originates early in post natal development because cell density is normal at P0 but deficient by P6 [25]. This deficit is associated with a decreased rate of mitosis in granule cell precursors and a reduced response to the mitogenic stimulus of the Sonic hedgehog growth factor. A single injection at P0 of an agonist of the hedgehog pathway, SAG 1.1 [41], was sufficient to produce normal levels of granule cell precursors and cerebellar density at P6 [26]. This methodology has implications for increasing cell numbers in the hippocampus, but abnormal activation of the Sonic hedgehog pathway in many cancers presents challenges.
Piracetam is a cyclic derivative of GABA and considered a nootropic agent, a stimulant of cognitive function. Its mechanism of action is unknown but has been proposed to involve modulation of cholinergic and glutamatergic neurotransmission. Positive effects of piracetam on learning and memory have been reported in both rodent and human studies of aging and/or neurological disease [reviewed in 42]. However, treatment of Ts65Dn mice with piracetam for one month starting at six weeks of age actually exacerbated deficits in MWM performance [43].
The Ts65Dn mice show decreased densities of excitatory synapses but normal numbers of inhibitory synapses; consistent with this they also show impaired LTP [44,45]. Some aspects of this imbalance in excitatory versus inhibitory transmission are rescued by the GABAA receptor antagonist picrotoxin [46]. On the basis of these and other studies, an alternative GABAA receptor antagonist, pentylenetetrazole (PTZ), was used to treat Ts65Dn mice starting at three months of age [47]. After 30 days, performance in NOR and the T-Maze (TM) improved to that of controls; this performance was maintained for a further three months in the absence of PTZ treatment. This analysis was extended to show that PTZ rescued deficits in the MWM in Ts65Dn mice, but caution is required because it also caused impairment in some tests of equilibrium [37]. Furthermore, PTZ is known to induce seizures at higher doses.
Targeting pathway perturbations
Although information on the functions of HSA21q genes is incomplete, and has not shed light on direct causes of phenotypic features discussed above, connections with many pathways and cellular processes relevant to learning and memory can be made [8,9,48]. Functional associations of fifteen genes that influence the activities of MAP kinase, calcineurin, the NMDA receptor, serotonin and production of reactive oxygen species (ROS) are summarized in Table 4. It is important to note that these functional associations have been ascertained in individual gene-specific studies, often in vitro or in other artificial systems using cDNA constructs (devoid of genomic and developmental regulatory features), and often using knockout or knockdown rather than overexpression. Simultaneous overexpression of these genes in DS will integrate their synergistic and antagonist effects on common pathways and processes. A number of studies with Ts65Dn mice have successfully targeted predicted pathway perturbations.
Serotonin signaling
Decreased cell numbers in DS hippocampus could be caused by impaired adult neurogenesis which has been observed in Ts65Dn [49,50] and Ts1Cje [51] mice and, on the basis of data from other rodent models, may underlie learning and memory deficits. A connection to HSA21q is predicted through serotonin signaling. Activity of the serotonin receptor 1A (5HTR1A) is required for adult neurogenesis in the hippocampus [52] and is mediated by the potassium channel KCNJ6. Overexpression of KCNJ6, as in the Ts65Dn model, may over inhibit presynaptic 5HTR1A causing reduced levels of serotonin. Fluoxetine inhibits KCNJ6, increases presynaptic serotonin levels, and consistent with this, rescues the Ts65Dn deficit in neurogenesis [49]. However, fluoxetine also alters the functional properties of a second serotonin receptor, 2C (5HTR2C), by altering patterns of adenosine-to-inosine editing in the pre-mRNA. In DS, there will be an additional influence on 5HTR2C function because it is a substrate for ADAR2 [53], a pre-mRNA editing enzyme encoded by HSA21q, which is not trisomic in the Ts65Dn model. Fluoxetine effects may therefore be altered in full trisomy by overexpression of ADAR2.
