

Abstract
The pyruvate dehydrogenase complex (PDC) catalyzes the conversion of pyruvate to acetyl-CoA in mitochondria and is a key regulatory enzyme in the oxidation of glucose to acetyl-CoA. Phosphorylation of PDC by the pyruvate dehydrogenase kinases (PDK) inhibits its activity. The expression of the pyruvate dehydrogenase kinase 4 (PDK4) gene is increased in fasting and other conditions associated with the switch from the utilization of glucose to fatty acids as an energy source. Transcription of the PDK4 gene is elevated by glucocorticoids and inhibited by insulin. In this study, we have investigated the factors involved in the regulation of the PDK4 gene by these hormones. Glucocorticoids stimulate PDK4 through two glucocorticoid receptor (GR) binding sites located more than 6,000 base pairs upstream of the transcriptional start site. Insulin inhibits the glucocorticoid induction in part by causing dissociation of the GR from the promoter. Previously, we found that the estrogen related receptor alpha (ERRα) stimulates the expression of PDK4. Here, we determined that one of the ERRα binding sites contributes to the insulin inhibition of PDK4. A binding site for the forkhead transcription factor (FoxO1) is adjacent to the ERRα binding sites. FoxO1 participates in the glucocorticoid induction of PDK4 and the regulation of this gene by insulin. Our data demonstrate that glucocorticoids and insulin each modulate PDK4 gene expression through complex hormone response units that contain multiple factors.
Keywords: Pyruvate dehydrogenase kinase (PDK4), glucocorticoids, insulin
Introduction
The metabolism of pyruvate to acetyl-CoA is catalyzed by the pyruvate dehydrogenase complex (PDC) which is located in mitochondria [1]. PDC is a key step in the control of glucose oxidation to acetyl-CoA, and PDC activity is decreased in diabetes and fasting [2]. Although activity of the pyruvate dehydrogenase complex is negatively regulated short term by its reaction products acetyl-CoA and NADH, long-term regulation of PDC activity is mediated via pyruvate dehydrogenase kinase (PDK) dependent phosphorylation [2]. Three serine phosphorylation sites on the α-subunit of pyruvate dehydrogenase (E1) are targeted by PDKs leading to reduced activity of PDC [1]. Thus PDK is an important regulator of the pyruvate dehydrogenase complex. While four PDK isoforms (PDK1, 2, 3, 4) have been identified in mammalian tissues, the PDK2 and PDK4 isoforms are highly expressed in the liver, heart and skeletal muscle. PDK2 and PDK4 mRNA and protein levels are elevated in livers of diabetic, fasted and insulin resistant animals [3–5]. Expression of the PDK4 gene is increased by glucocorticoids, retinoic acid, prolactin (via STAT5), long chain fatty acids and fibrate compounds [5–8]. The induction of PDK4 by fibrates is mediated by the peroxisome proliferator activated receptor α (PPARα) [9,10]. Conversely, PDK4 and PDK2 gene expression is inhibited by insulin [8,11].
Given the central role of PDK4 in regulating PDC activity, it is important to understand the molecular mechanisms underlying the regulation of PDK4 expression by hormonal and nutritional factors. In this regard, several nuclear receptors and transcription factors including the estrogen related receptor (ERR), glucocorticoid receptor (GR), forkhead transcription factor (FoxO1) and hepatic nuclear factor 4 (HNF4) have been found to regulate PDK4 gene expression. ERRs are orphan nuclear receptors and three isoforms have been identified, α β and γ[12]. Several lines of evidence have suggested a role for ERRα and ERRγ in the control of metabolic genes. ERRα induces medium chain acyl-CoA dehydrogenase (MCAD) indicating a role in fatty acid oxidation [13]. In the heart, ERRα and ERRγ activate a network of genes involved in fatty acid transport, mitochondrial function and fatty acid oxidation [14]. ERRα contains a DNA binding domain and a ligand binding domain which is involved in dimerization, transcriptional activation and interactions with coactivators [15]. We found that ERRα induces PDK4 expression through two binding sites in the proximal promoter [16]. In addition, we have determined that HNF4 stimulates the PDK4 gene in hepatoma cells [17].
In these studies, we have explored the mechanisms by which glucocorticoids and insulin modulate the expression of PDK4. The induction of genes by glucocorticoids is mediated by the glucocorticoid receptor (GR) which is sequestered in the cytosol with heat shock proteins and moves to the nucleus in the presence of ligands to modulate gene expression [18]. The GR binds to glucocorticoid response elements (GRE) within genes and interacts with other transcription factors and coactivators to create glucocorticoid response units (GRU) which control gene expression [18,19]. Factors including CCAAT enhancer binding protein β (C/EBPβ), FoxO1 and HNF4 are found in the GRUs of gluconeogenic genes such as the PEPCK gene [20]. FoxO1 is also an important participant in the insulin inhibition of several genes. Insulin activates protein kinase B (PKB) which phosphorylates FoxO1 on serines 256 and 319 and threonine 29 leading to the nuclear exclusion of FoxO1 [21–23]. FoxO1 dissociation from promoters contributes to the insulin inhibition of gene expression [24,25].
In addition, numerous coactivators participate in the control of gene expression. In particular, we have focused on the peroxisome proliferator activated receptor γ coactivator (PGC-1α) [16,17]. PGC-1α was initially cloned from brown adipose tissue as a protein that interacts with the peroxisome proliferator activated receptor γ (PPARγ) [26]. PGC-1α promotes mitochondrial biogenesis and mitochondrial oxidation of long chain fatty acids [27]. In the liver, PGC-1α abundance is greatly increased following an overnight fast, and PGC-1α is induced by glucocorticoids and cAMP [28,29]. Hepatic PGC-1α levels are elevated in streptozotocin-induced diabetes several models of type II diabetes [29]. In addition to its known role in hepatic glucose production (gluconeogenesis), PGC-1α amplifies the cAMP and glucocorticoid dependent induction of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6 phosphatase (G6Pase) gene expression [28–30]. Previous studies from our laboratory and others have shown that PDK4 expression is induced by PGC-1α in primary rat hepatocytes and cardiomyocytes [16,17]. PGC-1α is recruited to the PDK4 gene through interactions with ERRα [31].
In these studies, we have investigated the regulation of the PDK4 gene by glucocorticoids and insulin. Here, we report that the glucocorticoid induction of PDK4 requires two distal GREs as well as FoxO1 in the proximal promoter. However, ERRα and PGC-1α, which induce the PDK4 gene, do not amplify the glucocorticoid induction. Insulin acts at multiple sites in the promoter to decrease PDK4 transcription including the GREs in distal promoter, the FoxO1 site and a binding site for ERRα. Our data indicate that both the glucocorticoid and insulin regulation of the PDK4 gene are mediated through complex hormone response units.
Materials and Methods
Transient transfections of luciferase reporter genes
Transient transfections were performed in McA-RH7777 cells using the calcium phosphate method as described previously [16]. McA cells were transfected and incubated overnight at 37°C in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 5% calf serum and 5% fetal calf serum. The following day the cells were washed with phosphate buffered saline and serum-free media containing 100 nM dexamethasone or 10 nM insulin was added as described in the figure legends. Cells were harvested after 24 h and luciferase assays were performed using Promega Dual Luciferase Kit (E1960) following the manufacturer’s protocol. Luciferase values were normalized for protein content and renilla luciferase activity to account for cell density and transfection efficiency, respectively [16].
