

Abstract
In both dogs and humans Leishmania infantum infection is more prevalent than disease, as infection often does not equate with clinical disease. Previous studies additively indicate that advanced clinical visceral leishmaniasis is characterized by increased production of anti-Leishmania antibodies, Leishmania-specific lymphoproliferative unresponsiveness, and decreased production of gamma interferon (IFN-γ) with a concomitant increase of interleukin-10 (IL-10). In order to differentiate infection versus progressive disease for better disease prognostication, we temporally evaluated humoral and cellular immunologic parameters of naturally infected dogs. The work presented here describes for the first time the temporal immune response to natural autochthonous L. infantum infection in foxhounds within the United States. Several key changes in immunological parameters should be considered when differentiating infection versus clinical disease, including a dramatic rise in IgG production, progressive increases in antigen-specific peripheral blood mononuclear cell proliferation, and IFN-γ production. Polysymptomatic disease is precluded by increased IL-10 production and consistent detection of parasite kinetoplast DNA in whole blood. This clinical presentation and the immuno-dysregulation mirror those observed in human patients, indicating that this animal model will be very useful for testing immunomodulatory anti-IL-10 and other therapies.
Leishmaniasis is a group of vector-borne diseases caused by intracellular protozoan parasites of the genus Leishmania. Disease manifestations can range from localized, self-healing cutaneous ulcers to disseminated disease, referred to as visceral leishmaniasis (VL). VL is fatal if left untreated. It is primarily caused by Leishmania donovani in Africa and India and by L. infantum (or L. chagasi) in the Mediterranean basin, Asia, and Central and South America.
VL, as caused by L. infantum infection, is zoonotic (4). Both dogs and humans are natural hosts (27), and in endemic regions, infected dogs are the primary domestic reservoir for zoonotic VL and the most significant risk factor predisposing humans to infection (9). L. infantum infection often does not equate with clinical disease (18). Typical clinical signs of VL include fever, weight loss, anemia, lymphadenopathy, and hepato- and splenomegaly (4, 22, 27).Clinical stages of infection can be classified by the severity of clinical signs, humoral and cell-mediated immune responses, and parasite load (33). We propose that these parameters can also be used to determine the best window for treatment and in some cases predict the appearance of clinical signs and prognosis (24).
Host protection against Leishmania infection requires a proinflammatory, TH1 immune response, as characterized by the production of interleukin-12 (IL-12) by antigen-presenting cells and gamma interferon (IFN-γ) by T cells (reviewed in reference 22). Advanced clinical VL in human patients is characterized by Leishmania-specific lymphoproliferative unresponsiveness and decreased production of IFN-γ following in vitro Leishmania antigen restimulation (11, 31). Active disease is associated with elevated IL-10 levels in serum and enhanced IL-10 mRNA in lesional tissues (reviewed in reference 22). Cured or subclinical individuals are able to mount antigen-specific IFN-γ responses following Leishmania antigen restimulation in vitro. Cured patients are also resistant to reinfection and are leishmanin skin test positive, suggesting no inherent defect in the antigen-dependent TH1 response (3, 7, 34).
Canine visceral leishmaniasis (CVL) in endemic areas mimics both the immunologic alterations and pathophysiology of human disease. Autochthonous L. infantum infection in the United States foxhound population has been recently described (5, 8). Despite a potentially different means of transmission, i.e., non-vector borne (12, 24, 29), symptomatic disease and pathological findings in naturally infected foxhounds parallel those observed in both canines and humans in endemic regions (12). For these studies, we hypothesized that the immunopathology of primarily non-vector-mediated L. infantum CVL would reflect the changes observed in humans, including increased anti-Leishmania antibodies in sera and decreased lymphoproliferative IFN-γ-mediated responses with increased IL-10 production. Here we followed a cohort of U.S.-born, naturally infected canines to determine their immunopathology and clinical presentation(s) of autochthonous L. infantum infection. Analysis of clinical signs, serology, and kinetoplast-specific quantitative PCR (qPCR) categorized these animals into four different groups: (i) noninfected, (ii) infected resistant, (iii) infected susceptible, and (iv) clinical, as previously described in reference 33. Animals in the fourth clinical state had increased production of IgG1 and IgG2, decreased lymphoproliferative responses and IFN-γ production, and increased IL-10 production. The appearance of any of these immunological parameters correlated with disease progression.