NMDA receptor signaling
Nine HSA21q genes (APP, TIAM1, BACH1, SOD1, SYNJ1, ITSN1, RCAN1, DYRK1A and PCP4) directly interact with or indirectly affect the activity of the NMDA receptor and/or the protein phosphatase calcineurin (CaN) (Table 4) [48]. Inhibition of CaN activity alters NMDA receptor activity and causes increased locomoter activity in response to the NMDA receptor antagonist MK-801 [54]. Both the Ts65Dn and the Ts1Cje models display this exaggerated hyperactivity, indirectly supporting inhibition of CaN and altered NMDA receptor activity [48]. Another antagonist of the NMDA receptor, memantine, rescued the deficit in Context Fear Conditioning (CFC) displayed by the Ts65Dn [55]. This is of considerable interest because memantine is approved for use in humans in the treatment of AD. Again, however, there may be additional gene contributions in a full HSA21q trisomy that affect memantine response. Success in CFC requires activation of the transcription factor ELK [56]. Nuclear localization, and thus ELK activity, is regulated by dynamic interactions of phosphorylation and sumoylation modifications [57]. Phosphorylation is carried out by MAPK and is abnormal in Ts65Dn [48]; sumoylation is carried out by the HSA21q-encoded gene SUMO3 (Small Ubiquitin-like Modifier protein 3) which is not trisomic in the Ts65Dn. Thus, perturbations of ELK activity may be different, and may respond differently to memantine effects, in full trisomy.
Neuroprotective pathways
NAP and SAL are eight- and nine-amino-acid peptides derived, respectively, from the glial proteins Activity Dependent NeuroProtective protein (ADNP) and Activity Dependent Neurotrophic Factor (ADNF). NAP has been shown to inhibit the neurotoxicity of Aβ and to reduce neuro-inflammation after head injury, and both peptides increased survival of DS-derived neurons in culture [58]. This mechanism involves protection from oxidative stress because NAP and SAL treatment protected control neurons from damage by H2O2. NAP plus SAL treatment of pregnant Ts65Dn mice prevented developmental delays in trisomic offspring in several, but not all, motor and sensory assessments [59,60]. This was accompanied by an increase to normal levels of mRNA encoding some NMDA and GABA receptor subunits.
Oxidative stress pathways
Elevated levels of oxidative stress in DS may contribute to neuronal degeneration. HSA21q candidate genes include SOD1, APP and BACH1 (Table 4). When Ts65Dn were fed a diet high in the antioxidant vitamin E, markers of oxidative stress were reduced and performance in the water-RAM test improved to levels of controls. Loss of the NGF receptor trkA protein in the BFCN was prevented, and the decrease in size of the BCFN was delayed [61]. However, these positive effects may be modulated in a full HSA21q trisomy by contributions of the S100B protein. S100B is a Ca2+-binding protein whose function is regulated by oxidative state, promoting neuronal survival in the oxidized state and apoptosis and neurotoxicity in the reduced state [62]. A transgenic mouse that overexpresses S100B displays learning and memory impairment and increased levels of apoptotic markers, but treatment of this mouse with vitamin E increased oxidative damage. S100B maps to MMU10 and therefore in full trisomy DS may alter the positive effects of vitamin E treatment seen in the Ts65Dn model. Consistent with this, vitamin E was found to be ineffective in treatment of DS [63].
The piracetam analogue, SGS-111, was shown to prevent oxidative damage and apoptosis in cultured neurons derived from individuals with DS [64]. Based on the hypothesis that it might therefore also decrease ROS in the Ts65Dn mice and result in improved learning and memory, mice were treated with SGS-111, starting both during gestation and postnatally. No improvement in MWM performance was observed [65].
Challenges in target identification and preclinical evaluation of pharmacotherapies
Functional information on HSA21q genes currently remains limited to a subset of RefSeqP genes, with the majority of genes completely unexamined. Efficient prediction of potential targets for pharmacotherapies requires identification of additional pathway perturbations in trisomy that potentially contribute to ID. In vitro studies of individual HSA21q cDNAs could determine interaction partners and transcriptome consequences of overexpression. In vivo studies could make use of available bacterial artificial chromosome (BAC) clone contigs to construct overexpressing transgenic mice for the majority of HSA21q genes, which would then be examined for deficits in learning and memory.
