

Abstract
Rationale
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional kinase involved in vital cellular processes such as Ca2+ handling and cell fate regulation. In mammalian heart, two primary CaMKII isoforms, δB and δC, localize in nuclear and cytosolic compartments, respectively. Although previous studies have established an essential role of CaMKII-δC in cardiomyocyte apoptosis, the functional role of the more abundant isoform, CaMKII-δB, remains elusive.
Objective
Here we determined the potential role of CaMKII-δB in regulating cardiomyocyte viability and explored the underlying mechanism.
Methods and Results
In cultured neonatal rat cardiomyocytes, the expression of CaMKII-δB and CaMKII-δC was inversely regulated in response to H2O2-induced oxidative stress with a profound reduction of the former and an increase of the later. Similarly, in vivo ischemia/repefusion (IR) led to an opposite regulation of these CaMKII isoforms in a rat myocardial IR model. Notably, overexpression of CaMKII-δB protected cardiomyocytes against oxidative stress-, hypoxia- and angiotensin II-induced apoptosis, whereas overexpression of its cytosolic counterpart promoted apoptosis. Using cDNA microarray, real time-PCR and Western blotting, we demonstrated that overexpression of CaMKII-δB but not CaMKII-δC elevated expression of heat shock protein 70 (HSP70) family members, including inducible HSP70 (iHSP70) and its homologous (Hst70). Moreover, overexpression of CaMKII-δB led to phosphorylation and activation of heat shock factor 1 (HSF1), the primary transcription factor responsible for HSP70 gene regulation. Importantly, gene silencing of iHSP70, but not Hst70, abolished CaMKII-δB-mediated protective effect, indicating that only iHSP70 was required for CaMKII-δB elicited anti-apoptotic signaling.
Conclusions
We conclude that cardiac CaMKII-δB and CaMKII-δC were inversely regulated in response to oxidative stress and IR injury, and that in contrast to CaMKII-δC, CaMKII-δB serves as a potent suppressor of cardiomyocyte apoptosis triggered by multiple death-inducing stimuli via phosphorylation of HSF1 and subsequent induction of iHSP70, marking both CaMKII-δ isoforms as promising therapeutic targets for the treatment of ischemic heart disease.
Keywords: CaMKII isoforms, CaMKII-δB, oxidative stress, hypoxia, cardiomyocyte apoptosis, iHSP70, HSF1
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a ubiquitous and multifunctional serine/threonine protein kinase family, which consists of 30 distinct members encoded by four different genes (α, β, δ, and γ). The δ isoform of CaMKII is predominantly expressed in the heart of many mammalian species, including rat, mouse and human 1-4. Two primary splicing variants of the δ isoform, CaMKII-δB and CaMKII-δC, have been cloned from rat heart 1. The only difference between these isoforms is the additional 11-amino acids conferring a nuclear targeting signal of CaMKII-δB 5. As a result, CaMKII-δB and CaMKII-δC localize to the nucleus and the cytoplasm, respectively, although they share many common biochemical properties 5.
CaMKII is activated upon the binding of Ca2+ and calmodulin complex 6, 7. In addition to the widely-accepted Ca2+/calmodulin-mediated activation, recent studies have demonstrated that CaMKII can be activated by reactive oxygen species (ROS)-induced oxidation in a Ca2+-independent fashion 8. Activation of CaMKII leads to phosphorylation of a penal of target proteins involved in multiple important physiological and pathological processes from Ca2+ handling, muscle contraction to memory encoding 9-11.
With respect to isoform-specific functions in the heart, most studies over the past decade have been focused on the role of CaMKII-δC in regulating cardiac excitation-contraction coupling 12-14 and maladaptive cardiac remodeling 15. In addition, recent studies have shown that CaMKII-δC is a common intermediate of cardiac cell apoptosis induced by diverse death-inducing stimuli, including excessive β1-adrenergic receptor (β1-AR) stimulation, H2O2, intracellular high Ca2+, and acidosis 16-18. Despite the higher abundance of CaMKII-δB, it has received far less attention than its cytosolic counterpart. The functional role of CaMKII-δB in the heart remains largely unknown, although emerging evidence suggests that overexpression of CaMKII-δB induces cardiac hypertrophy 19, 20. In the current study, we demonstrate that the expression of the major cardiac CaMKII isoforms are inversely regulated in response to oxidative stress and ischemia/reperfusion (IR) injury, and that enhanced CaMKII-δB signaling potently protects cardiomyocytes against oxidative stress-, hypoxia-, and angiotensin II (Ang II)-induced apoptosis, while activation of CaMKII-δC is apoptotic. The anti-apoptotic effect is mediated by CaMKII-δB-induced phosphorylation of an important transcriptional factor, heat shock factor 1 (HSF1), which positively regulates the expression of inducible heat shock protein70 (iHSP70, also known as HSP72). These findings not only define a novel cardiac protective function of CaMKII-δB, but also imply that CaMKII-δB downregulation or malfunction may be a crucial pathogenic element and a potential therapeutic target for various forms of heart disease, particularly ischemic heart disease.
METHODS
Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich. An expanded Methods section is available in the online data supplement at http://circres.ahajournals.org.
Isolation, Culture and Adenoviral Infection of Rat Ventricular Myocytes
Neonatal or adult rat ventricular myocytes were isolated, cultured and infected as previously described 16, 21. See the online data supplement for further details.
Western Blotting Analysis, and Confocal Immunocytochemical Imaging of Hemagglutinin (HA)-tagged CaMKII-δB or CaMKII-δC
Separation of cytosolic and nuclear protein was achieved with a Nuclear/Cytosolic Fractionation Kit (Biovision Research Products, CA, USA) following the manufacturer's instructions. Western blot to assay the abundance of CaMKII-δ in cytosolic and nuclear extracts was performed with an anti-CaMKII-δ antibody (Santa Cruz Biotechnology, Inc., sc-5392), as previously described 17. Confocal immunocytochemical imaging was performed as previously described 16. See the online data supplement for further details.
