

Abstract
The microtubule-associated protein tau is important to normal neuronal activity in the mammalian nervous system. Aggregated tau is the major component of neurofibrillary tangles (NFTs), structures present in the brains of people affected by neurodegenerative diseases called tauopathies. Tauopathies include Alzheimer's disease (AD), frontotemporal dementia with Parkinsonism (FTDP) and the early onset dementia observed in Down syndrome (DS; trisomy 21). Splicing misregulation of adult-specific exon 10 results in expression of abnormal ratios of tau isoforms, leading to FTDP. Positions +3 to +19 of the intron downstream of exon 10 define a hotspot: point mutations in it result in tauopathies. All these mutations increase exon 10 inclusion except for mutation +19, which almost entirely excludes exon 10. To investigate the tau connection between DS and AD, we examined splicing factors located on chromosome 21 for their effect on tau exon 10. By co-transfections, co-immunoprecipitations and RNAi constructs, we discovered that one of them, hnRNPE3 (PCBP3), modestly activates splicing of exon 10 by interacting with its proximal downstream intron around position +19. These results, coupled with the developmental profile of hnRNPE3, suggest a pathogenic role for splicing factors on chromosome 21 in neurodegenerative diseases with tangles and create a connection between tau splicing and the early-onset dementia of Down syndrome.
Keywords: MAP tau, Isoform ratios, Alternative splicing regulation, Heterogeneous nuclear ribonucleoprotein E3, tangle dementia, Down syndrome
1. Introduction
Down syndrome (DS) is the most common genetic defect, estimated to affect about 1 in 800 live births. DS is caused by partial or complete trisomy of the long arm of chromosome 21 and its phenotype is complex and varies in severity. It includes defects in the heart and gastrointestinal system, increased susceptibility to leukemia, immune system deficiencies and skeletal abnormalities (Gardiner et al., 2002).
The feature shared by all DS sufferers is mental retardation characterized by specific cognitive and behavioral deficits. DS brains show decrease in weight and neuron number, abnormal neuronal differentiation and structural changes in synapses. People with DS develop early cognitive decline and neuropathological features characteristic of Alzheimer Disease (AD), including the presence of neurofibrillary tangles (NFTs; Goedert and Jakes, 2005).
Tau is a microtubule-associated protein (MAP) enriched in axons of mature and growing neurons. Hyperphosphorylated, microtubule-dissociated tau protein is the major component of NFTs, a hallmark of many neurodegenerative diseases (Goedert and Jakes, 2005). Null tau mice, though viable, show morphological and cognitive defects (Ikegami et al., 2000). Additionally, human pedigrees that contain microdeletions and microduplications in the tau locus result in developmental defects and learning disabilities (Shaw-Smith et al., 2006; Kirchoff et al., 2007).
The human tau gene undergoes extensive alternative splicing that is regulated spatially and temporally (Andreadis, 2006; Liu and Gong, 2008). Exon 10 modulates the C-terminus of the tau protein and encodes an additional microtubule binding domain. Exon 10 is adult-specific in rodents and humans but with a crucial difference relevant to neurodegeneration: in adult rodents, exon 10 becomes constitutive. In contrast, in adult humans exon 10 remains regulated in the central nervous system where the 10+ and 10− isoforms are present in a 1:1 ratio.
Misregulation of tau exon 10 splicing that disturbs the 1:1 ratio causes neurodegeneration whether the cause is cis or trans: Mutations in exon 10 that produce wild-type tau nevertheless result in tangle-only dementias grouped under the term “tauopathies” (represented by inherited frontotemporal dementia with Parkinsonism, FTDP-17; Goedert and Jakes, 2005); changes in factors that influence exon 10 splicing result in the cognitive defects that arise in myotonic dystrophy 1 (DM1; Jiang et al., 2004). The correct ratio of tau exon 10 is also disturbed in AD (Glatz et al., 2005; Conrad et al., 2007) and DS (Mehta et al., 1999; Shi et al., 2008).
Bioinformatics analysis of human genome indicates that almost all human genes are alternatively spliced (Pan et al., 2008). Alternative splicing plays a critical role in controlling differentiation and development (Stamm et al., 2005), and misregulation of alternative splicing is the cause of many life-threatening human diseases (Tazi et al., 2009). Despite the high fidelity of exon recognition in vivo, it is currently impossible to accurately predict alternative exons; it appears that combinatorial control and “weighing” of splice element strength are used to enable precise recognition of the short and degenerate splice sites (Hertel, 2008).
Exonic and intronic enhancers and silencers are involved in splicing regulation (Wang and Burge, 2008). These cis elements are regulated by trans splicing factors that mostly belong to two superfamilies, the SR/SR-like and hnRNP proteins (Long and Caceres, 2009; Martinez-Contreras et al., 2007). Several mammalian splicing factors are enhanced in or restricted to neurons. Nevertheless, it appears that the exquisite calibration of mammalian alternative splicing is primarily achieved by spatial and temporal variation in the expression and activity levels of quasi-ubiquitous splicing regulators (Hertel, 2008).
Exon 10 splicing is affected by exonic and intronic enhancers and silencers as well as by several trans factors and their phosphorylation (Andreadis, 2006; Liu and Gong 2008). Investigations of dementia pedigrees established that the proximal downstream intron of exon 10 is a hotspot for tauopathy mutations. We previously showed that SR protein 9G8 inhibits exon 10 splicing by interacting with this region (specifically residue +14; Gao et al., 2007). In this report we show that hnRNPE3 (also known as poly(rC) binding protein 3) modestly activates splicing of exon 10 by interacting with a C triplet in positions +19 to +21 of the intron. This extends our earlier findings of similar action by hnRNPE2, a close relative of hnRNPE3 (Broderick et al., 2004). Because hnRNPE3 is located on chromosome 21, the results of this work suggest a connection between tau splicing regulation and the early-onset dementia in DS.
2. Materials and Methods
2.1. Plasmid construction and mutagenesis
The starting point for mutant constructs was SP/10L (Fig. 1B; Wang et al., 2003) which contains human tau exon 10 plus 471 bp of its upstream intron and 408 bp of its downstream intron inserted into the EcoRI site of vector pSPL3 (Invitrogen). Deletions within the 30 bp downstream of exon 10 (I10-Δ3/10, I10-Δ11/18, I10-Δ19/26 and I10-Δ23/29) and point mutations reproducing several FTDP pedigree mutations (I10-M11, I10-M12, I10-M13, I10-M14, I10-M16 and I10-M19) were previously described (Gao et al., 2007; Wang et al., 2003, 2005). The mutations are diagrammed in Fig. 2A (deletions) and Fig. 3A (point mutations).