A major challenge for preclinical assessment of potential pharmacotherapies remains the choice of model system. Data from the new MMU17 segmental trisomy, the Ts1Yah model, emphasize that the contributions of genes not trisomic in the Ts65Dn model must be considered. As illustrated by examples in Tables 2 and 4, trisomy of nonoverlapping sets of genes can produce similar behavioral deficits and genes mapping to all three mouse chromosomal segments functionally interact. Thus, a full trisomy model system is needed. The likely insurmountable limitations of mouse models suggest that alternatives are needed.
Mammalian cell culture offers some possibilities. Lymphoblastoid cell lines derived from individuals with DS and typical controls are readily available and molecular phenotypes can be studied, although they lack neuronal features. An alternative cell culture system might come from recent advances in developing stem cell (iPS) lines from human fibroblasts followed by their differentiation to neural cell types [66]. If protocols and cell lines prove robust, creating sets of iPS lines would allow determination of baseline differences in neuronal characteristics, such as spine densities or axon development, dynamic responses to drug treatments and inter-individual variations in these features. Drug-treatment-induced correction of abnormalities in DS iPS lines could be used to predict drug efficacy and potentially to identify additional targets.
Understanding abnormalities documented in in vitro and in vivo systems, and effectively targeting them for correction, will be facilitated by analysis of pathway perturbations at the protein level. This includes protein modification, such as phosphorylation, acetylation and sumoylation, among others, that more directly assesses protein activity, versus mere gene expression. While challenging, protein measurements can provide stronger preliminary support for effective pharmacotherapies and potentially identify those that are likely to fail prior to human trials.
Concluding remarks
Recent successes in rescuing learning and memory deficits in the Ts65Dn mice, especially with PTZ and memantine, are causes for legitimate optimism that pharmacotherapies for cognitive deficits in DS are within reach. Nevertheless, the desire for immediate extrapolation to efficacy in DS must be tempered by recognizing and addressing the limitations of current model systems described above, that are specific to the genetic basis of DS. In addition further thought must be given to challenges that are generally applicable to studies of ID: (i) effective assessment in mouse models of the complexities of learning and memory in humans, (ii) assessment of genetic variability and its contributions to phenotypic variability and drug responses, and (iii) the potential for adverse drug effects or interactions specific to the developing brains of infants and children vs. the mature brains of adults.
Acknowledgments
This work was supported by the Sie Foundation, the Fondation Jerome Lejeune and the National Institute of Child Health and Human Development (NICHD) HD056235.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.CDC Centers for Disease Control and Prevention. Improved National Prevalence Estimates for 18 Major Birth Defects. MMWR. 2006;54:6–12. [Google Scholar]
- 2.Nadel L. Down's syndrome: a genetic disorder in biobehavioral perspective. Genes Brain Behav. 2003;2:156–166. doi: 10.1034/j.1601-183x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 3.Benavides-Piccione R, et al. On dendrites in Down syndrome and DS murine models: a spiny way to learn. Prog Neurobiol. 2004;74:111–126. doi: 10.1016/j.pneurobio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Pennington BF, et al. The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev. 2003;74:75–93. doi: 10.1111/1467-8624.00522. [DOI] [PubMed] [Google Scholar]
- 5.Abbeduto L, et al. Language development in Down syndrome: from the prelinguistic period to the acquisition of literacy. Ment Retard Dev Disabil Res Rev. 2007;13:247–261. doi: 10.1002/mrdd.20158. [DOI] [PubMed] [Google Scholar]
- 6.Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13:237–246. doi: 10.1002/mrdd.20163. [DOI] [PubMed] [Google Scholar]
- 7.Hattori M, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 8.Gardiner K, Costa AC. The genes of human chr21: choosing candidates for relevance to intellectual disability. Am J Med Genet. 2006;142:196–205. [Google Scholar]
- 9.Pritchard M, et al. Down syndrome and the genes of human chromosome 21: current knowledge and future potentials. Cyto Genet Genome Res. 2008;121:67–77. doi: 10.1159/000124384. [DOI] [PubMed] [Google Scholar]