Cell Viability
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or DNA laddering/Hoechst staining was used as cell viability or apoptosis index, respectively, as described previously 21. See the online data supplement for further details.
Small Interfering RNA (siRNA)
Cardiomyocytes were transfected with siRNA targeted specifically to iHSP70 or Hst70 by Lipofectamine™ RNAiMAX (Invitrogen) following the manufacturer's instructions. See the online data supplement for further details.
Statistical Analysis
Data are expressed as the mean ± S.E.. Statistical comparisons used one-way analysis of variance, followed by Bonferroni's procedure for multiple-group comparisons. P<0.05 was considered statistically significant.
RESULTS
Oxidative Stress Oppositely Regulates the Expression Profile of CaMKII-δB and CaMKII-δC in Cultured Neonatal Rat Cardiomyocytes
To determine the potential functional role of CaMKII-δB in regulating cardiomyocyte viability, we first examined the expression of the major cardiac CaMKII isoforms, CaMKII-δB and CaMKII-δC, in response to oxidative stress. H2O2 treatment elevated CaMKII-δC gene expression in a concentration- and time-dependent manner (Figure 1 A&B). In contrast, CaMKII-δB gene expression was profoundly suppressed in response to H2O2 treatment (Figure 1 A&B). To determine whether these CaMKII isoforms were also oppositely regulated at their protein levels by oxidative stress, we fractionalized cardiomyocyte proteins and detected CaMKII-δB and CaMKII-δC abundance by assaying CaMKII-δ in the nuclear and the cytosolic fractions, respectively, using the same antibody reacting with both CaMKII-δ isoforms. CaMKII-δB protein abundance was rapidly and profoundly reduced by half at 4 h after H2O2 (200 μM) treatment and reached the minimal level at 24 h after treatment, whereas the protein level of CaMKII-δC was significantly elevated after H2O2 exposure (Figure 1C).
Figure 1. CaMKII-δB and CaMKII-δC expression in neonatal rat cardiomyocytes in response to oxidative stress or in rat myocardium subjected to ischemia/reperfusion (IR) injury.
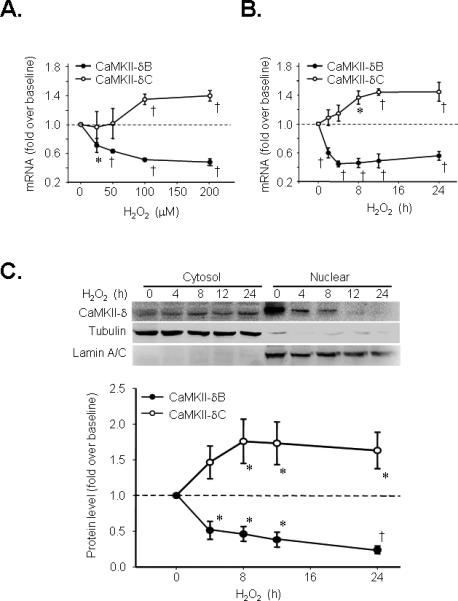
A. CaMKII-δB and CaMKII-δC gene expression assayed by real-time PCR in response to different concentrations of H2O2 treatment for 8 h in cultured neonatal cardiomyocytes (n = 6 for each data point). B. CaMKII-δB and CaMKII-δC gene expression assayed by real-time PCR in response to H2O2 (200 μM) treatment for different period of time in cultured neonatal cardiomyocytes (n = 6 for each data point). C. Typical Western blots and the statistic data of the time course of CaMKII-δ protein abundance in nuclear and cytosolic fractions from neonatal cardiomyocytes treated with H2O2 (200 μM) (n = 4 for each time point). D. shows CaMKII-δB and CaMKII-δC mRNA levels in ischemic area of rat hearts subjected to 45 min ischemia followed by different periods of reperfusion assayed by real-time PCR (n = 6 for each time point). E. CaMKII-δB and CaMKII-δC protein levels assayed by Western blotting in nuclear and cytosolic fractions from ischemic myocardium of rat hearts (n = 3-5 for each time point). For all panels, * P<0.05; † P<0.01. vs. baseline.
IR Oppositely Regulates CaMKII-δB and CaMKII-δC Expression In Vivo
To investigate whether CaMKII-δB and CaMKII-δC were oppositely regulated in vivo during cardiac IR injury, we utilized a rat myocardial IR model. The experimental protocol and cardiac damage readouts, including myocardial infarction and lactate dehydrogenase (LDH) release, were illustrated in Online Figure I (online data supplements). Importantly, IR led to a marked increase in CaMKII-δC and a decrease in CaMKII-δB expression at both mRNA and protein levels (Figure 1 D-E). These_results indicate there is an inverse regulation of the expression of CaMKII-δB and CaMKII-δC by oxidative stress or IR injury in in vivo as well as in cultured cardiomyocytes, highlighting that these CaMKII isoforms may elicit distinctly different, even opposing functional roles in regulating cardiomyocyte viability.