Fig. 1.

Two splicing factors located on chromosome 21 activate splicing of tau exon 10. (A) Alignment of human (H) and mouse (M) of tau exon 10 and its downstream proximal intron, up to position +30. The exon is in uppercase, the intron in lowercase. The boundaries of the three riboprobe constructs are indicated. The C triplet that is the likely site of the hnRNPE3 interaction is shaded. (B) Schematic representation of construct SP/10L. P and T represent the vector promoter and terminator. The numbers on each side of exon 10 show (above) how many kilobases of flanking introns are present and (below) the extent of native introns flanking exon 10. The major splicing product is shown by solid, the minor one by dashed lines. (C) RT-PCR of SP/10L in COS cells in the presence of chromosome 21 splicing factors. The RT-PCR products come from 1:1 co-transfections of SP/10L and the factors. The asterisks identify the two factors that influence exon 10 splicing. Primer pair: SPL-LS/SPL-LN.
Fig. 2.

Deletion of the C triplet in positions 19-21 of the downstream intron abolishes the ability of hnRNPE3 to activate exon 10 splicing and to bind to the tau pre-mRNA. (A) Nucleotides +1 to +30 of the intron downstream of tau exon 10. The deletions are demarcated by bars above or below the sequence. The C triplet that is the likely site of the hnRNPE3 interaction is shaded. (B) RT-PCR of wild-type and deleted SP/10L in COS cells in the absence and presence of hnRNPE3. The RT-PCR products come from 1:1 co-transfections of tau constructs and FLAG- hnRNPE3. Primers and graph conventions are as in Fig.1C. (C) 32P-labeled riboprobes containing wild-type or deleted exon 10 were incubated with extracts from COS cells transfected with FLAG- hnRNPE3 and were immunoprecipitated by anti-FLAG monoclonal antibody M2. Amounts of riboprobe bound to the hnRNPE3 protein were calculated relative to the nonspecific binding of the FLAG vector transfection by measuring the counts retained after washing (means ± SD of three analyses). Equal amounts of FLAG- hnRNPE3 were present in each experiment.
Fig. 3.
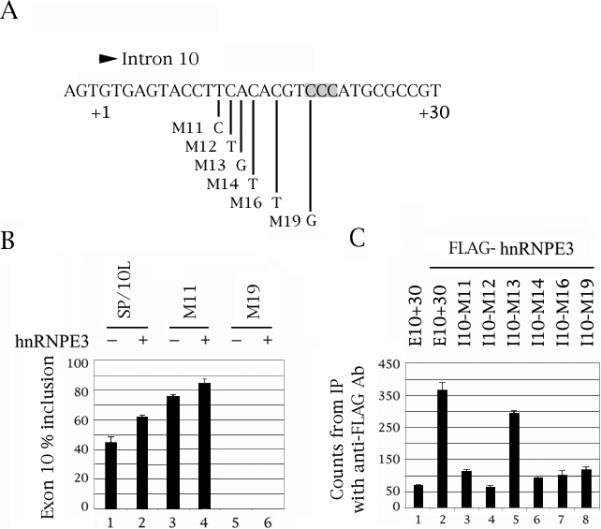
Mutation of C residue +19 of the intron downstream of tau exon 10, which affects an intronic enhancer, abolishes hnRNPE3 action and binding. (A) The last three nucleotides of exon 10 and nucleotides +1 to +30 of the intron downstream of tau exon 10. The point mutations are indicated. The C triplet that is the likely site of the hnRNPE3 interaction is shaded. (B) RT-PCR of wild-type and point-mutated SP/10L in COS cells in the absence and presence of hnRNPE3. The RT-PCR products come from 1:1 co-transfections of tau constructs and FLAG-hnRNPE3. Co-transfections were not done with mutants M12-16, because they exhibit 100% exon 10 inclusion and would not show additional activation by hnRNPE3. Primers and graph conventions are as in Fig.1C. (C) 32P-labeled riboprobes containing wild-type or point-mutated exon 10 were incubated with extracts from COS cells transfected with FLAG-hnRNPE3 and were immunoprecipitated by anti-FLAG monoclonal antibody M2. Amounts of riboprobe bound to the hnRNPE3 protein were calculated relative to the nonspecific binding of the FLAG vector transfection by measuring the counts retained after washing (means ± SD of three analyses). Equal amounts of FLAG- hnRNPE3 were present in each experiment.
To generate riboprobes, we inserted a human tau genomic fragment into vector pGEM-TE (Promega) to create construct E10+30. This contains human tau exon 10 plus 30 bp of its downstream intron. Two constructs were subsequently created from E10+30: E10-80 contains the 80 5'most bp of exon 10. E10-13+30 contains the 13 3'most bp of exon 10 plus 30 bp of its downstream intron (Gao et al., 2007; Fig. 1A). The deletions and point mutations created in SP/10L were also recreated in E10+30 as previously described (Gao et al., 2007).
We obtained cDNAs for hRNPE3, RBM11, U2AF35 and SRA4 from the I.M.A.G.E. Consortium. To express splicing factors in eukaryotic cells, we inserted the following cDNAs into N-terminal FLAG vectors: U2AF35 into Tag2C (Stratagene), hnRNPE3 and hRNPE2 into Tag2B (Stratagene; Broderick et al., 2004), SRp75 and SRA4 into Tag2A (Stratagene), 9G8 (Gao et al., 2007) into CMV-6c and Nova1 into CMV-6a (Sigma).
We also placed hnRNPE3 into the N-terminal 3xMyc vector Tag3A (Stratagene) for co-precipitations. We created two domain deletion variants of hnRNPE3 by PCR and cloned them into FLAG vector CMV-6c (Sigma). HnRNPE3N (primers E3-S/E3-KH2N) contains the first two and hnRNPE3C ((primers E3-KH2S/E3-N) contains the last two KH domains of hnRNPE3 (Fig 4A). The primers used to create the hnRNPE3 variants are listed in Table 1.
Fig. 4.

The N-terminal KH domain of hnRNPE3 is required for activation of exon 10 splicing. (A) Diagram of hnRNPE3 variants used in this study. The three KH domains and the amino acids that code for them are indicated. Total length of hnRNPE3 is 314 amino acids. (B) The hnRNPE3 variants express as FLAG fusion proteins in COS cells. The proteins on the Western blot were detected by anti-FLAG monoclonal antibody M2. Protein markers are indicated on the right of the panel. The cell lysates were normalized for protein content. (C) RT-PCR of SP/10L in COS cells in the presence of full-length and deletion variants of hnRNPE3. The RT-PCR products come from 1:1 co-transfections of SP/10L and FLAG-hnRNPE3 fusion constructs. Primers and graph conventions are as in Fig. 1C.