- 10.Korenberg JR, et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci U S A. 1994;91:4997–5001. doi: 10.1073/pnas.91.11.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korbel JO, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyle R, et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet. 2009;2017:454–466. doi: 10.1038/ejhg.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seregaza Z, et al. Mouse models of cognitive disorders in trisomy 21: a review. Behav Genet. 2006;36:387–404. doi: 10.1007/s10519-006-9056-9. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, et al. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- 15.Olson LE, et al. Trisomy for the Down syndrome ‘critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Hum Mol Genet. 2007;16:774–82. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- 16.Lopes Pereira P, et al. A new mouse model for trisomy of the Abcg1-U2af1 regions reveals the complexity of the combinatorial genetic code of Down syndrome. Hum Mol Genet. 2009 Sep 26; doi: 10.1093/hmg/ddp438. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Doherty A, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309:2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardiner K, et al. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. GENE. 2003;318:137–147. doi: 10.1016/s0378-1119(03)00769-8. [DOI] [PubMed] [Google Scholar]
- 19.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bimonte HA, et al. In two species, females exhibit superior working memory and inferior reference memory on the radial-arm maze. Physiology Behav. 2000;70:311–317. doi: 10.1016/s0031-9384(00)00259-6. [DOI] [PubMed] [Google Scholar]
- 21.Hughes RN. Neurosci Biobehav Rev. 2004;28:297–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez F, Garner CC. Object recognition memory is conserved in Ts1Cje, a mouse model of Down syndrome. Neurosci Lett. 2007;421:137–41. doi: 10.1016/j.neulet.2007.04.075. [DOI] [PubMed] [Google Scholar]
- 23.Belichenko NP, et al. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci. 2009;29:5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morice E, et al. Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tc1 mouse model of Down syndrome. Learn Mem. 2008;15:492–500. doi: 10.1101/lm.969608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roper RJ, et al. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci U S A. 2006;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baxter LL, et al. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet. 2000;9:195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- 27.Wiseman FK, et al. Down syndrome--recent progress and future prospects. Hum Mol Genet. 2009;18(R1):R75–83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cataldo AM, et al. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci. 2003;23:6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salehi A, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Bain J, et al. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie W, et al. Promotion of neuronal plasticity by (-)-epigallocatechin-3-gallate. Neurochem Res. 2008;33:776–783. doi: 10.1007/s11064-007-9494-7. [DOI] [PubMed] [Google Scholar]
- 32.Guedj F, et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS One. 2009;4:e4606. doi: 10.1371/journal.pone.0004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holtzman DM, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc Natl Acad Sci U S A. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granholm AC, et al. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down's syndrome. Exp Neurol. 2000;161:647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- 35.Cooper JD, et al. Failed retrograde transport of NGF in a mouse model of Down's syndrome: Reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc Natl Acad Sci USA. 2001;98:10439–14444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kishnani PS, et al. The efficacy, safety, and tolerability of donepezil for the treatment of young adults with Down syndrome. Am J Med Genet A. 2009;149A:1641–1654. doi: 10.1002/ajmg.a.32953. [DOI] [PubMed] [Google Scholar]
- 37.Rueda N, et al. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci Lett. 2008a;433:22–27. doi: 10.1016/j.neulet.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 38.Yuede CM, et al. Anti-dementia drugs and hippocampal-dependent memory in rodents. Behav Pharmacol. 2007;18:347–363. doi: 10.1097/FBP.0b013e3282da278d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunter CL, et al. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann Neurol. 2004;56:675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- 40.Blum D, et al. Clinical potential of minocycline for neurodegenerative disorders. Neurobiol Dis. 2004;17:359–366. doi: 10.1016/j.nbd.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Chen W, et al. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science. 2004;306:2257–2260. doi: 10.1126/science.1104135. [DOI] [PubMed] [Google Scholar]