Overexpression of CaMKII-δB Promotes Cardiomyocyte Viability
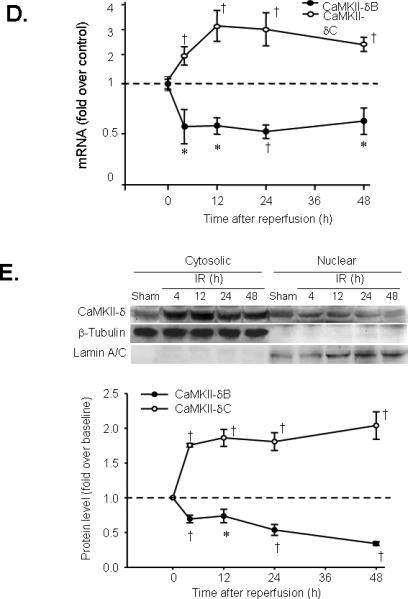
To test the above hypothesis, we investigated whether activation of the nuclear isoform, CaMKII-δB, regulates cardiac cell viability and, if so, whether the effect of CaMKII-δB differs from that of CaMKII-δC. Using adenoviral gene transfer, we overexpressed either CaMKII-δB or CaMKII-δC in cultured neonatal or adult rat cardiomyocytes in the presence or absence of H2O2 (200 μM). Overexpressed CaMKII-δB was enriched in the nuclear compartment while CaMKII-δC in the cytosol in neonatal cardiomyocytes (Figure 2A), as is the case in adult rodent cardiac myocytes 16. Expression levels and phosphorylation status of both CaMKII isoforms were elevated in a titer-dependent manner in neonatal myocytes infected with Adv-CaMKII-δB or Adv-CaMKII-δC for 24 h (Figure 2B). H2O2 exposure triggered robust cardiomyocyte apoptosis, as manifested by DNA laddering (Figure 2 C&D) and an increase in Hoechst staining-positive cells (Figure 2E) compared with non-treated cells. Remarkably, overexpression of CaMKII-δB, but not CaMKII-δC or β-gal, protected both neonatal and adult rat cardiac myocytes from H2O2-mediated apoptosis (Figure 2 C&D) as well as necrosis (Online Figure II, online data supplements). H2O2-induced DNA fragmentation assayed by DNA laddering (Figure 2 C&D) and apoptotic nuclear morphological changes visualized by Hoechst staining (Figure 2E) were markedly blunted in cells infected by Adv-CaMKII-δB. In contrast, overexpression CaMKII-δC promoted apoptosis in cultured neonatal cardiac myocytes even in the absence of H2O2 treatment, and overtly enhanced apoptosis induced by H2O2 (100 μM) (Figure 2 E). CaMKII-δC-induced apoptosis was abolished by overexpression of CaMKII-δB (Online Figure III, online data supplements).
Figure 2. Overexpression of CaMKII-δB protects both neonatal and adult rat cardiomyocytes against H2O2-triggered apoptosis.

A. Immunofluorescent imaging with an anti-HA antibody to visualize the intracellular distribution of HA-tagged CaMKII-δB and CaMKII-δC in neonatal rat cardiomyocytes infected with Adv-CaMKII-δB or Adv-CaMKII-δC (both at 20 m.o.i. for 24 h). B. Typical Western blot with a site-specific antibody reacting with CaMKII-d or phosphorylated CaMKII-d in lysate from myocytes infected with Adv-β-gal, Adv-CaMKII-δB, or Adv-CaMKII-δC at m.o.i. as indicated for 24 h. C. DNA laddering in neonatal rat cardiomyocytes infected with Adv-CaMKII-δB, Adv-CaMKII-δC or Adv-β-gal (all at 20 m.o.i. for 24 h) then treated with H2O2 (200 μM) for another 24 h. Similar results were obtained in three independent experiments. D. Representative DNA laddering in adult rat cardiomyocytes infected with Adv-CaMKII-δB or Adv-β-gal (both at 100 m.o.i. for 24 h) then treated with H2O2 (10 μM) for another 24 h. Similar results were obtained in three independent experiments. E. Average data of Hoechst staining in neonatal cardiomyocytes infected with Adv-CaMKII-δB, Adv-CaMKII-δC or Adv-β-gal (all at 20 m.o.i. for 24 h) then subjected to H2O2 (200 μM) for 24 h. F. Average data of Hoechst staining in neonatal cardiomyocytes infected with Adv-CaMKII-δB or Adv-β-gal (at 20 m.o.i. for 24 h) then subjected to hypoxia for 9 h. G. Average data of Hoechst staining in neonatal cardiomyocytes infected with Adv-CaMKII-δB or Adv-β-gal (at 20 m.o.i. for 24 h then subjected to Ang II (1 μM for 48 h). For E-G, each data point shows the result from 5000-8000 cells in four independent experiments for Hoechst staining (* P<0.01 vs. Adv-β-gal in the absence of H2O2; † P<0.01 vs. the corresponding Adv-β-gal in each group of different concentrations of H2O2 or as indicated).
Next, we determined whether CaMKII-δB can protect cardiomyocytes from other death-inducing stimuli, in addition to oxidative stress. Overexpression of CaMKII-δB abolished hypoxia- and Ang II-induced myocyte apoptosis (Figure 2 F-G, respectively). These results provided the first documentation that activation of the most abundant cardiac CaMKII isoform, CaMKII-δB, potently protects cardiomyocytes against apoptosis triggered by multiple death-inducing stimuli, whereas its cytosolic counterpart promotes apoptosis.
An Essential Role of iHSP70 in CaMKII-δB-Mediated Cardiomyocyte Protection
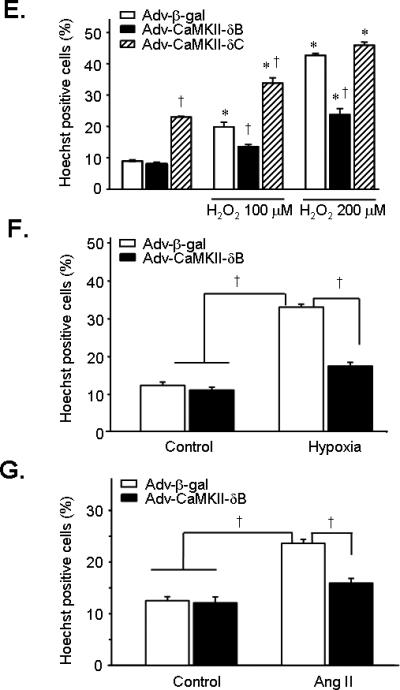
To delineate the mechanism underlying the protective effect of CaMKII-δB, we examined possible differential gene expression profiles with cDNA microarray analysis in neonatal cardiomyocytes infected with Adv-CaMKII-δB, Adv-CaMKII-δC, or Adv-β-gal (all at 20 m.o.i for 36 h). The expression levels of 174 genes were significantly altered in neonatal cardiomyocytes infected with Adv-CaMKII-δB relative to those in cells infected with Adv-β-gal (>1.5-fold or < 0.5-fold, n = 3). Among them, 17 genes were specifically upregulated or downregulated by overexpression of CaMKII-δB but not CaMKII-δC (>1.5 fold or <0.5 fold, n = 3) (Online Table I and Online Figure IV, online data supplements), although the majority of the 174 genes were similarly regulated by both CaMKII isoforms. Importantly, among the 17 differentially regulated genes, we identified an important cell protective factor, iHSP70 (also known as HSP72), which was selectively upregulated in response to overexpression of CaMKII-δB but not CaMKII-δC. The cDNA microarray analysis revealed a 1.7-fold increase in iHSP70 gene expression in cells infected with Adv-CaMKII-δB but not Adv-CaMKII-δC relative to those infected by Adv-β-gal (Figure 3A). Using real-time PCR, we validated the CaMKII-δB-dependent elevation of iHSP70 gene expression at either 36 h or 48 h after adenoviral infection (Figure 3B). Likewise, iHSP70 protein abundance was also augmented by 2.4-folds only in cells infected with Adv-CaMKII-δB (m.o.i. 20 for 48 h) (Figure 3C).