Table 1.
Primers
| Name | Nt | Strand Location | Sequence | |
|---|---|---|---|---|
|
hnRNPE3 deletion constructs | ||||
| E3-S | 29 | Start of ORF | CGGAATTCATGGAGTCCAAGGTCTCAGAA | |
| E3-N | 28 | A | End of ORF | GCGTCGACTTACAGCGTGCCCATCCCGG |
| E3-KH2S | 29 | KH2 domain start | CGGAATTCCCAGTGACGCTGAGGCTGGTG | |
| E3-KH2N | 29 | A | KH2 domain end | GCGTCGACCCCCGAGATGGTCACCGCTCG |
|
PCR of endogenous and transfected hnRNPE3 | ||||
| E3-PS | 30 |  |
Start of ORF | ATGGAGTCCAAGGTCTCAGAAGGTGGCCTG |
| E3-DN | 30 | A | End of ORF | TTACAGCGTGCCCATCCCGGTGACCTCGGA |
| E3-KH2NewS | 30 |  |
KH2 domain start | TGACGCTGAGGCTGGTGGTGCCTGCCAG |
| E3-KH2NewN | 30 | A | KH2 domain end | CCCCGAGATGGTCACCGCTCGCTCCGTG |
|
PCR of endogenous and transfected tau 10 | ||||
| HT7S3 | 24 | S | In tau exon 7 | CAACGCCACCAGGATTCCAGCAAA |
| HT11N | 24 | A | In tau exon 11 | ATGTTGCCTAATGAGCCACACTTG |
| SPL3-LS | 27 | S | In SPL3 vector | TCTGAGTCACCTGGACAACCTCAAAGG |
| SPL3-LN | 27 | A | In SPL3 vector | ATCTCAGTGGTATTTGTGAGCCAGGGC |
|
hnRNPE3 RNAi constructs | ||||
| In pFIV | ||||
| E3i-1S | 23 | S | Just before KH1 | AAAGGTCCAAGGTCTCAGAAGGT |
| E3i-1N | 23 | A | Just before KH1 | AAAAACCTTCTGAGACCTTGGAC |
| E3i-2S | 23 | S | End of KH2 | AAAGGCAGATCTGTGTGGTCATG |
| E3i-2N | 23 | A | End of KH2 | AAAACATGACCACACAGATCTGC |
| E3i-3S | 23 | S | Before ORF end | AAAGCGTCCGAGGTCACCGGGAT |
| E3i-3N | 23 | A | Before ORF end | AAAAATCCCGGTGACCTCGGACG |
|
In SM2c | ||||
| SME3-1 | 21 | S | In 3' UTR | CCTGCCTCACAGATACCAATA (Open Biosystems V2HS_65894) |
| SME3-2 | 22 | S | In 3' UTR | ACGGCATGCAGTGGTAATTATT (Open Biosystems V2HS_65889) |
| SME3-3 | 22 | S | In 3' UTR | CCCGGCATGCAGTGGTAATTAT (Open Biosystems V2HS_247015) |
Nt=Length in nucleotides. S=sense, A=antisense.
Three pairs of siRNA oligonucleotides for hnRNPE3 were designed and synthesized (Table 1). Each pair was annealed and cloned into the pFIV-H1/U6 vector (SBI). We also obtained three plasmids containing shRNA for hnRNPE3 from the UMMS SiRNA Core (Table 1).
2.2 Cell culture and transfection
We cultured monkey kidney (COS), human epithelioma (HeLa) and human epithelial kidney (HEK) cells as previously described (Gao et al., 2007). The cells were grown on 60 mm plates and transfected when they reached confluence of 40% (if using LT1 from Mirus) or 80% (if using lipofectamine 2000 from Invitrogen). The medium was changed 16 hours after transfection and the cells were harvested 48 hours after transfection.
2.3. RNA preparation, reverse transcription and PCR reaction
Total RNA was isolated from transfected cells by the TRIzol method (Invitrogen). We performed reverse transcription and PCR as previously described (Gao et al., 2007; Wang et al., 2005). For analysis of endogenous hnRNPE3 levels, we used the Ambion Quantum kit with a ratio of 2:8 of 18S primers to 18S competimers as the internal control. Vectors pFIV and SM2c-GFP acted as the negative controls for the RNAi experiments.
For PCR, we used the following primer pairs (shown in Table 1): SPL-LS/SPL-LN for transfected tau, HT7S3/HT11N for endogenous tau, E3-KH2NewS/E3-KH2NewN for endogenous hnRNPE3. We did 27 PCR cycles if analyzing transfected constructs (denaturation 94 oC/1 min, annealing 62 oC/1 min, extension 72 oC/1 min); 30 cycles if analyzing endogenous hnRNPE3 in cells (denaturation 94 oC/1 min, annealing 58 oC/1 min, extension 72 oC/1 min); and 21 cycles if analyzing endogenous hnRNPE3 in polyA+ RNA (fetal brain, adult whole brain, cerebellum, hippocampus, spinal cord, skeletal muscle, heart, and liver; Clontech).
We calculated RNA ratios by scanning the bands from three independent transfections and measuring them using the ImageJ software. The exception was endogenous hnRNPE3 in polyA+ RNA, which we performed only once.
2.4. Cell lysate preparation, Western blots and co-immunoprecipitations (co-IPs)
We verified the expression of all the factor constructs by using anti-FLAG M2 monoclonal (Sigma) or rabbit anti-myc (GeneScript) antibodies on Westerns of protein lysates from cells transfected with the constructs (Fig. 4B, 5A, 5B).
Fig. 5.

HnRNPE3 interacts with hnRNPE2 and SRA4, both also moderate activators of exon 10 splicing, and the three activators act additively. Western blots of (A, B) protein lysates and (C) co-IPs from 1:1 co-tranfections of myc-hnRNPE3 and FLAG-factors in HEK293 cells. Protein markers markers are indicated on the right of each panel. The positions of factors are indicated on the left. (A) Input, factors detected by anti-FLAG monoclonal antibody M2. (B) Input and (C) co-IP, hnRNPE3 detected by anti-myc polyclonal antibody. (D) RT-PCR of SP/10L in COS cells co-transfected with various combinations of FLAG constructs of hnRNPE3, SRA4 and hnRNPE2. Primers and graph conventions are as in Fig.1C.