- 42.Winblad B. Piracetam: a review of pharmacological properties and clinical uses. CNS Drug Rev. 2005;11:169–82. doi: 10.1111/j.1527-3458.2005.tb00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moran TH, et al. The effects of piracetam on cognitive performance in a mouse model of Down's syndrome. Physiol Behav. 2002;77:403–409. doi: 10.1016/s0031-9384(02)00873-9. [DOI] [PubMed] [Google Scholar]
- 44.Kurt MA, et al. Synaptic deficit in the temporal cortex of Ts65Dn mice. Brain Res. 2000;858:191–197. doi: 10.1016/s0006-8993(00)01984-3. [DOI] [PubMed] [Google Scholar]
- 45.Kleschevnikov AM, et al. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costa AC, Grybko MJ. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci Lett. 2005;382:317–322. doi: 10.1016/j.neulet.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez F, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10:411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 48.Siddiqui A, et al. Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK-801. Genes Brain Behav. 2008;7:810–820. doi: 10.1111/j.1601-183X.2008.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clark S, et al. Fluoxetine rescues deficient neurogenesis in hippocampus of the mouse model for Down syndrome Ts65Dn. Exp Neurol. 2006;200:256–261. doi: 10.1016/j.expneurol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Rueda N, et al. Cell proliferation is reduced in the dentate gyrus of aged but not young Ts65Dn mice, a model of Down syndrome. Neurosci Lett. 2005;380:197–201. doi: 10.1016/j.neulet.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 51.Ishihara K, et al. Enlarged Brain Ventricles and Impaired Neurogenesis in the Ts1Cje and Ts2Cje Mouse Models of Down Syndrome. Cereb Cortex. 2009 Aug 26; doi: 10.1093/cercor/bhp176. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 52.Banasr M, et al. Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology. 2004;29:450–460. doi: 10.1038/sj.npp.1300320. [DOI] [PubMed] [Google Scholar]
- 53.Werry TD, et al. RNA editing of the serotonin 5HT2C receptor and its effects on cell signalling, pharmacology and brain function. Pharmacol Ther. 2008;119:7–23. doi: 10.1016/j.pharmthera.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Miyakawa T, et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci U S A. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa AC, et al. Acute Injections of the NMDA Receptor Antagonist Memantine Rescue Performance Deficits of the Ts65Dn Mouse Model of Down Syndrome on a Fear Conditioning Test. Neuropsycopharmacology. 2008;33:1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- 56.Sananbenesi F, et al. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- 57.Salinas S, et al. SUMOylation regulates nucleo-cytoplasmic shuttling of Elk-1. J Cell Biol. 2004;165:767–773. doi: 10.1083/jcb.200310136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Busciglio J, et al. NAP and ADNF-9 protect normal and Down's syndrome cortical neurons from oxidative damage and apoptosis. Curr Pharm Des. 2007;13:1091–1098. doi: 10.2174/138161207780618957. [DOI] [PubMed] [Google Scholar]
- 59.Toso L, et al. Prevention of developmental delays in a Down syndrome mouse model. Obstet Gynecol. 2008;112:1242–1251. doi: 10.1097/AOG.0b013e31818c91dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vink J, et al. Prenatal NAP+SAL prevents developmental delay in a mouse model of Down syndrome through effects on N-methyl-D-aspartic acid and gamma-aminobutyric acid receptors. Am J Obstet Gynecol. 2009;200:524.e1–4. doi: 10.1016/j.ajog.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lockrow J, et al. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp Neurol. 2009;216:278–289. doi: 10.1016/j.expneurol.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bialowas-McGoey LA, et al. Vitamin E increases S100B-mediated microglial activation in an S100B-overexpressing mouse model of pathological aging. Glia. 2008;56:1780–1790. doi: 10.1002/glia.20727. [DOI] [PubMed] [Google Scholar]
- 63.Ellis JM, et al. Supplementation with antioxidants and folinic acid for children with Down's syndrome: randomised controlled trial. BMJ. 2008;336:594–597. doi: 10.1136/bmj.39465.544028.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelsman A, et al. GVS-111 prevents oxidative damage and apoptosis in normal and Down's syndrome human cortical neurons. Int J Dev Neurosci. 2003;21:117–24. doi: 10.1016/s0736-5748(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 65.Rueda N, et al. Effects of chronic administration of SGS-111 during adulthood and during the pre- and post-natal periods on the cognitive deficits of Ts65Dn mice, a model of Down syndrome. Behav Brain Res. 2008;188:355–367. doi: 10.1016/j.bbr.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 66.Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gardiner K. Transcriptional deregulation in Down syndrome: predicting effects on CREB, ELK, GR and ER. Behav Genet. 2006;36:439–453. doi: 10.1007/s10519-006-9051-1. [DOI] [PubMed] [Google Scholar]