Construction of PDK4-luciferase reporter genes
Cloning of the rat −2989/+78 PDK4 promoter region and ligation to a luciferase-reporter construct was described previously [16,17]. The −1529/+78 PDK4-luciferase vector was generated by a Kpn I digestion of −2989/+78 PDK4-luciferase to remove the −2989 to −1529 fragment. Mutations in the promoter were introduced using the Stratagene QuikChange Site-Directed Mutagenesis kit (200518). The following primers were used: PDK4 Mut338, 5′-agatggctcctgagttgtaaacaaaaacaagtctgggcggg-3′; Mut347, 5′- tgacattgagatggctcctgagtttgcaacaaggacaagtctgggc-3′; M370 5′-cgaggaatgcgtcttcgcgagatggctcct -3′. We had previously shown that these mutations disrupted the binding of ERR and FoxO1 to their respective sites [16].
To construct the −6529/+78 PDK4-luciferase vector, an Asc I site was introduced at nucleotide −2872 into both the −6529/−2829 and the −2989/+78 segments of the PDK4 promoter. The two promoter fragments were ligated together using the Asc I site to generate a −6529 to +78 promoter region. The −6.5K/+78 PDK4 promoter was then ligated into pGL3 basic luciferase using the Sac I and Bgl II restriction sites. To introduce the ERRα and FoxO1 mutations into the −6.5/+78 PDK4-luciferase, the −1529/+78 PDK4-luciferase vectors containing the disrupted sites were digested with Kpn I and Bgl II. This promoter region was substituted into the Kpn I and Bgl II sites of the 6.5/+78 wild type PDK4-luciferase vector. All sequences were verified using the Molecular Resource Center at the University of Tennessee Health Science Center.
The −7009/+2829 region of the PDK4 gene was ligated in front of SV40-luciferase. The putative glucocorticoid response elements (GREs) were disrupted by site directed mutagenesis using the QuikChange site-directed mutagenesis kit. The following primers were used: GRE Mut- 6460 5′-ctttgaactagaacgactgatccagtacagggac-3′; Mut-6386 5′- gttattatgaaggtttgtgtcgcactacagttcttccacaaaggg-3′; Mut-6010 5′-gtcattttccttactatggggagcaggggag-3′. The altered nucleotides are underlined.
Real time PCR analysis
For real time PCR, cDNA was prepared using RNA isolated from McA cells or primary rat hepatocytes. The RNA was isolated with RNA-Stat-60 (Tel-test) [17]. The RNA was then treated with DNase I (2 units) at 37 °C for 1 hour followed by addition of DNase Inactivation Reagent (Ambion). The concentration of each sample containing DNA free RNA was measured at a wavelength of 260 nm using UV/Vis spectrophotometer. Equal amounts of DNA-free RNA were used for first-strand cDNA synthesis. RNA was mixed with 1 μl of 10 mM dNTP mix and 1 μl of random hexamers (50 ng/μl). The conditions for the reverse transcription to generate cDNA have been described previously [17]. The parameters for real time PCR were as follows: 95 °C for 10 min, 40 cycles of 95 °C 30 s and 60 °C 1 min. A DNA dissociation curve was executed for each sample that consisted of 100 cycles of 10 s each from 60 to 100 °C in 0.4 °C increments. The first derivative of this DNA dissociation curve was plotted to show peaks for each target amplified. The final concentration of primers in each well in the PCR plates was 0.1 μM. Oligonucleotide standards were synthesized and quantified by HPLC. These standards were diluted to 10−9, 10−8, 10−7, 10−6 and 10−5 μM and used as templates for each standard curve. A 1:50 dilution of each cDNA as template was used to measure 18S ribosomal RNA (rRNA), and 1 μl of 1:10 or 1:20 dilution of each cDNA was used as a template to assess target genes. The 18S rRNA was used to normalize all results. The following forward (FP) and reverse primers (RP) were used for real time PCR to quantity mRNA abundance: PDK4 forward primer, 5′-ggattactgaccgcctctttagtt; PDK4 reverse primer, 5′-gcattccgtgaattgtccatc; 18S forward primer, 5′-cggctaccacatccaaggaa; 18S reverse primer, 5′-ttttcgtcactacctccccg.
Adenoviral infection
The shRNA for rat ERRα was designed for the coding sequence gagcatcccaggcttctcc. Hairpin siRNA template DNA oligomers were inserted into pSilencer 3.0-H1 plasmid (Ambion 7209) driven by H1 promoter. The pSilencer H1-ERRα RNAi was inserted into promoterless pAdTrack (ATCC cat # JHU-23) and this modified pAdTrack was called pAdTrack- H1-RNAi. The pAdTrack-H1-RNAi was transformed into BJ5183 cells which contained the pAdEasy-1 adenovirus backbone (Stratagene). Positive clones were selected and confirmed by DNA sequencing. The correct plasmids were amplified following transformation into DH5 cells. The resulting adenoviral DNA Ad-H1-RNAi or Ad-H1-negative control vectors were linearized with PacI. The linearized adenoviral DNA was used to transfect AD-293 packaging cells. Adenovirus was amplified according to Stratagene AdEasy protocol and purified by cesium chloride method. McA-RH7777 cells were infected with adenovirus expressing short hairpin RNA against ERRα (Ad-shERRα). Adenovirus expressing a non-specific shRNA (Ad-NC) was used as a control. McA cells were maintained in DMEM media in the absence of serum. Media was changed 16 h after infection and hormones added for 24 hours. The McA cells were harvested and RNA was isolated using RNA Stat 60 (Tel-test).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were conducted with slight modifications following the protocol given by the Millipore Magna ChIP kit (17–610). Rat primary hepatocytes were maintained for 18 hours in RPMI 1640 media containing 5% fetal bovine serum and 5% calf serum. Cells were treated with 100nM dexamethasone or 100nM insulin for 4 hours in serum-free media. After treatment, cross-linking was performed with 1% formaldehyde for 10 minutes at room temperature and quenched by the addition of glycine. Cross-linked cells were harvested and placed in cell lysis buffer for 15 minutes followed by homogenization using a Dounce homogenizer. The homogenate was pelleted, resuspended in nuclear lysis buffer, and sonicated to 400–600bp 9 × 30 seconds in ice water. Chromatin preparations were diluted with dilution buffer and protease inhibitor cocktail and designated as input samples (no antibody), used for immunoprecipitation with the desired antibody, or with the control antibody rabbit IgG (Santa Cruz # sc-2027). Antibodies used were anti-FKHR (Santa Cruz, sc-11350), anti-GR (Santa Cruz, sc-8992), and anti-PGC1α (Santa Cruz, sc-13067). Samples were left rotating overnight at 4°C with the antibody and the magnetic protein A beads. The magnetic beads were pelleted and washed with a low-salt immune complex wash buffer, high-salt immune complex wash buffer, lithium-chloride immune complex wash buffer, and TE buffer. DNA was eluted using the ChIP elution buffer and treated with proteinase K for 4 hours at 65°C followed by 10 minutes at 95°C. DNA was purified using the PCR purification kit (Qiagen 28104). Eluted DNA was subjected to 35 cycles of PCR using 3–6 μL of DNA. PCR products were analyzed on 2% Nusieve 3:1 agarose (Lonza 50090) and visualized with MultiImage Light Cabinet with AlphaImager EP software. The following primers were used to amplify: the proximal region (−591/−338) forward primer 5′- taaggctatttaggcagttt and reverse primer 5′-ccagacttgtccttgtttac, the glucocorticoid response region (−6634/−6377) forward primer 5′-tatgagaagtgctgcaataa and reverse primer 5′-atgttaccacaaaccttcat, and the region (−1535/−1228) forward primer 5′-agtgtctccaccagattgt and reverse primer 5′-ctaagagagctaacctagt.