Figure 3. Upregulation of iHSP70 and Hst70 by overexpression of CaMKII-δB but not CaMKII-δC in cultured neonatal rat cardiomyocytes.
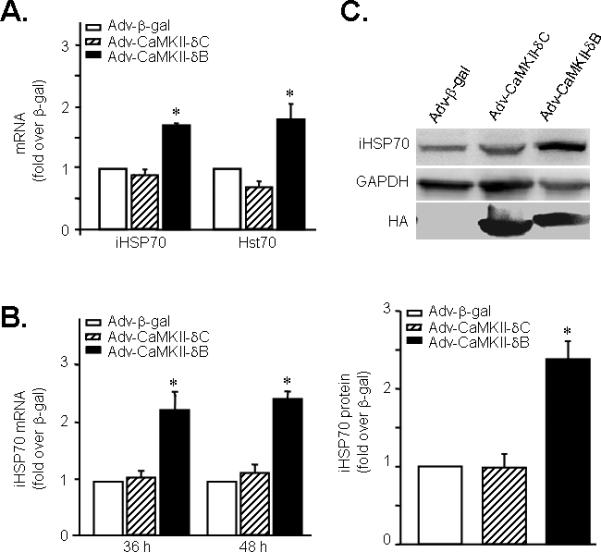
A. iHSP70 and Hst70 gene expression assayed by cDNA microarray analysis in cells infected with Adv-CaMKII-δB, Adv-CaMKII-δC, or Adv-β-gal. B. iHSP70 gene expression assayed by real-time PCR in neonatal cardiomyocytes after adenoviral infection for 36 h or 48 h. (n = 4). C. Typical Western blot with an anti-iHSP70 antibody and the average data in cells infected with Adv-CaMKII-δB, Adv-CaMKII-δC, or Adv-β-gal (at 20 m.o.i.) for 48 h. (n = 4). For all panels, * P<0.05 vs. Adv-CaMKII-δC and Adv-β-gal.
To determine whether the elevation in iHSP70 expression is causatively linked to CaMKII-δB-induced anti-apoptotic effect, we utilized siRNA-mediated iHSP70 gene silencing. siRNAs (including siRNA1 and siRNA2) effectively reduced the expression of iHSP70 at mRNA (Figure 4A) and protein levels (Figure 4B). Notably, iHSP70 gene silencing substantially abolished CaMKII-δB-mediated anti-apoptotic effect, as assayed by DNA laddering in cardiomyocytes treated with either siRNA1 or siRNA2 of iHSP70, regardless of CaMKII-δB overexpression (Figure 4C). Moreover, MTT assay further revealed that overexpression of CaMKII-δB could not protect cardiomyocytes from H2O2-induced cell death in the presence of iHSP70 siRNA1 or siRNA2 (Figure 4D). These results provide strong evidence to indicate that upregulation of iHSP70 is essentially involved in CaMKII-δB-elicited anti-apoptotic signaling.
Figure 4. Gene silencing of iHSP70 blocks CaMKII-δB-mediated anti-apoptotic effect.
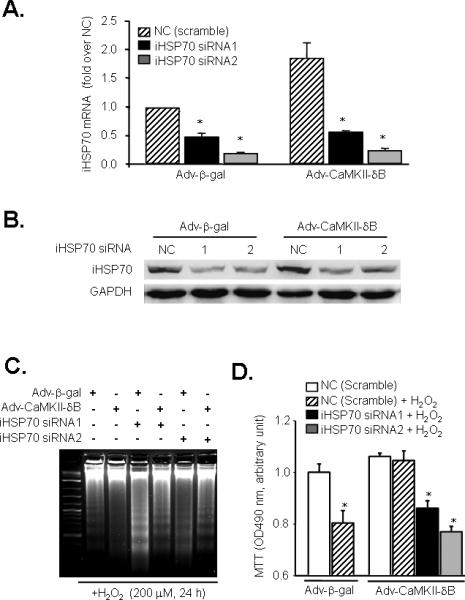
A. siRNA-induced reduction of iHSP70 at mRNA level assayed by real-time PCR in neonatal cardiomyocytes infected with Adv-β-gal or Adv-CaMKII-δB (n = 3 for each group * P<0.05 vs. the NC of each group). B. siRNA-induced reduction of iHSP70 at protein level in cells infected with Adv-β-gal or Adv-CaMKII-δB. Similar results were observed in three independent experiments. C. DNA laddering in cells infected with Adv-β-gal or Adv-CaMKII-δB in the absence or presence of iHSP70 siRNA after H2O2 (200 μM for 24 h) treatment. Similar results were obtained in three independent experiments. D. MTT cell viability assay in cells infected with Adv-β-gal or Adv-CaMKII-δB in the absence or presence of iHSP70 siRNA after H2O2 (200 μM for 24 h) treatment (n = 3, * P<0.05 vs. the NC of each group).