The work presented here describes for the first time the temporal immune response to natural autochthonous L. infantum canine infection in the United States. We show that even in the likely absence of vector-mediated transmission (32), clinical presentation and immuno-dysregulation mirror those observed in dogs and humans infected in regions of endemicity (1, 22). The ongoing antigen-specific immune response to L. infantum infection wanes as disease progresses, and production of anti-Leishmania antibodies and IL-10 are key immunologic features of disease manifestation and progression.
MATERIALS AND METHODS
Description of animals.
Although VL is not endemic in the United States, canine visceral leishmaniasis has recently been described as an epidemic within the foxhound population in this country. The first report of a foxhound CVL epidemic in the United States was in 1999, in a foxhound kennel in New York (8). By 2005, it was reported that 60 kennels in 22 states and two Canadian provinces had L. infantum-seropositive foxhounds and that autochthonous infection in canines was for the most part limited to foxhounds (5).
Dogs used in this study, all foxhounds, ranged in age from 6 months to 7 years. These animals or their tissues were donated to Iowa State University College of Veterinary Medicine by two different midwestern foxhound kennels. Nine of the dogs were donated based on positive serological indirect immunofluorescence assay test (IFAT) results (>1:64) and presentation of clinical signs. The remaining four dogs were born to an IFAT-positive (>1:256) female. All animals were housed at Iowa State University Veterinary College, and the Institutional Animal Care and Use Committee at Iowa State approved all protocols involving animals. Prior to arrival, all dogs were vaccinated for core canine diseases. Once under the care of laboratory animal resources (LAR) at Iowa State University, blood samples were obtained for complete blood count (CBC) and chemistry, and stool samples were collected for enteric parasite assessment. All animals were treated for ectoparasites and intestinal parasites (Giardia, roundworms, and Coccidia) via treatment with Strongid (5 mg/kg of body weight), Baytril (1/4 tablet), Albon (55 mg/kg), Panacur (2 ml/kg), Clavamox (13.75 mg/kg), and Cephalexin (25 mg/kg).
Clinical evaluation.
Upon arrival at Iowa State University College of Veterinary Medicine, all animals were clinically assessed via physical examination, complete blood count, chemistry panel analysis, L. infantum kinetoplast DNA (kDNA)-specific qPCR, and IFAT serologic analysis. Based on these parameters, animals were classified into four distinct categories: noninfected, showing no clinical signs of disease and qPCR and IFAT negative; infected resistant, showing no to mild clinical signs and IFAT and qPCR positive or negative; infected susceptible, showing mild to moderate clinical signs and qPCR and IFAT positive; clinical, showing severe, disseminated disease and IFAT and qPCR positive.
Parasites.
Leishmania infantum (LIVT-2) (30) was grown to stationary phase in complete Grace's medium (incomplete Grace's supplemented with 20% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine). Freeze-thawed whole antigen was prepared as described previously (13).
PBMC isolation and CFSE staining.
All animals were allowed to acclimate for 1 week prior to immunological studies. Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood samples using Ficoll-Histopaque 1077 (Sigma, St. Louis, MO) gradient centrifugation. Red blood cells were removed using ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2EDTA, pH 7.4). PBMC were labeled with carboxyfluorescein succinyl ester (CFSE; Molecular Probes, Eugene, OR) as described previously (14). PBMC were washed twice in phosphate-buffered saline (PBS) and resuspended in complete medium (RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 25 mM HEPES buffer). PMBC were counted and adjusted to 4 × 106/ml for further analysis.
PBMC proliferation assay.