Results
Our initial experiments investigated whether glucocorticoids and insulin would regulate the expression of PDK4 gene expression in McA-RH7777 rat hepatoma cells. Addition of dexamethasone increased PDK4 mRNA abundance 13 ± 1.1 fold and this induction was inhibited 56% by the addition of insulin (Fig. 1A). Insulin alone reduced the expression of the PDK4 gene about 10% but this change was not statistically significant (Fig. 1A). These data indicate that PDK4 gene expression is regulated in a physiologic manner in McA cells. Our next experiments were designed to localize the glucocorticoid response element (GRE) in the PDK4 gene. Previously, we had cloned the promoter of the rat PDK4 gene [17]. A PDK4-luciferase vector (−2989/+78) containing 3000 base pairs of the promoter was transfected into McA cells in the presence or absence of dexamethasone. However, this region of the promoter did not respond to glucocorticoids (data not shown). We cloned an additional upstream region of the promoter containing the promoter sequences from −7009/−2829 (−7/−2.8) and ligated this segment of the promoter in front of the enhancer-less SV40-luciferase gene. This construct was induced 7.8 ± 0.3 fold by dexamethasone and this stimulation was decreased 25% by insulin (Fig. 1B). The glucocorticoid responsiveness was fully retained in the −6529/−2829 (−6.5/−2.8) SV40-luciferase vector. The expression of the −6.5/−2.8 SV40-luciferase vector was 0.93 ± 0.2 fold relative to the control suggesting that insulin was primarily decreasing the induction by glucocorticoids through this region (data not shown). Three potential glucocorticoid response elements (GRE) sequences were identified in the −6529/−2829 region of the PDK4 promoter and each was disrupted by site directed mutagenesis. Mutation of GRE −6386 decreased the glucocorticoid induction by 50%. A double mutation of the GREs at −6460 and −6386 completely eliminated the dexamethasone responsiveness (Fig. 1B). Mutation of the GREs at −6386 and −6010 had no additional effect over mutation of −6386 alone (Fig. 1B). These results indicated that induction of PDK4 by glucocorticoids is mediated by two GREs in this region of the gene and that insulin partially inhibited the dexamethasone induction through the distal promoter.
Figure 1. Identification of glucocorticoid receptor binding sites in the PDK4 promoter.
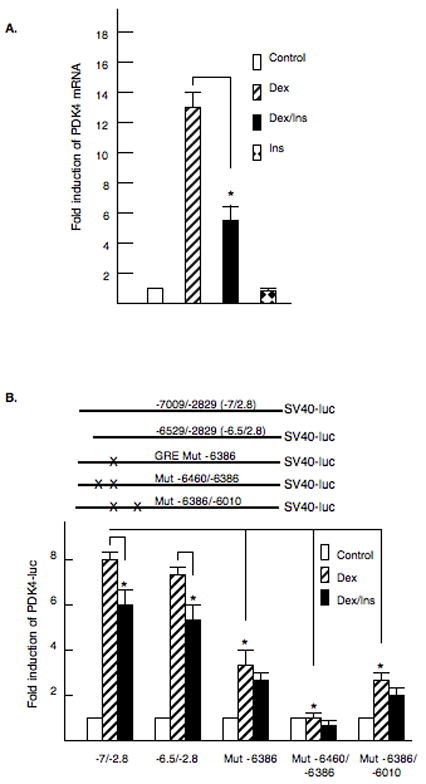
A. McA-RH7777 hepatoma cells were treated with dexamethasone (100 nM), dexamethasone and insulin (100 nM) or insulin alone (100 nM) for 24 hours. RNA was harvested and PDK4 mRNA abundance measured by real time PCR. The data are presented as the fold induction of PDK4 mRNA abundance. The values are the average of three repeats. B. A model of the SV40-luciferase vectors and the site of the mutations are shown. McA-RH7777 hepatoma cells were cotransfected with the −7009/−2829 (−7/−2.8) SV40-luciferase reporter and mammalian expression vector for glucocorticoid receptor. Dexamethasone (100 nM) or dexamethasone plus insulin (100 nM) were added for 24 hrs. Luciferase activity was normalized by protein content and renilla activity. Site directed mutagenesis was used to disrupt GR binding sites at position −6460, −6386 and −6010 in the −7/−2.8 SV40-luciferase construct. The data are presented as the relative luciferase activity ± S.E. The induction of luciferase activity was corrected for renilla and protein content. All transfections were done in duplicate and repeated four times. The p< 0.01 is indicated by the * asterisk.
To demonstrate that the glucocorticoid receptor (GR) was bound to this region of the promoter in vivo, we conducted ChIP assays in primary rat hepatocytes using the antibody for the GR. Primer sets were constructed for three regions of the gene including the region containing the GREs (−6634/−6377), a central region of the promoter (1535/−1228) and the proximal promoter (−591/−338) (Fig. 2A). Primary rat hepatocytes were crosslinked with 1% formaldehyde four hours after the addition of dexamethasone or dexamethasone plus insulin. Addition of dexamethasone increased the association of GR with the GRE region 9.1 ± 2.3 fold (Fig 2B and 2C). Interestingly, addition of insulin decreased the association of the GR with the upstream promoter region suggesting that insulin inhibits PDK4 gene expression in part by decreasing association of the GR with the promoter. Unexpectedly, a weak association of the GR with the proximal promoter was observed (Figure 2, panel B). Although the proximal (−591/−338) PDK4 promoter does not have an identified GRE, the addition of glucocorticoids increased the association of the GR with the proximal promoter 2.6 ± 0.6 fold (Fig 2C). One possible explanation for this apparent weak association of the GR with the proximal PDK4 promoter is that it contacts the basal transcription machinery through DNA looping. Consistent with this interpretation, glucocorticoids do not induce the PDK4-luciferase vectors driven by this proximal region of the PDK4 promoter alone (data not shown). We did not observe association of GR with the −1535/1228 region of the promoter.
Figure 2. Glucocorticoid receptor binds to the PDK4 promoter.