Interestingly, another closely related member of the HSP70 family, namely Hst70 in rats (but called HSP70.2 in mouse), was also significantly increased by 1.8-fold in cells overexpressing CaMKII-δB but not in those overexpressing CaMKII-δC (Figure 3A and Online Table I, online data supplements). However, gene silencing of Hst70 had no detectable effect on CaMKII-δB-induced protection. Specifically, two out of three tested siRNA sequences (Hst70 siRNA1 and 3) suppressed Hst70 mRNA level by 60-70 % (Figure 5A), but could not abolish the protective effect of CaMKII-δB assayed by DNA laddering (Figure 5B) and MTT (Figure 5C), indicating that Hst70, unlike HSP72, is not involved in CaMKII-δB-mediated anti-apoptotic signaling.
Figure 5. Gene silencing of Hst70 does not affect CaMKII-δB-mediated anti-apoptotic effect.
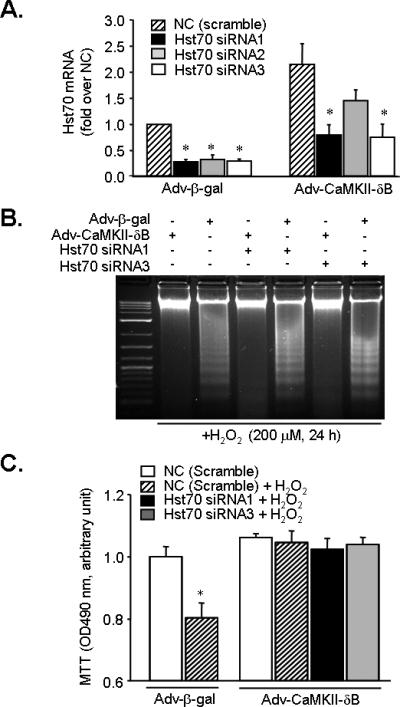
A. siRNA-induced reduction of Hst70 at mRNA level assayed by real-time PCR in neonatal cardiomyocytes infected with Adv-β-gal or Adv-CaMKII-δB (n = 3 for each group, * P<0.05 vs. the NC of each group). B. DNA laddering in myocytes infected with Adv-β-gal or Adv-CaMKII-δB in the absence or presence of Hst70 siRNA after H2O2 (200 μM for 24 h) treatment. C. MTT cell viability assay in cells infected with Adv-β-gal or Adv-CaMKII-δB in the absence or presence of Hst70 siRNA after H2O2 (200 μM for 24 h) treatment (n = 3, * P<0.05 vs. the NC of each group).
CaMKII-δB Overexpression Increases Phosphorylation of Nuclear HSF1 at Ser230
To investigate the mechanism underlying CaMKII-δB-mediated upregulation of iHSP70, we analyzed the expression and phosphorylation status of HSF1, the transcription factor primarily responsible for the regulation of iHSP70 expression. Immunofluorescent imaging revealed that CaMKII-δB and HSF1 were co-localized at the nuclear compartment in cardiomyocytes infected with Adv-CaMKII-δB (Figure 6A). Using a site-specific antibody reacting with phosphorylated HSF1 at Ser230, which has been shown to positively contribute to the transcriptional activity of HSF1 22, we found that overexpression of CaMKII-δB but not CaMKII-δC or β-gal elevated phosphorylation level of HSF1 at Ser230 relative to the control group (Figure 6B). In contrast, overexpression of CaMKII-δB had no detectable effect on phosphorylation of HSF1 at Ser303, a constitutively phosphorylated residue, indicating that CaMKII-δB selectively phosphorylates HSF1 at Ser230 (Figure 6B). It is noteworthy that H2O2 (200 μM for 15 min) treatment transiently increased CaMKII-δB mediated phosphorylation of HSF1-Ser230 (Figure 6C) and HSF1 nuclear translocation (Online Figure V, online data supplements).
Figure 6. CaMKII-δB co-localizes with HSF1 and selectively increases phosphorylation of HSF1 at Ser230.
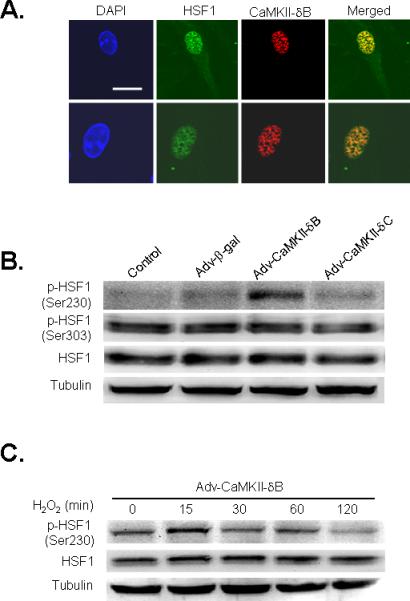
A. Confocal immunofluorescent imaging to visualize the nucleus by DAPI staining (blue) and intracellular distribution of HSF1 (green) and HA-tagged CaMKII-δB (red) in cardiomyocytes infected with Adv-CaMKII-δB (scale bar is 20 μm). The merged imaging (yellow) shows HSF1 and HA-tagged CaMKII-δB were co-localized in the nuclear compartment. B. Typical western blots with an antibody reacting with total or Ser230- or Ser303-phosphorylated HSF1 in uninfected cells (Control) or those infected with Adv-CaMKII-δB, Adv-CaMKII-δC, or Adv-β-gal (at 20 m.o.i.). Similar results were obtained in other three independent experiments. C. Phosphorylation of HSF1 at Ser230 in neonatal cardiomyocytes infected with Adv-CaMKII-δB for 36 h and then treated with H2O2 (200 μM) for various time periods.