CFSE-labeled PBMC (2 × 105/well) were plated on 96-well plates and incubated with medium alone or stimulated with concanavalin A (ConA; 5 μg/ml) for 4 days, with freeze-thawed whole L. infantum antigen (10 μg/ml) for 7 days, or with distemper vaccine (Vanguard Plus 5; Pfizer) control for 10 days, at 37°C with 5% CO2. Cells were harvested, washed in fluorescence-activated cell sorter (FACS) buffer (0.1% albumin, 0.1% sodium azide in PBS), and labeled with phycoerythrin (PE)-conjugated anti-canine CD4 antibody (Serotec, Raleigh, NC). Cells were fixed in 1% paraformaldehyde and analyzed using a FACSCanto flow cytometer (BD Pharmingen, San Diego, CA). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
IFN-γ and IL-10 ELISA.
Unlabeled PMBC (2 × 105) were plated and incubated as described above. Supernatants were collected at the indicated time points and stored at −20°C until analysis. IFN-γ and IL-10 production levels were measured using enzyme-linked immunosorbent assay (ELISA) kits from R&D Systems (Minneapolis, MN) according to the manufacturer's recommendations.
Serology and real-time qPCR.
Serum samples were collected from all animals, stored at −20°C, and sent to the Centers for Disease Control and Prevention for IFAT testing for antibodies to Leishmania spp. as previously described (5). DNA from whole blood samples collected in heparinized tubes (BD Pharmingen, San Diego, CA) was isolated using the Qiagen blood DNA isolation kit according to the manufacturer's instructions. DNA quality and quantity were measured using a NanoDrop ND1000 spectrophotometer (Wilmington, DE). L. infantum kDNA-specific primers and probe (F, 5′-CCGCCCGCCTCAAGAC; R, 5′-TGCTGAATATTGGTGGTTTTGG [Integrated DNA Technologies, Coralville, IA]; probe, 5′-6-carboxyfluorescein [6-FAM]-AGCCGCGAGGACC-3′ minor groove binder nonfluorescent quencher [Applied Biosystems, Foster City, CA]) were used. (FAM is a laser-activated reporter dye.) Blood DNA samples were assayed via qPCR in duplicates of three dilutions (straight, 1:10, and 1:20) using a Stratagene Mx3005P qPCR system via a 96-well format and Platinum qPCR SuperMix-UDG master mix (Invitrogen, Carlsbad, CA). Primers were used at 775 nM and probe at 150 nM, with thermocyling at 50°C for 2 min, 95°C for 2 min, and 50 cycles of 95°C for 30 s, 57°C for 1 min, and 60°C for 1 min. Results were analyzed via MxPro QPCR software version 4.01 in conjunction with Microsoft Excel.
L. infantum-specific IgG ELISA.
High-affinity plates were coated overnight at 4°C with 10 μg/well of freeze-thawed L. infantum antigen in 50 mM carbonate-bicarbonate buffer. Plates were blocked with 200 μl of blocking buffer for 1 h at room temperature and washed. Serum samples (100 μl) were diluted 1:100 and incubated for 2 h at room temperature. Plates were developed with horseradish peroxidase-conjugated anti-canine IgG1 or IgG2 (1:20,000; Bethyl Laboratories, Montgomery, TX) for 1 h, and absorbance was read at 405 nm using a microplate reader (Molecular Devices, Sunnyvale, CA).
Statistical analysis.
Statistical significance was analyzed using Prism4 (GraphPad Software Inc., La Jolla, CA). Differences between groups were determined using the Mann-Whitney U-test. P values below 0.05 were considered significantly different.
RESULTS
Clinical evaluation.
Clinical assessment included a CBC and chemistry panel (Table 1). Following euthanasia, necropsy was performed by a veterinary pathologist and a complete set of tissues was collected for each animal and evaluated histologically (Table 1). Lymphocytosis (elevated lymphocyte numbers in the blood) was consistently present in all dogs tested. Persistent lymphocytosis is indicative of chronic antigen stimulation, which we would attribute to the presence of Leishmania parasites in infected animals. However, since noninfected dogs also showed lymphocytosis, we cannot rule out the possibility of an increased circulating lymphocyte number due to other infections, including gastrointestinal or ectoparasitism, which had been observed in these foxhounds previously (data not shown). In all infected animals we observed a moderate to marked hyperglobulinemia. Serum chemistry and histopathologic findings in the infected susceptible dogs indicated the onset of systemic disease consistent with visceral leishmaniasis, including elevated blood urea nitrogen (BUN), creatinine, and phosphorous, anemia, lymphoplasmacytic portal hepatitis, histiocytic splenitis, and membranous glomerulonephritis. CBC and serum chemistry evaluations of clinical dogs indicated these animals had signs of nonregenerative anemia, renal compromise (elevated BUN, creatine, and phosphorus), and hepatic injury (elevated alanine transferase). Despite their clinical state, lymphocytopenia was not observed in clinical dogs. Histopathologic examination confirmed these findings and also showed systemic histiocytic inflammation with a myriad of intracellular Leishmania amastigotes.