We prepared cell lysates from transfected cells using RIPA lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Nonidet P-40 [NP-40], 0.5% deoxycholate, 0.1% SDS) containing 1x Protease Inhibitor Cocktail Tablets (Roche Molecular Biochemicals). Lysates were nutated at 4 oC for 1 h, cleared by centrifugation at 13,200 rpm for 15 min at 4 oC and boiled for 10 min prior to running on SDS-PAGE gels.
For co-IPs, we prepared protein lysates from cells co-transfected with Myc-hnRNPE3 plus FLAG-factors (9G8, Nova1, SRp75, hnRNPE2, SRA4). After the clearing step, we added anti-FLAG-antibody agarose beads (Sigma) to the supernatant, nutated it at 4 oC overnight, rinsed the beads 4x with wash buffer (50 mM Tris pH 7.4, 150 mM NaCl) and analyzed the proteins retained on the beads by Western blotting with the rabbit anti-myc antibody.
We performed Western blotting, transfer and detection as detailed in Gao et al. (2007). For detection we used chemiluminescence reagents from either Perkin Elmer (Lightning Plus) or Roche (Opti-4CN). All antibodies, primary and secondary, were used at 1:5,000 dilution.
2.5. Riboprobe preparation and RNA-protein co-IP
We linearized E10+30 and its variants at the unique MluI site downstream of the inserts and phenol-extracted them. We made radiolabeled riboprobes from these templates using T7 RNA polymerase (Promega) with a Promega or Ambion in vitro transcription kit and [32P]-CTP plus [32P]-UTP (Amersham).
For RNA-protein immunoprecipitation, cell lysates transfected with FLAG-hnNRPE3 were mixed with riboprobes for 20 min at RT. Then anti-FLAG-antibody agarose beads (Sigma) were added and incubated at 4 °C overnight. The beads were washed four times with wash buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl). After washing, the radioactivity retained by beads was counted in a beta scintillation counter. We did all the IPs in triplicate and averaged the results.
3. Results
3.1. HnRNPE3 moderately activates splicing of exon 10
Chromosome 21 contains four known or putative splicing factors: U2AF35, RBM11, SRA4 (SFRS15, KIAA 1172) and hnRNPE3 (poly-(rC) binding protein 3, PCBP3). All but RBM11 are located within the Down syndrome critical region between 21q22.1 to 21q22.3 (Gardiner et al., 2002). RBM11 was already in a eukaryotic expression vector (pCMV-SPORT6), and we placed the other three into N-terminal FLAG vectors. Our Westerns show that all three express in cells (Fig. 4B shows FLAG-hnRNPE3; data are not shown for U2AF35 and SRA4).
Co-transfection of the factors with tau exon 10 showed that U2AF35 and RBM11 do not affect exon 10 splicing, but hnRNPE3 and SRA4 moderately increase exon 10 inclusion (Fig. 1C). Previous work from our laboratory had shown that exon 10 splicing is also activated by hnRNPE2, a close relative of hnRNPE3 (Broderick et al., 2004). Given these results, we decided to concentrate on hnRNPE3.
When hnRNP proteins influence splicing by direct binding to the pre-mRNA, they invariably bind to intronic elements (Martinez-Contreras et al., 2007). Additionally, hnRNPE proteins bind poly-(rC) sequences, hence their alternative appellation (PCBP; Makayev and Liebhaber, 2002). There is one C triplet just downstream of exon 10, at positions +19 to +21 (Fig. 1A-3A, shaded). To find out which region hnRNPE3 interacts with, we did co-transfections and RNA-protein pulldowns of deletion and point mutants of the downstream intron of exon 10 with hnRNPE3 (Fig. 2C, 3C). We did not do co-tranfections of hnRNPE3 with point mutants M12, M13, M14 and M16, because they produce almost exclusively 10+ (Gao et al., 2007; Wang et al., 2003, 2005) and thus would not show additional activation by hnRNPE3.
3.2. HnRNPE3 activates splicing of exon 10 by binding to the C triplet in its downstream proximal intron
As we previously described (Gao et al., 2007; Wang et al., 2003) and also show here (Figs. 2B, 3B, odd-numbered lanes), the splicing behavior of our mutants defines region 11-18 as an intronic splicing silencer (ISS) and region 19-26 as an intronic splicing enhancer (ISE), in agreement with FTDP pedigrees and results from other laboratories (Andreadis, 2006; Liu and Gong, 2008).
The co-transfections show that hnRNPE3 can no longer activate splicing of exon 10 in mutant Δ19/26, which lacks the C triplet (Fig. 2B, lane 7 versus 8). HnRNPE3 is also less effective in activating mutant Δ23/29 (Fig. 2B, lane 9 versus 10). Additionally, hnRNPE3 is unable to activate splicing of mutant M19, in which the first C of the triplet has become a G (Fig. 3B, lane 5 versus 6). The M19 mutation completely abolishes inclusion of exon 10 in human FTDP pedigrees and transfection assays (Broderick et al., 2004; Stanford et al., 2003).
The pulldowns of the equivalent constructs with FLAG-hnRNPE3 show that hnRNPE3 interacts directly with exon 10 (Fig. 2C, lane 1 versus 2). HnRNPE3 binds to the intronic construct, E13+30, nearly as strongly as it binds to E10+30, and much more strongly than it binds to the exonic one, E80 (Fig. 2C, lanes 2 to 4).
In the pulldowns of the deletions, hnRNPE3 binds as strongly to deletion Δ11/18 as it does to E13+30 (Fig. 2C, lane 6). It binds weakly to deletion Δ19/26 that lacks the C triplet (Fig. 2C, lane 7) but also binds weakly to deletions Δ3/10 and Δ23/29 that contain it (Fig. 2C, lanes 5 and 8). In the pulldowns of the point mutations, hnRNPE3 binds to M13 as strongly as it does to E13+30 (Fig. 3C, lane 5). It binds very weakly to M19 which no longer has a C triplet (Fig. 3C, lane 8), but binds equally weakly to M11, 12, 14 and 16 (Fig. 3C, lanes 3, 4, 6 and 7).
These results indicate that hnRNPE3 regulates splicing of exon 10 by binding to the C triplet, but that its binding is influenced by the details and configuration of the local sequence.