DISCUSSION
Cardiac myocyte apoptosis plays a pivotal role in the development of a variety forms of heart disease, including ischemic heart disease, congestive heart failure, and acute myocardial infarction 23, 24. ROS has been defined as a major contributor in triggering cardiomyocyte apoptosis during myocardial ischemia/reperfusion injury, dilated cardiomyopathy and the progression of heart failure 25. Recent studies have established a link between oxidative stress and activation of CaMKII and defined an essential role of CaMKII signaling in oxidative stress-triggered cardiomyocyte apoptosis 8, 17. However, isoform-specific expression regulation and function of cardiac CaMKII-δB and CaMKII-δC in response to oxidative stress, IR injury, or hypoxia have not been well characterized until now. Given their distinct intracellular distributions, these cardiac CaMKII isoforms may be differentially regulated and exhibit different actions. Indeed, in this study, we have revealed following important differences between these CaMKII-δ isoforms in their responses to oxidative stress or IR injury and their functional roles in regulating cardiac muscle cell viability. First, the expression profile of CaMKII-δB and CaMKII-δC is inversely regulated in cardiomyocytes subjected to oxidative stress with a downregulation of CaMKII-δB and an upregulation of CaMKII-δC. Similar opposite regulation of these cardiac CaMKII-δ isoforms is induced by myocardial IR injury in vivo. Second, overexpression of CaMKII-δB markedly attenuates H2O2-, hypoxia-, and AngII-induced apoptotic cell death, whereas overexpression of CaMKII-δC promotes cardiomyocyte apoptosis. Moreover, overexpression of CaMKII-δB, but not CaMKII-δC, selectively elevates phosphorylation of HSF1 at Ser230, resulting in increased expression of the major inducible HSP70, iHSP70, that is obligated to CaMKII-δB-induced cardiomyocyte protection. These present findings support the perception that the nuclear-located isoform, CaMKII-δB, constitutes an important cardiac protective factor against multiple insulting stimuli-induced myocardial damage, and that a reduction in this beneficial isoform in conjunction with a simultaneous increase in the detrimental isoform, CaMKII-δC, is a potential cause factor of various cardiac disorders, including cardiac myocyte apoptosis, IR injury, cardiac arrhythmia, and heart failure 26-29.
Although the present findings underscore a potentially important anti-apoptotic effect of the nuclear-located isoform, CaMKII-dB, it is noteworthy that overexpression of CaMKII-δB activates hypertrophic gene expression and produces hypertrophy in cultured cardiomyocytes 30. Similarly, transgenic mice with cardiac-specific overexpression of the nuclear-targeted cardiac CaMKII-isoform develop cardiac hypertrophy, cardiomyocyte enlargement and increases in hypertrophic gene expression 20. Thus, exaggerated upregulation of either cardiac CaMKII isoform or an imbalance between these two major isoforms might cause pathological consequences in the heart.
Isoform-Specific Function and Regulation of Cardiac CaMKII-δB and CaMKII-δC
Differences between these CaMKII-δ isoforms are manifested by the distinct phenotypes of transgenic mouse models with cardiac-specific overexpression of CaMKII-δB or CaMKII-δC. Although both transgenic models can develop cardiac hypertrophy, CaMKII-δC transgenic mice display much more accelerated cardiac functional deterioration and shortened life span as compared to CaMKII-δB transgenic mice even with a matched increase in their total cellular CaMKII activity 15, 20. Additionally, transgenic overexpression of the cytosolic isoform, CaMKII-δC, leads to hyper-phosphorylation of substrates involved in cardiac excitation-contraction coupling such as ryanodine receptor (RyR) and phospholamban (PLB), resulting in increased SR Ca2+ sparks and decreased SR Ca2+ load 12, 15, whereas none of these parameters are changed in age-matched transgenic mice with the nuclear targeted CaMKII-δB 20. The isoform-specific phenotypes of CaMKII-δB and CaMKII-δC transgenic mouse models can be explained, at least in part, by our previous notion that overexpression of CaMKII-δC, instead of CaMKII-δB, exaggerates the adult mouse cardiomyocyte apoptotic death induced by β1-AR stimulation and relays apoptotic signal for multiple death-inducing stimuli 16, 17. Here, we have further demonstrated that CaMKII-δB, opposite to CaMKII-δC, protects cardiomyocytes against apoptosis induced by multiple insulting stimuli such as oxidative stress, hypoxia and AngII stimulation. It is also noteworthy that in response to oxidative stress and IR injury, expression of CaMKII-δB and CaMKII-δC are inversely regulated at gene and protein levels. The downregulation of the protective factor, CaMKII-δB, as well as the upregulation of the deleterious factor, CaMKII-δC, are expected to contribute to oxidative stress- or IR-induced cell injury and cell death. Taken together, there are distinct differences between these cardiac CaMKII isoforms in their function and regulation under various physiological and pathological circumstances.
Cardioprotection by CaMKII-δB Is Mediated by HSF1 Phosphorylation and Subsequent Upregulation of iHSP70
HSP belong to an important family of endogenous protective proteins that rapidly respond to a wide variety of stress stimuli, such as heat shock, hypoxia, ischemia, increased ROS, and inflammation. Among various members of HSP family, HSP70 has been implicated as cardiac protective 31-33. The HSP70 family consists of, at least, eight highly homologous members, including constitutively expressed (or cognate) and highly inducible proteins that differ from each other by the intracellular localization and expression pattern 34. In particular, Hst70 (encoded by Hspa2 gene) is expressed at high levels in testis and at considerably lower or undetectable levels in other tissues 35, 36. In contrast, iHSP70 (also named HSP72, encoded by Hspa1b gene) is expressed at low or undetectable levels in most unstressed normal cells and tissues, but its expression is rapidly induced by multiple physical and chemical stresses, and therefore it is often called the major stress-inducible HSP70. Nevertheless, both iHSP70 and Hst70 can be induced under diverse cell stress conditions 37, 38, and have been implicated in promoting cell survival 39, 40.
In searching for potential mechanisms responsible for the protective effect of CaMKII-δB, we have found that both iHSP70 and Hst70 are specifically upregulated by overexpression of CaMKII-δB but not its cytosolic counterpart. Importantly, gene silencing of iHSP70 blocks the protective effect of CaMKII-δB, establishing an essential role of iHSP70 in relaying CaMKII-δB anti-apoptotic signaling. In contrast, gene silencing of the closely related HSP70 family member, Hst70, has no detectable impact on CaMKII-δB-mediated protection. Thus, iHSP70 but not its homologous Hst70 plays an essential role in delivering CaMKII-δB activated anti-apoptotic signaling.