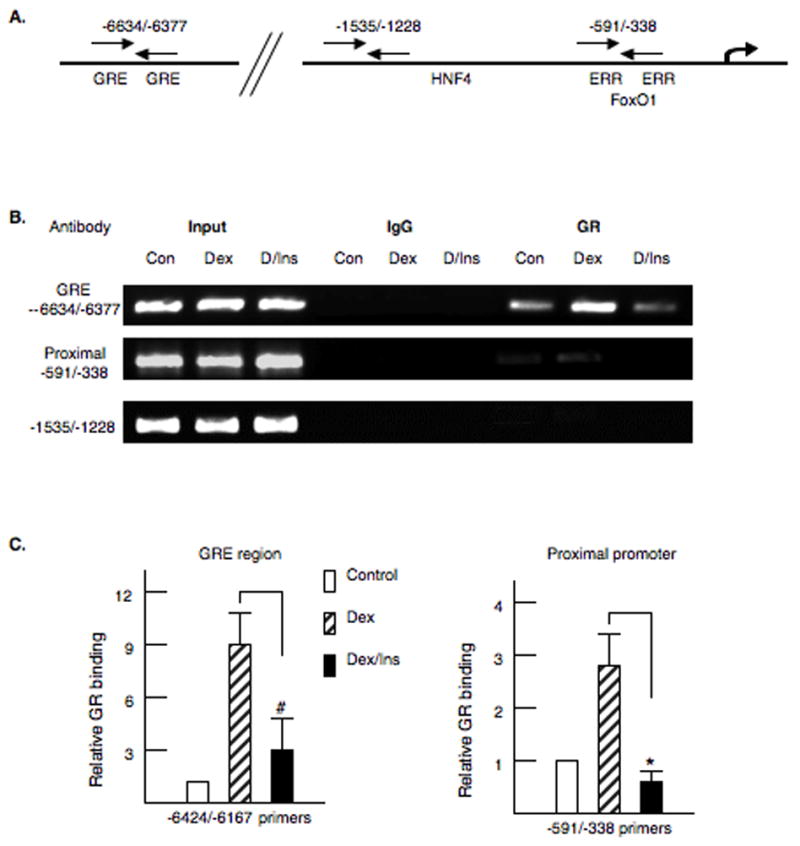
Chromatin immunoprecipitation (ChIP) assays were conducted with the PDK4 gene and antibodies to the glucocorticoid receptor (GR). Primary rat hepatocytes were treated with dexamethasone (100 nM) or dexamethasone plus insulin (100 nM). After 4 hours of hormone treatment, the cells were crosslinked with 1% formaldehyde and the ChIP assays conducted as described in the materials and methods. A. The location of the primer sets used for PCR amplification is indicated in the model. B. Representative PCR products are shown for each primer set. Also shown are the PCR amplifications for input DNA and the IgG control. C. The PCR products were quantified with Quantity One software. The changes in receptor binding are shown. The ChIP assays were repeated four times on independent preparations of primary rat hepatocytes. The p< 0.01 for the insulin inhibition is indicated by the * asterisk and p< 0.05 indicated by #.
Previously, we reported that PGC-1α induced the PDK4 gene through interactions with ERRα in the proximal promoter [17]. Previous studies suggested that PGC-1α would amplify the induction of gluconeogenic genes by dexamethasone and cAMP [32]. Therefore, we tested whether overexpression of PGC-1α would increase the expression the −7/−2.8 SV40-luciferase vector or increase the glucocorticoid induction. Our data indicate that PGC-1α does not enhance the induction of PDK4 through this region of the promoter or increase the glucocorticoid induction (Fig 3A). In addition, we tested whether PGC-1α could enhance glucocorticoid responsiveness by co-transfecting the −6529/+78 PDK4-luciferase reporter with PGC-1α. Basal expression of this reporter was elevated 2.1 fold by PGC-1α, but the glucocorticoid responsiveness was not enhanced (Fig 3A). To determine if PGC-1α association with the PDK4 gene was altered by dexamethasone, we conducted ChIP assays in primary hepatocytes treated with dexamethasone or dexamethasone plus insulin for four hours (Fig. 3B). Our data demonstrated that PGC-1α is associated with the proximal promoter of the PDK4 gene. However, no change in PGC-1α association with the proximal promoter was observed at 4 hours in response to either dexamethasone or insulin. In the ChIP assays, PGC-1α was found to be associated with the GRE region of the PDK4 gene in some of the PGC-1 αpulldowns although this association was inconsistent (data not shown). Given that PGC-1α cannot induce through the −7009/−2829 region of the PDK4 gene, we speculate that any interactions of PGC-1α with distal region of the PDK4 gene must reflect looping of the proximal promoter to the distal promoter.
Figure 3. PGC-1α is associated with the PDK4 promoter.
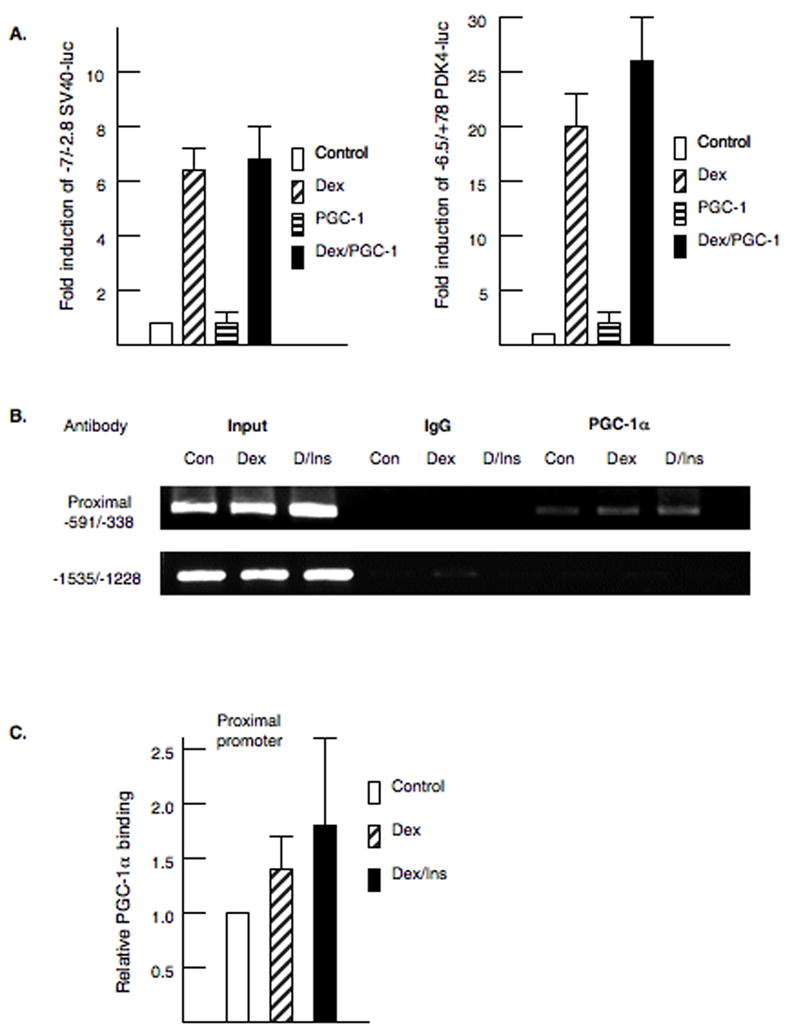
A. As shown in the left panel, McA cells were transfected with −7/−2.8 SV40-luciferase, the glucocorticoid receptor and pSV or pSV-PGC-1α exactly as described in figure 1. Dexamethasone (100 nM) was added for 24 hrs. On the right panel, McA cells were transfected with −6.5/+78 PDK4-luciferase. B. Chromatin immunoprecipitation (ChIP) assays were conducted in primary rat hepatocytes using an antibody to PGC-1α. Cells were treated with dexamethasone (100 nM) or dexamethasone plus insulin (100 nM) for 4 hours. The ChIP assays were repeated four times on independent preparations of primary rat hepatocytes. Representative PCR products are shown for each primer set. Also shown are the PCR amplifications for input DNA and the IgG control. C. The PCR products were quantified as described in the materials and methods and the legend to figure 2.