3.3. The N-terminal KH domain of hnRNPE3 is essential for activation of exon 10 splicing
To establish which domains of hnRNPE3 are required for regulation of exon 10, we created two hnRNPE3 deletion variants as FLAG fusions: hnRNPE3N and hnRNPE3C contain the first and last two KH domains of hnRNPE3, respectively (Fig. 4A, 4B). In co-transfections, hnRNPE3N is as effective as full-length hnRNPE3 in increasing exon 10 splicing (Fig. 4C, lane 3), whereas hnRNPE3C has essentially no effect (Fig. 4C, lane 4). The results strongly suggest that the N-terminal KH domain of hnRNPE3 is required for activation of exon 10 splicing.
3.4. HnRNPE3 interacts with hnRNPE2, another modest activator of exon 10 splicing and the two activators act additively
We tested the interaction of hnRNPE3 with SRA4 and with factors known (9G8, hnRNPE2) or suspected (SRp75, Nova1) to bind to the proximal downstream intron of exon 10. Of these, SRA4 and hnRNPE2, a member of the hnNRPE family, also modestly activate splicing of exon 10 (Broderick et al., 2004; Fig. 1C), whereas the other three factors inhibit splicing of exon 10 strongly (9G8) or moderately (SRp75, Nova1; Gao et al., 2007; Wang et al., 2003).
The co-IPs show that hnRNPE3 does not interact with 9G8, Nova1 or SRp75 (Fig. 5C, lanes 2-4). However, it co-precipitates with hnRNPE2 (Fig. 5C, lane 5). HnRNPE proteins are known to interact (Kim et al., 2000). Our result suggests that the two proteins could potentially act as a heterodimer. HnRNPE3 also interacts weakly with SRA4 (Fig. 5C, lane 6).
To test whether the three factors that seem to interact by co-IP also act additively as exon 10 splicing activators, we co-transfected SP/10L with hnRNPE3, SRA4 and hnRNPE2 combinations (Fig. 5D). The results indicate that the three factors act additively (Fig. 5D, lanes 4-6). The three together (Fig. 5D, lane 6) are as active as hnRNPE3+hnRNPE2 (Fig. 5D, lane 5) and slightly less active than hnRNPE3+SRA4 (Fig. 5D, lane 4). This may reflect either a problem with co-expressing so many plasmids or steric interference between the three factors.
The co-IP and co-transfection results suggest the possibility that the three activators might act as a heterotrimer, though definitive proof will require additional interaction experiments.
3.5. HnRPNE3 shRNA reverses the activation of exon 10 splicing
To establish that hnRNPE3 is a native activator of tau exon 10 splicing, we tested three siRNA and three shRNA constructs against it (E3i-1, -2 and -3 and SME3-1, -2 and -3; Table 1 shows the sequences and their location within hnRNPE3). We chose HeLa because it expresses hnRNPE3 and tau and its tau is 40% 10+. Neuroblastoma cells are useless for testing RNAi against splicing activators, because their tau almost entirely lacks exon 10.
Three of the siRNAs and two of the shRNAs showed no influence on endogenous hnRNPE3 in HeLa cells. The exception was SME3-1, which decreased the expression level of endogenous hnRNPE3 significantly (Fig. 6A, 6B, lane 3 versus 4). SME3-1 had an extremely slight effect on endogenous tau exon 10. In contrast, it strongly decreased inclusion of exon 10 in SP/10L co-transfected in a 1:1 ratio with the SME3-1 plasmid (Fig 6A, 6B, lane 1 versus 2).
Fig. 6.
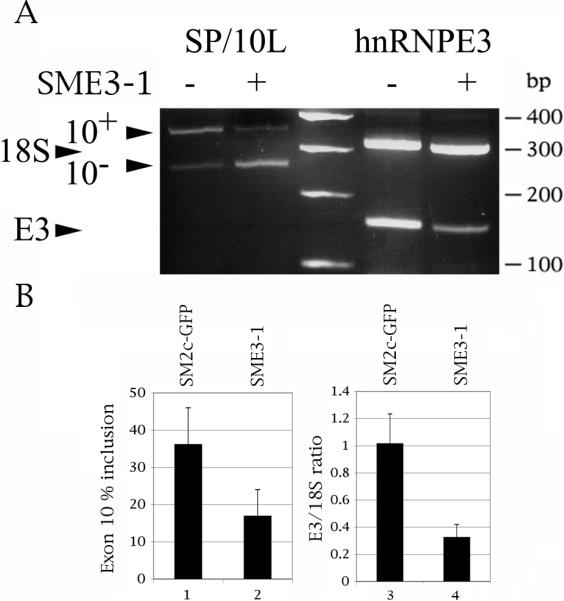
Suppression of hnRNPE3 by shRNA decreases inclusion of exon 10. 1 ug of SP/10L was transfected into HeLa cells together with 1 ug of shRNA construct SME3-1 or control vector SM2c-GFP. (A) A representative result of the effect of SME3-1 on transfected tau (left) and endogenous hnRNPE3 (right). DNA markers are indicated on the right of the panel. The positions of the exon 10, 18S and hnNRPE3 PCR products are indicated on the left. (B, Left panel) Quantitative RT-PCR of tau exon 10. Primers and graph conventions are as in Fig.1C. (B, right panel) Quantitative RT-PCR of endogenous hnRNPE3. The ratio of hnRNPE3 to 18S is indicated. Primer pair: E3-KH2NewS/E3-KH2NewN. The 18S:competimer ratio was 2:8.
Tau mRNA is known to have a long half-life (Aronov et al., 1999). This, plus the fact that hnRNPE3 is a modest activator, may explain the inability of SME3-1 to affect endogenous tau. Nevertheless, decrease of endogenous hnRNPE3 results in decrease of inclusion of exon 10 in a transfected construct which undergoes active transcription concommitant with the shRNA. The result strengthens the conclusion that hnRNPE3 is an endogenous activator of tau exon 10 splicing, though it falls just short of providing definitive proof on its own.
3.6. HnRNPE3 is preferentially expressed in neuronal tissues
Given the effect of hnRNPE3 on exon 10 splicing, we wanted to determine its expression profile. Quantitative PCR with competing 18S primers shows that hnRNPE3 is exclusively expressed in neuronal tissues and is also adult-specific (Fig. 7A). It is entirely absent in skeletal muscle, heart and liver (Fig. 7B, lanes 6-8) and barely present in fetal brain (Fig. 7B, lane 1). However, it is present in all the compartments of adult brain. Its relative expression levels are cortex>>cerebellum≈spinal cord>hippocampus (Fig. 7B, lanes 2, 3, 5 and 4, respectively).