HSF1 is the transcription factor responsible for stress-induced expression of HSP70. In unstressed cells, HSF1 is present in both the cytoplasm and nucleus in a monomeric form that has no DNA binding activity. In response to heat shock and other stress stimuli, HSF1 assembles into a trimer, accumulates within the nucleus and binds with the proximal promoter heat shock element (HSE) on the HSP70 gene to induce the transcription of HSP70 41-43. Previous studies have shown that hyper-phosphorylation of HSF1 is required for the transcriptional competence of the factor 44-46. There are two kinds of phosphorylation sites on HSF1, i.e. inducible residue (Ser 230), which positively contributes to the transcriptional competence of HSF1 22, and constitutive residues (Ser 303, Ser 307 and Ser363), which repress the transcriptional activity of HSF1 47, 48.
In the present study, we have demonstrated that CaMKII-δB and HSF1 are co-localized in the nuclear compartment and that phosphorylation of HSF1 at Ser230, but not a constitutive residue Ser303, is clearly elevated in myocytes infected with Adv-CaMKII-δB in the presence or absence of H2O2 treatment (Online Figure V, online data supplements). Our data indicate that CaMKII-δB specifically phosphorylates the inducible residue of HSF1-Ser230, and subsequently enhances its transcriptional activity, leading to increased expression of iHSP70 which is obligated to CaMKII-δB-mediated cardioprotection.
In summary, we have demonstrated, for the first time, oxidative stress and IR injury leads to a downregulation of CaMKII-δB and an upregulation of CaMKII-δC, and that the two major cardiac CaMKII isoforms exhibit opposing effects on cardiomyocyte viability with CaMKII-δB anti-apoptotic and CaMKII-δC pro-apoptotic (Figure 7). Phosphorylation of HSF1 and subsequent induction of iHSP70 are essentially involved in CaMKII-δB-mediated cell survival signaling (Figure 7). These findings mark both cardiac CaMKII isoforms as promising therapeutic targets for preventing or alleviating cardiomyocyte loss caused by myocardial infarction, IR injury, heart failure, and many other etiologies.
Figure 7. Schematic presentation to show oxidative stress- or IR-induced opposite regulation of the expression and functional consequence of the two major cardiac CaMKII isoforms, CaMKII-δB and CaMKII-δC.
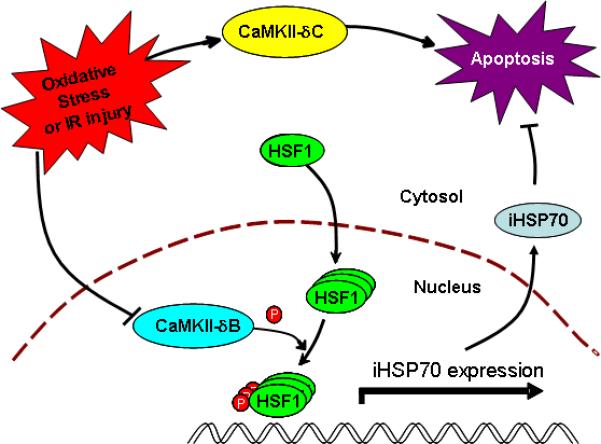
Oxidative stress or IR injury leads to a downregulation of the anti-apoptotic isoform, CaMKII-δB, and an upregulation of the apoptotic isoform, CaMKII-δC, resulting in robust cardiomyocyte apoptosis. The anti-apoptotic effect of CaMKII-δB is attributable to the kinase-mediated phosphorylation of HSF1 at Ser230 and subsequent induction of iHSP70 gene expression.
Supplementary Material
Acknowledgements
We would like to thank LP Wei, CY Li, ZY Peng, WJ Xie, HQ Fang, H Huang, and HL Zhang for their excellent technical support.
Sources of Funding
This work was supported by Peking University 985 Project (Wei Peng, Yan Zhang, Ming Zheng, Heping Cheng, Chun-Mei Cao, and Rui-Ping Xiao) and 973 Program (2007CB512100) (Chun-Mei Cao, Ming Zheng and Heping Cheng) and, in part, by the Intramural Research Program of the NIH, National Institute on Aging (Weizhong Zhu and Rui-Ping Xiao).
Non-standard Abbreviations and Acronyms
- β1-AR
β1-adrenergic receptor
- Adv
Adenovirus
- AngII
Angiotensin II
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- HA
Hemagglutinin
- HSE
Heat shock element
- HSF1
Heat shock factor 1
- Hst70
Testis-specific heat shock protein 70
- iHSP70
Inducible heat shock protein 70
- IR
Ischemia/reperfusion
- LDH
Lactate dehydrogenase
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide
- PLB
Phospholamban
- ROS
Reactive oxygen species
- RyR
Ryanodine receptor
- siRNA
Small interfering RNA
Footnotes
Disclosures: None.