Our next experiments investigated the role of the ERRα sites in the proximal promoter of the PDK4 gene in glucocorticoid and insulin responsiveness. Each ERR site was disrupted by site directed mutagenesis. Since ERRα stimulates transcription in conjunction with the coactivator PGC-1α, we cotransfected mammalian expression vectors for ERRα and PGC-1α with the −1529/+78 PDK4-luciferase reporter [33,34]. ERRα and PGC-1α induced PDK4-luciferase 9.7 ± 1.2 fold and this induction was inhibited 62% by insulin (Fig. 4A). Mutation of the 5′ ERR binding site (Mut-370) decreased the ERRα and PGC-1α stimulation, but insulin still inhibited the induction by 48%. Interestingly, mutation of the 3′ ERR site (Mut-338) decreased the ERRα/PGC-1α induction and reduced the inhibition by insulin to 32% which is significantly less than the insulin inhibition of the wild type vector. These data suggest that the 3′ ERRα binding site contributes to the inhibition of PDK4 by insulin. In addition, we tested whether insulin might act in part by disrupting the ERRα and PGC-1α interactions. Transfection of ERRα alone induced the −1529/+78 PDK4-luc vector (8.5 fold), and this stimulation was inhibited by insulin indicating that insulin can directly inhibit the induction by ERRα (Fig 4B). PGC-1α also induced PDK (3-fold) and this response was also inhibited by insulin (Fig 4B). Co-transfection of ERRα and PGC-1α resulted in a synergistic (22- fold) activation of PDK4-luc (Fig 4B) and this effect was attenuated by insulin. ERRα recruits PGC-1α to promoters via interactions with both the L2 and L3 motifs in PGC-1α [35]. We tested whether mutation of the L2/L3 motifs in PGC-1α (PGC-1α L2/L3) would alter the insulin inhibition. Mutation of these motifs decreased the induction by ERRα and PGC-1α, but the insulin inhibition was not changed (Fig 4B). The alteration of L2 to L3 (L2/L9), which makes PGC-1α more ERRα selective also did not modify the insulin inhibition [35]. These data suggest that insulin does not affect PDK4 expression by directly disrupting the interactions of ERRα and PGC-1α.
Figure 4. Role of ERRα in the insulin inhibition of the PDK4 gene.
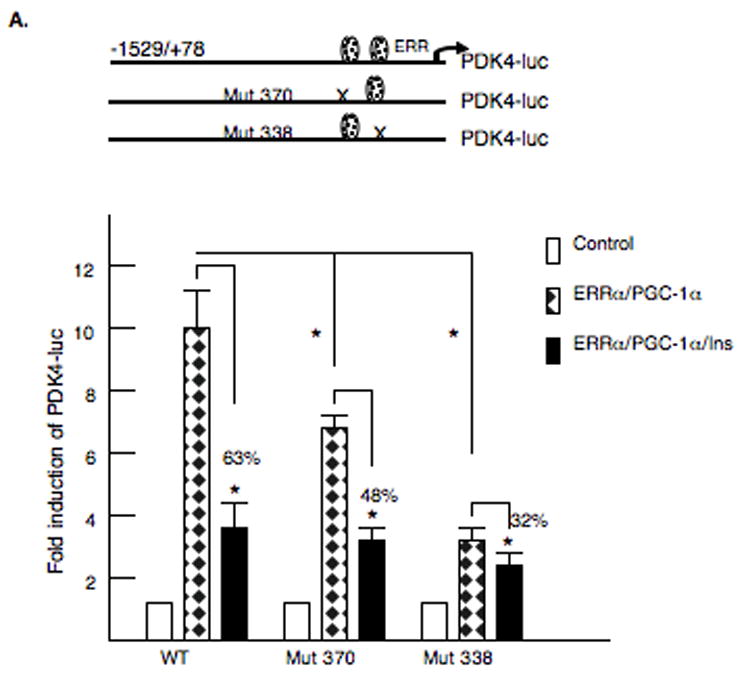

A. McA-RH7777 hepatoma cells were transfected with 2 μg of rat PDK4-luciferase reporter gene −1529/+78 region or rat PDK4-luciferase reporter gene −1529/+78 with the ERR sites mutated. A model of the mutated PDK4 promoters is shown. The expression vectors for ERRα (250 ng) and PGC-1α (1 μg) were cotransfected and TK-Renilla (0.2 μg) was included as a transfection control. Cells were harvested 36 h after transfection. The luciferase activity was corrected for protein content and Renilla activity. All transfections were done in duplicate and repeated three or four times. Insulin was used at a concentration of 100 nM and was added for 24 h. The data are expressed as the mean of the fold induction ± S.E. in the presence or absence of insulin. All transfections were performed in duplicate and repeated four times. The p< 0.01 is indicated by the * asterisk. B. A model of the PGC-1α mammalian expression vectors is shown. The −1529/+78 PDK4 luciferase reporter gene was transfected into McA-RH7777 hepatoma cells along with PGC-1α vectors where the L2 and L3 domains were altered. Luciferase assays were carried out after 40 h and values were normalized by protein content and Renilla activity. The luciferase activity observed following transfection with the mutant vectors was plotted as a percentage of the wild-type PGC-1α vector. The data are presented as the average percentage activation of luciferase ± S.E. All transfections were performed in duplicate and repeated four times.
To determine if the ERRα sites participate in the glucocorticoid induction of PDK4, we introduced these mutations into the −6529/+78 PDK4-luciferase vector (Fig 5). These plasmids were cotransfected with the glucocorticoid receptor into McA-RH7777 cells. The full-length vector was induced 22.7 ± 4.2 fold by dexamethasone and this induction was inhibited 70% by insulin (Fig 5A). Mutation of the −370 ERR site or the −338 ERR site had no effect on the glucocorticoid induction. However, disruption of the −338 ERR site reduced the insulin inhibition from 71% to 42% and this was significantly less than the insulin inhibition of the wild type promoter. These data indicate that the proximal ERRα site is involved in the insulin inhibition. We also tested whether mutation of the FoxO1 site (Mut 347) would alter glucocorticoid responsiveness. The dexamethasone induction was reduced from 22 fold to 13 fold, and the ability of insulin to inhibit the PDK4 gene was eliminated (Fig 5A). To further examine the role of ERRα in PDK4 expression, we tested whether knocking down the ERRα isoform would reduce the mRNA abundance of the PDK4 gene. McA-RH777 cells were infected adenoviruses expressing a short hairpin siRNA specific for the ERRα mRNA. Basal expression of the PDK4 gene was decreased significantly by 35% (Fig. 5B). However, the induction of PDK4 by glucocorticoids was not altered suggesting that ERRα is not involved in the glucocorticoid induction of PDK4. The ERRα mRNA levels were decreased 60% by the siRNA. Our data suggest that ERRα stimulates the basal expression of PDK4 but is not involved in the glucocorticoid induction of this gene.
Figure 5. ERRα participates in regulation of PDK4 expression.
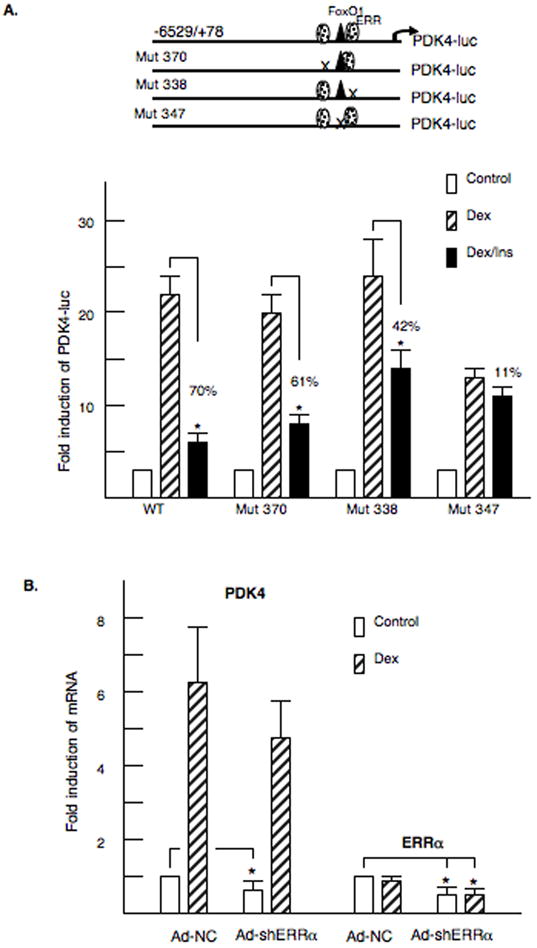
A. At the top, models of the −6529/+78 PDK4-luciferase vectors are shown. McA-RH7777 cells were transfected with PDK4- luciferase vectors and 100 nM dexamethasone or 100 nM insulin were added for 24 hours. The luciferase activity was corrected for renilla and protein content. Transfections were repeated four to six times in duplicate. B. McA-Rh7777 cells were infected with adenoviruses expressing shRNA for ERRα (Ad-shERRα) or the (Ad-NC). The infection conditions are described in the Materials and Methods. RNA was isolated, and the mRNA abundance of PDK4 and ERRα was measured by real time PCR. The data are expressed as the mean of the -fold induction ± S.E. of mRNA abundance of the shRNA ERRα infected cells relative to the NC-infected cells. The data represent the average of four independent experiments.