Fig. 7.
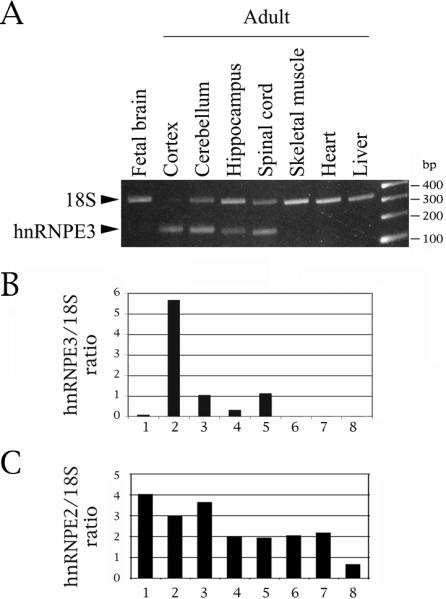
HnRNPE3 is restricted to the central nervous system and is adult-specific. (A) Quantitative RT-PCR reactions were done on polyA+ RNA. Primer pair: E3-KH2NewS/E3-KH2NewN. 18S:competimer ratio was 2:8. (B) Expression of hnRNPE2 relative to 18S. (C) Expression of hnRNPE2 relative to 18S, obtained with the same common reagents and techniques, for comparison purposes (adapted from Broderick et al., 2004).
This profile is totally different from that of hnRNPE2 (Fig. 7C, adapted from Broderick et al., 2004). Our results for both mRNAs are compatible with results from Affymetrix arrays and eNorthern results, though the latter show expression of the two mRNAs in additional tissues (http://www.genecards.org/cgi-bin/carddisp.pl?gene=PCBP3; http://www.genecards.org/cgi-bin/carddisp.pl?gene=PCBP2). The expression profile of hnRNPE3 makes it a good candidate for being an endogenous activator of exon 10 splicing, given that the exon is adult-specific.
4. Discussion
4.1. HnRNPE3, like hnRNPE2, moderately activates splicing of tau exon 10 by binding to the C triplet in the intronic splicing enhancer downstream of the exon
Previous work showed that tau exon 10 contains several splicing silencers and enhancers (reviewed in Andreadis, 2006). A particularly important and interesting regulatory region of the exon is its proximal downstream intron, which diverges considerably between human and mouse (Fig. 1A). This contrasts with the near-total conservation of the exon itself, and strongly suggests that the species-specific difference in the expression of exon 10 arises from regulation of this portion of the pre-mRNA. The region known to affect splicing of exon 10 consists of a silencer (+3 to +16) followed by an enhancer (+19 to +30). This region is a hotspot for point mutations that cause tauopathies by either strongly increasing (M3, 11, 12, 13, 14, 16) or strongly decreasing (M19, M29) inclusion of exon 10 (reviewed in Andreadis, 2006).
The co-transfection and pull-down results indicate that hnRNPE3 exerts its influence on exon 10 by directly binding to the C triplet at positions +19 to +21. This is congruent with the finding that hnRNPE3, like all members of its family, binds to C tracts (Makayev and Liebhaber, 2002). It is also consistent with the observation that M19, which changes the C at +19 to a G thereby entirely abolishing exon 10 inclusion, is impervious to activation by hnRNPE3 (Fig. 3B).
SR protein 9G8, which strongly inhibits splicing of exon 10, binds upstream of the C triplet at the +14 residue (Gao et al., 2007; Wang et al., 2003). However, whereas hnRNPE2 interacts with 9G8 in yeast two-hybrid assays (Funke et al., 1996), hnRNPE3 does not interact with 9G8 in our co-IP experiments, although it interacts with hnRNPE2 (Fig. 5C).
HnRNPE3 by itself is a modest activator of exon 10 splicing, but its effect increases in the presence of SRA4 and/or hnRNPE2 (Fig. 5D). The activation of exon 10 splicing by SRA4 and the interaction of SRA4 with hnRNPE3 suggest it may also be part of the hnRNPE3/hnRNPE2 complex, but further experiments are needed to prove this definitively.
Based on our results, we think that splicing regulation in this region may arise partly from steric interference between 9G8 (inhibitor) and a hnRNPE2/hnRNPE3 complex (activator), with hnRNPE2 the ligand that touches 9G8. This is supported by the observation that hnRNPE3 binds strongly to mutant Δ11/18, in which the binding site for 9G8 has been deleted (Fig 2C).
Fig. 8 shows a speculative model that summarizes our cumulative knowledge of the splicing regulation at the proximal downstream intron of tau exon 10. The binding sites of 9G8 and hnRNPE3 and the interactions between 9G8, hnRNPE3 and hnRNPE2 are well-defined (Broderick et al., 2004; Gao et al., 2007; this work). Future work will allow us to determine the identities, binding sites and interactions of additional factors that we think may be involved in regulating splicing of exon 10 in this critical region.
Fig. 8.
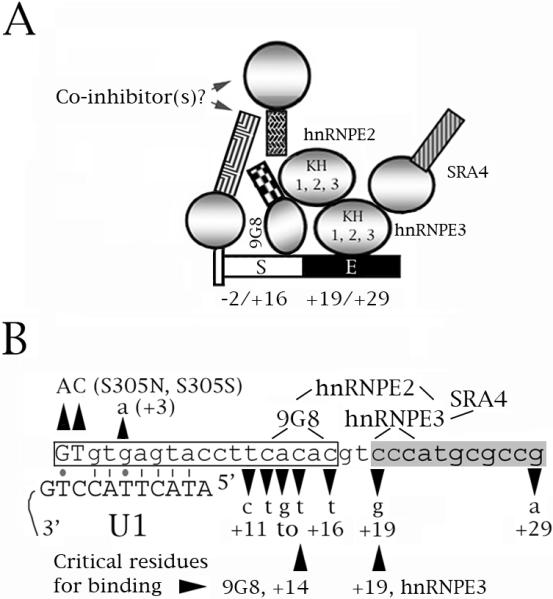
A speculative model of the role of hnRNPE3 in the regulation of tau exon 10 splicing. The model comes from this study, Broderick et al. (2004) and Gao et al. (2007). (A, B) Diagrams of the proximal downstream intron of exon 10. The numbers below the sequence correspond to the residue number within the exon (− numbers) and the intron (+ numbers). 9G8 inhibits exon 10 splicing whereas hnRNPE2, hnRNPE3 and SRA4 activate it. 9G8 and hnRNPE3 bind to the tau pre-mRNA at sites around residues +14 and +19, respectively. hnRNPE2 interacts with 9G8 and hnRNPE3, hnRNPE3 interacts with SRA4. (A) The end of the exon is shown as a narrow tall rectangle, the intron as a narrower rectangle. S=intronic splicing silencer (white), E=intronic splicing enhancer (black). For the factors, circles represent RRM or KH domains, squares represent RS domains. (B) The boxed region is the intronic silencer, the shaded region the intronic enhancer. The exon is shown in uppercase, the intron in lowercase letters. The FTDP mutations are indicated. The lines show which factors interact and the regions of the tau pre-mRNA that interact with 9G8 and hnRNPE3. The two residues critical for binding of 9G8 (+14) and hnRNPE3 (+19) are indicated. Also shown is the base paring of the exon 10 5' splice site with the U1 snRNA. Lines are Crick-Watson pairs, dots G-T base pairs.