References
- 1.Edman CF, Schulman H. Identification and characterization of delta B-CaM kinase and delta C-CaM kinase from rat heart, two new multifunctional Ca2+/calmodulin-dependent protein kinase isoforms. Biochim Biophys Acta. 1994;1221:89–101. doi: 10.1016/0167-4889(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 2.Baltas LG, Karczewski P, Krause EG. The cardiac sarcoplasmic reticulum phospholamban kinase is a distinct delta-CaM kinase isozyme. FEBS Lett. 1995;373:71–75. doi: 10.1016/0014-5793(95)00981-e. [DOI] [PubMed] [Google Scholar]
- 3.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 4.Hoch B, Wobus AM, Krause EG, Karczewski P. delta-Ca(2+)/calmodulin-dependent protein kinase II expression pattern in adult mouse heart and cardiogenic differentiation of embryonic stem cells. J Cell Biochem. 2000;79:293–300. [PubMed] [Google Scholar]
- 5.Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol. 1994;126:839–852. doi: 10.1083/jcb.126.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 8.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda, Md. 2008;23:151–159. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- 10.Wright SC, Schellenberger U, Ji L, Wang H, Larrick JW. Calmodulin-dependent protein kinase II mediates signal transduction in apoptosis. FASEB J. 1997;11:843–849. doi: 10.1096/fasebj.11.11.9285482. [DOI] [PubMed] [Google Scholar]
- 11.Lisman J, Malenka RC, Nicoll RA, Malinow R. Learning mechanisms: the case for CaM-KII. Science (New York, N.Y. 1997;276:2001–2002. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- 12.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 13.Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, Xiao RP, Cheng H. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr., Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 16.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282:10833–10839. doi: 10.1074/jbc.M611507200. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. 2005;97:1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]
- 19.Ramirez MT, Zhao XL, Schulman H, Brown JH. The nuclear deltaB isoform of Ca2+/calmodulin-dependent protein kinase II regulates atrial natriuretic factor gene expression in ventricular myocytes. J Biol Chem. 1997;272:31203–31208. doi: 10.1074/jbc.272.49.31203. [DOI] [PubMed] [Google Scholar]
- 20.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Jr., Brown JH. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–1267. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 21.Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- 22.Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M, Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson JE, Sistonen L. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. The EMBO journal. 2001;20:3800–3810. doi: 10.1093/emboj/20.14.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 24.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–323. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 25.Takano H, Zou Y, Hasegawa H, Akazawa H, Nagai T, Komuro I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid Redox Signal. 2003;5:789–794. doi: 10.1089/152308603770380098. [DOI] [PubMed] [Google Scholar]
- 26.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundina-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc. Res. 2007;73:689–698. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 28.Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ, Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation. 2006;114:1352–1359. doi: 10.1161/CIRCULATIONAHA.106.644583. [DOI] [PubMed] [Google Scholar]
- 29.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. The Journal of clinical investigation. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong KY, Lai CC, Lille S, Chang C, Su CY. Stable overexpression of the constitutive form of heat shock protein 70 confers oxidative protection. J Mol Cell Cardiol. 1998;30:599–608. doi: 10.1006/jmcc.1997.0623. [DOI] [PubMed] [Google Scholar]
- 33.Okubo S, Wildner O, Shah MR, Chelliah JC, Hess ML, Kukreja RC. Gene transfer of heat-shock protein 70 reduces infarct size in vivo after ischemia/reperfusion in the rabbit heart. Circulation. 2001;103:877–881. doi: 10.1161/01.cir.103.6.877. [DOI] [PubMed] [Google Scholar]
- 34.Tavaria M, Gabriele T, Kola I, Anderson RL. A hitchhiker's guide to the human Hsp70 family. Cell Stress Chaperones. 1996;1:23–28. doi: 10.1379/1466-1268(1996)001<0023:ahsgtt>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Son WY, Hwang SH, Han CT, Lee JH, Kim S, Kim YC. Specific expression of heat shock protein HspA2 in human male germ cells. Mol Hum Reprod. 1999;5:1122–1126. doi: 10.1093/molehr/5.12.1122. [DOI] [PubMed] [Google Scholar]
- 36.Fourie AM, Peterson PA, Yang Y. Characterization and regulation of the major histocompatibility complex-encoded proteins Hsp70-Hom and Hsp70-1/2. Cell Stress Chaperones. 2001;6:282–295. doi: 10.1379/1466-1268(2001)006<0282:carotm>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner TT, Miller DW. On the synthesis and secretion of rat seminiferous tubule proteins in vivo after ischemia and germ cell loss. Biol Reprod. 1997;57:1275–1284. doi: 10.1095/biolreprod57.6.1275. [DOI] [PubMed] [Google Scholar]
- 38.Gabai VL, Meriin AB, Yaglom JA, Wei JY, Mosser DD, Sherman MY. Suppression of stress kinase JNK is involved in HSP72-mediated protection of myogenic cells from transient energy deprivation. HSP72 alleviates the stress-induced inhibition of JNK dephosphorylation. J Biol Chem. 2000;275:38088–38094. doi: 10.1074/jbc.M006632200. [DOI] [PubMed] [Google Scholar]
- 39.Nakano M, Mann DL, Knowlton AA. Blocking the endogenous increase in HSP 72 increases susceptibility to hypoxia and reoxygenation in isolated adult feline cardiocytes. Circulation. 1997;95:1523–1531. doi: 10.1161/01.cir.95.6.1523. [DOI] [PubMed] [Google Scholar]
- 40.Schomisch SJ, Murdock DG, Hedayati N, Carino JL, Lesnefsky EJ, Cmolik BL. Cardioplegia prevents ischemia-induced transcriptional alterations of cytoprotective genes in rat hearts: a DNA microarray study. J Thorac Cardiovasc Surg. 2005;130:1151. doi: 10.1016/j.jtcvs.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 41.Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Molecular and cellular biology. 1993;13:2486–2496. doi: 10.1128/mcb.13.4.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green M, Schuetz TJ, Sullivan EK, Kingston RE. A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Molecular and cellular biology. 1995;15:3354–3362. doi: 10.1128/mcb.15.6.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuo J, Rungger D, Voellmy R. Multiple layers of regulation of human heat shock transcription factor 1. Molecular and cellular biology. 1995;15:4319–4330. doi: 10.1128/mcb.15.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress-induced serine phosphorylation. Evidence for a multistep pathway of regulation. J Biol Chem. 1996;271:3355–3358. doi: 10.1074/jbc.271.7.3355. [DOI] [PubMed] [Google Scholar]
- 45.Bruce JL, Price BD, Coleman CN, Calderwood SK. Oxidative injury rapidly activates the heat shock transcription factor but fails to increase levels of heat shock proteins. Cancer research. 1993;53:12–15. [PubMed] [Google Scholar]
- 46.Xia W, Voellmy R. Hyperphosphorylation of heat shock transcription factor 1 is correlated with transcriptional competence and slow dissociation of active factor trimers. J Biol Chem. 1997;272:4094–4102. doi: 10.1074/jbc.272.7.4094. [DOI] [PubMed] [Google Scholar]
- 47.Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Molecular and cellular biology. 1997;17:2107–2115. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xavier IJ, Mercier PA, McLoughlin CM, Ali A, Woodgett JR, Ovsenek N. Glycogen synthase kinase 3beta negatively regulates both DNA-binding and transcriptional activities of heat shock factor 1. J Biol Chem. 2000;275:29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.