Finally, FoxO1 has been identified as an important transcription factor in the insulin inhibition of several genes [25,36]. We disrupted the FoxO1 binding site in the context of the −1529/+78 PDK4-luciferase gene. Mutation of this site decreased the induction by ERRα and PGC-1α by 50% (Fig 6A). However, the ability of insulin to decrease PDK4 gene expression was greatly reduced indicating that FoxO1 has an important role in the insulin regulation of PDK4 expression. FoxO1 binding was also examined by ChIP analysis. Primary rat hepatocytes were treated with glucocorticoids or glucocorticoids plus insulin for four hours and then crosslinked with 1% formaldehyde. The association of FoxO1 was not significantly altered by glucocorticoids but insulin decreased FoxO1 binding (Fig 6B and 6C). These data suggest that FoxO1 is a key participant in the insulin regulation of PDK4 gene expression.
Figure 6. Foxo1 participates in the insulin inhibition of PDK4 gene expression.
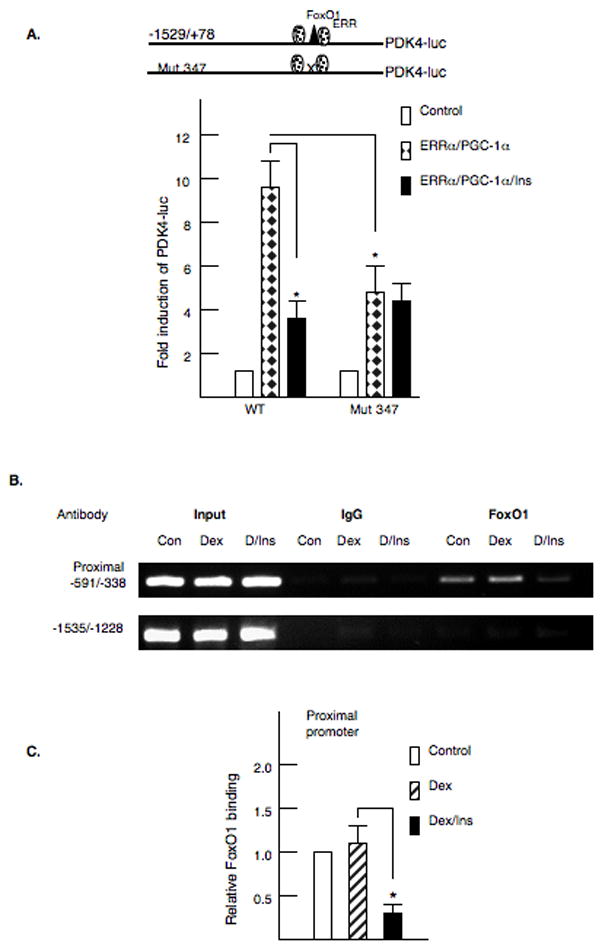
A. The FoxO1 site in the −1529/+78 site in the PDK4 promoter was disrupted by site directed mutagenesis. The mutant vector was transfected into McA cells with ERRα and PGC-1α as described in the legend to figure 4. Transfections were repeated four times in duplicate. B. Primary rat hepatocytes were treated with dexamethasone or insulin for 4 hours prior to crosslinking with 1% formaldehyde. Chromatin immunoprecipitation (ChIP) assays were conducted with the PDK4 promoter and an antibody to FoxO1 essentially as described in the legend to figure 2. C. The PCR products were quantified as described in the materials and methods. The values represent the average of four independent preparations of hepatocytes.
Discussion
PDK4 is a key regulator of PDC activity, pyruvate oxidation and glucose homeostasis [1,2]. The importance of PDK4 in metabolic regulation has been confirmed in PDK4 knock-out mice which exhibit lower blood glucose levels and decreased hepatic gluconeogenesis due to elevated glucose utilization [37]. In addition, PDC activity was increased leading to elevated glucose oxidation to acetyl-CoA [37]. Analysis of these mice supported the previously proposed concept that pharmacologic inhibition of PDK4 activity ameliorated hyperglycemia [38,39]. Prior investigations demonstrated that PDK4 abundance is controlled by changes in gene expression [5,6,8]. In these studies, we investigated the transcriptional mechanisms underlying the regulation of the rat PDK4 gene by glucocorticoids and insulin. Our data demonstrate that induction of PDK4 transcription by glucocorticoids is mediated via two GREs in the PDK4 promoter that are located more than 6,000 base pairs 5′ to the start site of transcription. Moreover FoxO1, which is bound to the proximal promoter, participates in the glucocorticoid responsiveness of the PDK4 gene. Insulin decreases expression of the PDK4 gene through three promoter elements including the GREs, the FoxO1 site and an ERR element. Interestingly, PGC-1α, which stimulates PDK4 gene expression, does not appear to be necessary for the acute regulation of PDK4 by glucocorticoids or insulin.
Glucocorticoids, which contribute to the induction of the PDK4 gene in fasting and diabetes, act in part via GREs in the distal promoter. Recent reports have indicated that subtle changes in the GRE profoundly alter transcriptional activation [40]. In addition, analysis of several glucocorticoid responsive genes has suggested that the GR functions in association with other transcription factors which constitute glucocorticoid response units (GRU) [19]. The involvement of these accessory factors allows additional gene specific modulation of glucocorticoid responsiveness. The PEPCK gene possesses a GRU that has been analyzed extensively. Studies with the PEPCK gene have identified two weak GREs which cannot confer a glucocorticoid response in isolation [41]. Four additional factors including HNF4, FoxO1, COUP-TF and C/EBPβ are required for the full glucocorticoid induction of the PEPCK gene [42,43]. Our studies showed that FoxO1 participated in the glucocorticoid induction of PDK4 although half of the glucocorticoid induction remained following mutation of the FoxO1 site. We and others have previously identified FoxO1 binding sites in the PDK4 promoters of mouse, human and rat [11,16,44]. It was reported that overexpression of a constitutively active FoxO1 strongly stimulated human PDK4 luciferase reporters and enhanced the induction by dexamethasone [11]. In our hands, overexpression of FoxO1 did not induce the rat PDK4 gene (data not shown). In contrast to the rat promoter with widely separated GREs and FoxO1 sites, the human PDK4 promoter contains three FoxO1 sites that are in close proximity to the GRE [11]. Our observation that the GR is associated with the proximal promoter of the PDK4 gene in our ChIP assay suggests that looping from the GRE region to the proximal promoter may occur. Overall, our data and that of others indicate that the GRU of the PDK4 gene includes at least FoxO1 and GR. The potential role of other factors including HNF4 and C/EBPβ which comprise the GRUs of several hepatic genes has not been examined [19].