4.2. Splicing regulation is a novel function for hnRNPE3
HnRNPs are involved in several RNA-related biological processes: transcription, pre-mRNA processing, mRNA transport and translation. Several of the hnRNP family members (A1, F, G, H, I/PTB, L) are involved in both constitutive and alternative splicing (Martinez-Contreras et al., 2007).
HnRNPE3 belongs to the hnRNP group which shuttles between the nucleus and the cytoplasm (Makeyev and Liebhaber, 2002; Ostareck-Lederer et al., 1998). These include hnRNP A, D, E, I/PTB and K. The E/K superfamily consists of five highly conserved siblings whose best-established function is modulation of mRNA stability and translation (Makeyev and Liebhaber, 2002). A relatively recent publication also demonstrated that hnRNPE3 represses the transcription of the mu opioid receptor (Choi et al., 2007). Our study places it among the hnRNPs which also influence splicing.
KH (hnRNPK homology) domains are found in several splicing regulators (SF1, KSRP, the Nova, FMR and STAR families, PSI; Valverde et al., 2008). In vitro assays show that the KH modules can bind to both RNA and protein. Their affinity for their binding partners is influenced by their detailed makeup as well as their context.
We had previously found that hnRNPE2 also moderately activates splicing of exon 10 (Broderick et al., 2004). That was the first time that a member of the hnRNPE/K family was shown to influence splicing. Both hnRNPE2 and hnRNPE3 activate exon 10 splicing and neither is able to activate mutant M19 (Fig. 1C, 3B; Broderick et al., 2004). Additionally, the two interact with each other in co-IPs (Fig. 5C). However, there are differences between them: hnRNPE2 is moderately expressed in non-neuronal tissues and more highly expressed in fetal than adult brain (Broderick et al., 2004; Fig. 7C). In contrast, hnRNPE3 is neuronal- and adult-specific (Fig. 7A). All 3 KH domains of hnRNPE2 are required for exon 10 splicing activation, whereas the N-terminal KH1 domain of hnRNPE3 suffices (Fig. 4C). Thus, of the two siblings, hnRNPE3 has an expression profile congruent with a native activator of exon 10.
4.3. Connections of tau exon splicing to dementia and DS
Tau exon 10 codes for an additional microtubule binding domain, and its addition increases affinity of tau protein for microtubules (Andreadis, 2006; Liu and Gong, 2008). The accumulation of abnormal tau filaments into tangles is a hallmark of many neurodegenerative diseases, including AD and DS. The number of NFTs correlates with disease severity, although the emerging consensus is that the true toxic species are tau oligomers that form earlier in the process (Jellinger, 2009) . In several neurodegenerative diseases, collectively termed tauopathies, tau pathology is solely and directly responsible for neuronal death and development of the clinical dementia manifestations (Goedert and Jakes, 2005).
The tauopathy pedigrees analyzed thus far predominantly show mutations in tau exon 10, although several pedigrees carry mutations in tau exons 1, 9, 11, 12 and 13 which influence either microtubule binding or protein conformation (Goedert and Jakes, 2005). The exon 10 mutations fall in two categories -- 50% influence microtubule binding and 50% alter the ratio of exon 10 isoforms (Andreadis, 2006; Goedert and Jakes, 2005). The latter category is effectively a subtle version of a dosage disease, as the disease results from ratio alterations even though the gene produces wild-type protein. In addition to tangle-only tauopathies, the ratio of exon 10 is also disturbed in sporadic AD (Glatz et al., 2005; Conrad et al., 2007) and DS (Mehta et al., 1999; Shi et al., 2008).
Although the amyloid precursor protein gene is located on chromosome 21, it is not within the Down syndrome critical region nor is it present in triplicate in the Ts1Cje segmental mouse model of DS (Gardiner et al., 2002, 2003). Additionally, amyloid levels do not seem to change in DS (Argellati et al., 2006). On the other hand, dual-specificity kinase DYRK1A, the molecule most closely linked to the mental defects of DS, influences tau post-transcriptionally, although analysis of its role is complicated by the fact that it seems to affect tau protein phosphorylation as well as tau splicing (Liu et al., 2008; Shi et al., 2008).
RT-PCR results from sporadic AD show that exon 10 increases 1.6-fold in cortex (Glatz et al., 2005; Conrad et al., 2007; the former used 15 AD samples, whereas the latter performed polony analysis). Our own RT-PCR analysis of one DS/AD and two DS cortices (Maryland Brain Bank) showed significant increase of exon 10 in the DS/AD sample, moderate increase in one DS sample and moderate decrease in the other (Andreadis laboratory, unpublished results). In contrast, Shi et al. (2008) report that the six DS samples they examined showed decreased inclusion of exon 10, although they performed their analysis at the protein level.
Because at least three factors located on chromosome 21 (DYRK1A, hnRNPE3 and SRA4) are now known to affect exon 10 splicing (Shi et al. 2008; this work), additional experiments are necessary to identify which of them are bona fide endogenous regulators of the process. Given that tauopathies ensue whether exon 10 inclusion decreases or increases, it is possible that this may also be the case for cases of DS or sporadic AD caused by exon 10 ratio imbalance. Thus, regardless of details, it appears increasingly likely that tau, via its splicing and the ensuing altered ratios of exon 10, is involved in the early-onset dementia of DS.
It is becoming clear that many finely-tuned biological processes achieve their exquisite calibration by combinatorial methods. The overlap of hnRNP functions and the regulation of alternative splicing both belong in this regulatory mode. This inherently complex regulation mode complicates the possibility of ameliorating or curing diseases caused through missplicing (such as FTDP) by tinkering with factor ratios. Nevertheless, work on the basic molecular biology of the tau molecule may give us the tools to comprehend and combat not only FTDP, but also other types of dementia, which is becoming increasingly prevalent as the human lifespan lengthens, as well as the common and pleiotropic DS and DM1.