PGC-1α stimulates the PDK4 gene in liver and skeletal muscle [17,31]. Our experiments did not indicate that PGC-1α was necessary for the acute induction of PDK4 by dexamethasone as overexpression of PGC-1α did not enhance the glucocorticoid induction of PDK4 in transient transfection assays. Also in our ChIP assays, we were unable to detect an increased association of PGC-1α with the PDK4 promoter following dexamethasone addition at the four-hour time point. Previous studies in H4IIE cells demonstrated that PGC-1α was not required for the glucocorticoid induction of the PEPCK gene although it was shown that PGC-1α acted as a transcriptional amplifier [45]. Likewise in hepatocytes from PGC-1α knockout mice, PEPCK and G6Pase were still induced by cAMP and dexamethasone [32]. We and others have found that PGC-1α is recruited to the PDK4 promoter by ERRα [17,31]. Our promoter mutagenesis studies suggested that ERRα was not involved in the glucocorticoid induction of PDK4, and this result was reinforced by the knockdown of ERRα in McA cells. Although ERR is not involved in the glucocorticoid induction, ERRα does increase basal expression of PDK4 and recruit PGC-1α to the PDK4 gene. Previously, we reported that FoxO1 was not involved in the recruitment of PGC-1α to the PDK4 promoter [16]. This observation was based on the mutation of the FoxO1 site in the context of short oligomers ligated in front of SV40-luciferase. Here, we observed that mutation of the FoxO1 site in the context of the PDK4 promoter did decrease the induction by PGC-1α suggesting that FoxO1 does recruit PGC-1α to the promoter. In addition, we had reported that PGC-1β does not induce PDK4 expression [46]. This observation is consistent with the fact that PGC-1α but not PGC-1β can interact with FoxO1 and may explain the PGC-1α selective induction of the PDK4 gene [47].
The ability of insulin to decrease PDK4 gene expression has been described in animal models and in cell systems [4]. Elevated insulin, whether from the refeeding of fasted animals or administration of insulin to diabetic animals, decreases PDK4 mRNA levels in heart, muscle, kidney and liver [8,48]. In addition, liver specific knock-out of IRS-2 led to elevated PDK4 gene expression suggesting that insulin represses PDK4 [49]. Multiple mechanisms have been described for the insulin inhibition of gene expression. First, FoxO1 stimulates genes involved in gluconeogenesis and inhibits glycolysis [24,50,51]. Insulin via the activation of PKB phosphorylates FoxO1 on serines 256 and 319 and threonine 29 leading to FoxO1 dissociation from genes such as PEPCK and G6Pase as well as the nuclear exclusion of FoxO1 [21,24,50]. FoxO1 clearly plays a significant role in the insulin inhibition of PDK4. In addition, insulin dissociates the GR from the distal PDK4 gene. However, the most unexpected observation was that mutation of the 3′ ERR site reduced the ability of insulin to inhibit PDK4 transcription. These results which were obtained in the context of the PDK4 promoter were slightly different than our previous observations using multiple copies of isolated elements driving the SV40-luciferase gene [16]. In that context, we did not identify a role for ERRα in the insulin regulation of PDK4. Our results implicating FoxO1 in the insulin regulation of gene expression are consistent with previous observations both for the human PDK4 gene and several other genes [21,36].
One surprising result from our experiments was that PGC-1α was not involved in the short-term regulation of PDK4 gene expression by insulin. It was reported that the association of PGC-1α with the PEPCK promoter was decreased following insulin addition [25]. In other studies, we observed that after 24 hours of insulin administration there was decreased association of PGC-1α with the PDK4 promoter [16]. However, after 4 hours, PGC-1α was still associated with the PDK4 gene even though there was a substantial decrease in the binding of FoxO1. Recent studies have shown that PKB can phosphorylate and inhibit PGC-1α suggesting a link of phosphorylation with insulin action [52]. Our results suggest that PGC-1α dissociation was not involved in the acute inhibition of PDK4 expression. We also observed by ChIP assay that insulin caused dissociation of the glucocorticoid receptor from the PDK4 promoter. We have not found that FoxO1 can regulate through the upstream region of the PDK4 gene suggesting that this portion of the insulin regulation is independent of FoxO1. Factor dissociation is a model for the insulin inhibition of gene expression as insulin decreases association of coactivators including the CREB binding protein (CBP/p300) and steroid receptor coactivator (SRC-1) from the PEPCK and G6Pase promoters [53,54]. Our results suggest that the GR and FoxO1 are dissociated from the PDK4 promoter by insulin. Numerous studies indicate that insulin does not inhibit gene expression through a single promoter element or transcription factor but rather through a combination of sites and factors. Having multiple inhibitory mechanisms may allow insulin to block a number of stimulatory signals. Additional factors including the sterol regulatory element binding protein (SREBP-1c), C/EBPβ and TORC2 have been correlated with insulin inhibition of hepatic gene expression [54–56].
In this study, our data indicate that glucocorticoids and insulin regulate PDK4 gene expression though complex hormone response units. In addition to the GR, FoxO1 participates in the glucocorticoid induction of PDK4. Insulin acts through at least three elements and several transcription factors to decrease the expression of the PDK4 gene. This complex regulation likely reflects the need for a subtle modulation of PDK4 expression under a variety of physiological conditions.
Acknowledgments
This work was supported by the NIH - R01DK059368 to EAP and RO1DK75504 to MBE.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors maybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284:E855–62. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- 2.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. 2002;42:249–59. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 3.Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys. 2000;381:1–7. doi: 10.1006/abbi.2000.1946. [DOI] [PubMed] [Google Scholar]
- 4.Harris RA, Huang B, Wu P. Control of pyruvate dehydrogenase kinase gene expression. Adv Enzyme Regul. 2001;41:269–88. doi: 10.1016/s0065-2571(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 5.Holness MJ, Bulmer K, Smith ND, Sugden MC. Investigation of potential mechanisms regulating protein expression of hepatic pyruvate dehydrogenase kinase isoforms 2 and 4 by fatty acids and thyroid hormone. Biochem J. 2003;369:687–95. doi: 10.1042/BJ20021509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang B, Wu P, Bowker-Kinley MM, Harris RA. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes. 2002;51:276–83. doi: 10.2337/diabetes.51.2.276. [DOI] [PubMed] [Google Scholar]
- 7.White UA, Coulter AA, Miles TK, Stephens JM. The STAT5A-mediated induction of pyruvate dehydrogenase kinase 4 expression by prolactin or growth hormone in adipocytes. Diabetes. 2007;56:1623–9. doi: 10.2337/db06-1286. [DOI] [PubMed] [Google Scholar]
- 8.Kwon HS, Harris RA. Mechanisms responsible for regulation of pyruvate dehydrogenase kinase 4 gene expression. Adv Enzyme Regul. 2004;44:109–21. doi: 10.1016/j.advenzreg.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 9.Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun. 2001;287:391–6. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]
- 10.Holness MJ, Smith ND, Bulmer K, Hopkins T, Gibbons GF, Sugden MC. Evaluation of the role of peroxisome-proliferator-activated receptor alpha in the regulation of cardiac pyruvate dehydrogenase kinase 4 protein expression in response to starvation, high-fat feeding and hyperthyroidism. Biochem J. 2002;364:687–94. doi: 10.1042/BJ20011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004;53:899–910. doi: 10.2337/diabetes.53.4.899. [DOI] [PubMed] [Google Scholar]
- 12.Heard DJ, Norby PL, Holloway J, Vissing H. Human ERRgamma, a third member of the estrogen receptor-related receptor (ERR) subfamily of orphan nuclear receptors: tissue-specific isoforms are expressed during development and in the adult. Mol Endocrinol. 2000;14:382–92. doi: 10.1210/mend.14.3.0431. [DOI] [PubMed] [Google Scholar]
- 13.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–74. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 14.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5:345–56. doi: 10.1016/j.cmet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Horard B, Vanacker JM. Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. J Mol Endocrinol. 2003;31:349–57. doi: 10.1677/jme.0.0310349. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Ma K, Sadana P, Chowdhury F, Gaillard S, Wang F, McDonnell DP, Unterman TG, Elam MB, Park EA. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J Biol Chem. 2006;281:39897–906. doi: 10.1074/jbc.M608657200. [DOI] [PubMed] [Google Scholar]
- 17.Ma K, Zhang Y, Elam MB, Cook GA, Park EA. Cloning of the Rat Pyruvate Dehydrogenase Kinase 4 Gene Promoter: ACTIVATION OF PYRUVATE DEHYDROGENASE KINASE 4 BY THE PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR {gamma} COACTIVATOR. J Biol Chem. 2005;280:29525–32. doi: 10.1074/jbc.M502236200. [DOI] [PubMed] [Google Scholar]
- 18.Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Schoneveld OJ, Gaemers IC, Lamers WH. Mechanisms of glucocorticoid signalling. Biochim Biophys Acta. 2004;1680:114–28. doi: 10.1016/j.bbaexp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol. 2005;40:129–54. doi: 10.1080/10409230590935479. [DOI] [PubMed] [Google Scholar]
- 21.Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002;277:45276–84. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
- 23.Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR, Cohen P. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. Embo J. 2002;21:2263–71. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vander Kooi BT, Streeper RS, Svitek CA, Oeser JK, Powell DR, O’Brien RM. The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J Biol Chem. 2003;278:11782–93. doi: 10.1074/jbc.M212570200. [DOI] [PubMed] [Google Scholar]
- 25.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–5. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 26.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–39. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 27.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–83. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 29.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–8. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 30.Boustead JN, Stadelmaier BT, Eeds AM, Wiebe PO, Svitek CA, Oeser JK, O’Brien RM. Hepatocyte nuclear factor-4 alpha mediates the stimulatory effect of peroxisome proliferator-activated receptor gamma co-activator-1 alpha (PGC-1 alpha) on glucose-6-phosphatase catalytic subunit gene transcription in H4IIE cells. Biochem J. 2003;369:17–22. doi: 10.1042/BJ20021382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP. PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRalpha: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol. 2005;25:10684–94. doi: 10.1128/MCB.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–35. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 33.Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–91. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Araki M, Motojima K. Identification of ERRalpha as a specific partner of PGC-1alpha for the activation of PDK4 gene expression in muscle. Febs J. 2006;273:1669–80. doi: 10.1111/j.1742-4658.2006.05183.x. [DOI] [PubMed] [Google Scholar]
- 35.Gaillard S, Grasfeder LL, Haeffele CL, Lobenhofer EK, Chu TM, Wolfinger R, Kazmin D, Koves TR, Muoio DM, Chang CY, McDonnell DP. Receptor-selective coactivators as tools to define the biology of specific receptor-coactivator pairs. Mol Cell. 2006;24:797–803. doi: 10.1016/j.molcel.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 36.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–6. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 37.Jeoung NH, Wu P, Joshi MA, Jaskiewicz J, Bock CB, Depaoli-Roach AA, Harris RA. Role of pyruvate dehydrogenase kinase 4 (PDK4) in glucose homeostasis during starvation. Biochem J. 2006 doi: 10.1042/BJ20060125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugden MC, Holness MJ. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:151–65. [PubMed] [Google Scholar]
- 39.Crabb DW, Yount EA, Harris RA. The metabolic effects of dichloroacetate. Metabolism. 1981;30:1024–39. doi: 10.1016/0026-0495(81)90105-0. [DOI] [PubMed] [Google Scholar]
- 40.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–10. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott DK, Stromstedt PE, Wang JC, Granner DK. Further characterization of the glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. The role of the glucocorticoid receptor-binding sites. Mol Endocrinol. 1998;12:482–91. doi: 10.1210/mend.12.4.0090. [DOI] [PubMed] [Google Scholar]
- 42.Wang JC, Stromstedt PE, Sugiyama T, Granner DK. The phosphoenolpyruvate carboxykinase gene glucocorticoid response unit: identification of the functional domains of accessory factors HNF3 beta (hepatic nuclear factor-3 beta) and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol Endocrinol. 1999;13:604–18. doi: 10.1210/mend.13.4.0269. [DOI] [PubMed] [Google Scholar]
- 43.Yamada K, Duong DT, Scott DK, Wang JC, Granner DK. CCAAT/enhancer-binding protein beta is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem. 1999;274:5880–7. doi: 10.1074/jbc.274.9.5880. [DOI] [PubMed] [Google Scholar]
- 44.Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;375:365–71. doi: 10.1042/BJ20030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herzog B, Hall RK, Wang XL, Waltner-Law M, Granner DK. Peroxisome proliferator-activated receptor gamma coactivator-1alpha, as a transcription amplifier, is not essential for basal and hormone-induced phosphoenolpyruvate carboxykinase gene expression. Mol Endocrinol. 2004;18:807–19. doi: 10.1210/me.2003-0384. [DOI] [PubMed] [Google Scholar]
- 46.Sadana P, Park EA. Characterization of the transactivation domain in the peroxisome proliferator activated receptor gamma coactivator (PGC-1) Biochem J. 2007 doi: 10.1042/BJ20061526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–8. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 48.Sugden MC, Bulmer K, Gibbons GF, Knight BL, Holness MJ. Peroxisome-proliferator-activated receptor-alpha (PPARalpha) deficiency leads to dysregulation of hepatic lipid and carbohydrate metabolism by fatty acids and insulin. Biochem J. 2002;364:361–8. doi: 10.1042/BJ20011699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest. 2006;116:101–14. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo SH, Montminy M, Unterman TG. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–17. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 51.Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–53. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 52.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–6. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 53.Hall RK, Wang XL, George L, Koch SR, Granner DK. Insulin Represses PEPCK Gene Transcription by Causing the Rapid Disruption of an Active Transcription Complex: A Potential Epigenetic Effect. Mol Endocrinol. 2006 doi: 10.1210/me.2006-0307. [DOI] [PubMed] [Google Scholar]
- 54.Duong DT, Waltner-Law ME, Sears R, Sealy L, Granner DK. Insulin inhibits hepatocellular glucose production by utilizing liver-enriched transcriptional inhibitory protein to disrupt the association of CREB-binding protein and RNA polymerase II with the phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem. 2002;277:32234–42. doi: 10.1074/jbc.M204873200. [DOI] [PubMed] [Google Scholar]
- 55.Chakravarty K, Wu SY, Chiang CM, Samols D, Hanson RW. SREBP-1c and Sp1 interact to regulate transcription of the gene for phosphoenolpyruvate carboxykinase (GTP) in the liver. J Biol Chem. 2004;279:15385–95. doi: 10.1074/jbc.M309905200. [DOI] [PubMed] [Google Scholar]
- 56.Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–9. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]