Acknowledgements
This work was supported by NIH/NICHD R21 grant HD056195. We want to thank Dr. Stefan Stamm for stirring our interest in this aspect of tau regulation and Dr. Alonso Ross for his generous subsidy of the shRNA clones via the UMMS SiRNA Core.
Abbreviations used
- AD
Alzheimer's disease
- DS
Down syndrome
- (k)bp
(kilo)base pairs
- co-IP
co-immunoprecipitation
- FTDP-17
frontotemporal dementia with Parkinsonism associated with chromosome 17
- hnRNP
heterogeneous nuclear ribonucleoprotein
- KH
hnRNPK homology
- MAP
microtubule-associated protein
- nt
nucleotides
- NFT
neurofibrillary tangle
- PCBP
poly-(rC) binding protein
- RRM
RNA recognition motif
- R/S
arginine/serine
- snRNPs
small nuclear ribonucleoproteins
- SR proteins
serine/arginine-rich proteins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreadis A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog. Mol. Subcell. Biol. 2006;44:89–107. doi: 10.1007/978-3-540-34449-0_5. [DOI] [PubMed] [Google Scholar]
- Argellati F, Massone S, d'Abramo C, Marinari UM, Pronzato MA, Domenicotti C, Ricciarelli R. Evidence against the overexpression of APP in Down syndrome. IUBMB Life. 2006;58:103–106. doi: 10.1080/15216540600644853. [DOI] [PubMed] [Google Scholar]
- Aronov S, Marx R, Ginzburg I. Identification of 3'UTR region implicated in tau mRNA stabilization in neuronal cells. J. Mol. Neurosci. 1999;12:131–145. doi: 10.1007/BF02736927. [DOI] [PubMed] [Google Scholar]
- Broderick JA, Wang J, Andreadis A. Heterogeneous nuclear ribonucleoprotein E2 binds to tau exon 10 and moderately activates its splicing. Gene. 2004;331:107–114. doi: 10.1016/j.gene.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Choi HS, Kim CS, Hwang CK, Song KY, Law PY, Wei LN, Loh HH. Novel function of the poly(C)-binding protein alpha CP3 as a transcriptional repressor of the mu opioid receptor gene. FASEB J. 2007;21:3963–3973. doi: 10.1096/fj.07-8561com. [DOI] [PubMed] [Google Scholar]
- Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, Stamm S, Church G, Hyman BT. Single molecule profiling of tau gene expression in Alzheimer's disease. J. Neurochem. 2007;103:1228–1236. doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- Funke B, Zuleger B, Benavente R, Schuster T, Goller M, Stevenin J, Horak I. The mouse polyC.-binding protein exists in multiple isoforms and interacts with several RNA-binding proteins. Nucl. Acids Res. 1996;24:3821–3828. doi: 10.1093/nar/24.19.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang J, Wang Y, Andreadis A. SR protein 9G8 modulates splicing of tau exon 10 via its proximal downstream intron, a clustering region for frontotemporal dementia mutations. Mol. Cell. Neurosci. 2007;34:48–58. doi: 10.1016/j.mcn.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner K, Slavov D, Bechtel L, Davisson M. Annotation of human chromosome 21 for relevance to Down syndrome: gene structure and expression analysis. Genomics. 2002;79:833–843. doi: 10.1006/geno.2002.6782. [DOI] [PubMed] [Google Scholar]
- Gardiner K, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003;318:137–147. doi: 10.1016/s0378-1119(03)00769-8. [DOI] [PubMed] [Google Scholar]
- Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, Rieder P, Moeller HJ, Andreadis A, Henkel K, Stamm S. The alternative splicing of tau exon 10 and its regulatory proteins clk2 and tra2-beta1 changes in sporadic Alzheimer's disease. J. Neurochem. 2005;96:635–644. doi: 10.1111/j.1471-4159.2005.03552.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Hertel KJ. Combinatorial control of exon recognition. J. Biol. Chem. 2008;283:1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 2000;279:129–132. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Recent advances in our understanding of neurodegeneration. J. Neural Transm. 2009;116:1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- Kim JH, Hahm B, Kim YK, Choi M, Jang SK. Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J. Mol. Biol. 2000;298:395–405. doi: 10.1006/jmbi.2000.3687. [DOI] [PubMed] [Google Scholar]
- Kirchhoff M, Bisgaard AM, Duno M, Hansen FJ, Schwartz M. A 17q21.31 microduplication, reciprocal to the newly described 17q21.31 microdeletion, in a girl with severe psychomotor developmental delay and dysmorphic craniofacial features. Eur. J. Med. Genet. 2007;50:256–263. doi: 10.1016/j.ejmg.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, Ramakrishna N, Gong CX. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–3233. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008;3:8. doi: 10.1186/1750-1326-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Cáceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA. The polyC.-binding proteins: a multiplicity of functions and a search for mechanisms. RNA. 2002;8:265–278. doi: 10.1017/s1355838202024627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- Mehta PD, Patrick BA, Dalton AJ, Aisen PS, Emmerling ME, Sersen EA, Wisniewski HM. Increased levels of tau-like protein in patients with Down syndrome. Neurosci. Lett. 1999;275:159–162. doi: 10.1016/s0304-3940(99)00754-5. [DOI] [PubMed] [Google Scholar]
- Ostareck-Lederer A, Ostareck DH, Hentze MW. Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem. Sci. 1998;23:409–411. doi: 10.1016/s0968-0004(98)01301-2. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, Porter K, Prigmore E, Krepischi-Santos AC, Varela MC, Koiffmann CP, Lees AJ, Rosenberg C, Firth HV, de Silva R, Carter NP. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, Zhou J, Hwang YW, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 2008;283:28660–28669. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, Martins RN, Kwok JB, Schofield PR. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain. 2003;126:814–826. doi: 10.1093/brain/awg090. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Stamm S. Alternative splicing and disease Biochem. Biophys. Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde R, Edwards L, Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J. Neurochem. 2003;88:1078–1090. doi: 10.1046/j.1471-4159.2003.02232.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang J, Gao L, Lafyatis R, Stamm S, Andreadis A. Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c / SRp55 complex that either recruits or antagonizes htra2beta1. J. Biol. Chem. 2005;280:14230–14239. doi: 10.1074/jbc.M413846200. [DOI] [PubMed] [Google Scholar]