TABLE 1.
Summary of kDNA qPCR, IFAT serology, CBC and blood chemistry, and gross and histopathology findings for L. infantum-infected dogs
| Clinical state (n) | No. PCR+/no. tested | IFAT titer | CBC and chemistry (no. with indicated finding/total no. evaluated) | Body condition (no. with condition/total no. tested) | Gross pathological findings (no. with finding/total no. tested) | Histopathologic finding(s) (no. with finding/total no. tested) |
|---|---|---|---|---|---|---|
| Noninfected (2) | 0/2 | Lymphocytosis (2/2) | Adequate (2/2) | Mild mesenteric and popliteal lymphadenomegaly (2/2) | No significant findings (2/2) | |
| Infected resistant (4) | 2/4 | 1:64 | Lymphocytosis (4/4), hyperglobulinemia (2/4) | Adequate (4/4) | Moderate systemic lymphadenomegaly (4/4), mild splenomegaly (2/4) | No significant findings (2/4), mild lymphoplasmacytic portal hepatitis (2/4), rare Leishmania amastigotes identified via splenic impression smear (1/4) |
| Infected susceptible (3) | 3/3 | 1:256 | Lymphocytosis (3/3), hyperglobulinemia (3/3), anemia (1/3), elevated BUN, creatinine, and phosphorus (1/3) | Adequate to thin (3/3) | Moderate systemic lymphadenomegaly (3/3), mild splenomegaly (2/3), hepatomegaly (1/3) | Lymphohistiocytic portal hepatitis (3/3), membranous glomerulonephritis (3/3), histiocytic splenitis (2/3), rare Leishmania amastigotes identified via splenic impression smear (1/3) |
| Clinical (4)a | 4/4 | 1:512 | Lymphocytosis (2/2), anemia (2/2), elevated BUN, creatinine, phosphorus,and alanine transferase (2/2), hyperglobulinemia (2/2) | Thin to emaciated (4/4) | Marked systemic lymphadenomegaly (2/2), splenomegaly (2/2), hepatomegaly (2/2) | Lymphohistiocytic portal hepatitis (2/2), membranous glomerulonephritis (2/2), histiocytic splenitis (2/2), systemic histiocytic inflammation with myriad intracellular Leishmania amastigotes (2/2) |
Only two of four dogs were assessed for CBC, chemistry, and necropsy changes.
Serology and qPCR.
To confirm infection and disease status, each dog was evaluated for L. infantum serostatus and qPCR for L. infantum kDNA. Serum samples from all foxhounds in the study were sent to the Centers for Disease Control and Prevention for IFAT analysis of antibodies against Leishmania spp. (Table 1). All dogs in the control (noninfected, nonfoxhound), noninfected group and two dogs from the infected resistant group were seronegative (titer of ≤1:16). The two remaining dogs within the infected resistant group had seropositive titers (1:64). Dogs within the infected susceptible group (three dogs) had titers of 1:256, and dogs within the clinical group (four dogs) had strong seroreactivity to Leishmania antigen (1:512).
L. infantum-specific kDNA amplification was observed in all clinical and infected susceptible dogs and in two of the infected resistant group. As expected, no amplification was observed in the noninfected foxhounds (Table 1) or in the control, nonfoxhound dogs. These data indicate that increased parasitemia is found during later stages of infection.
L. infantum-specific IgG1 and IgG2 production.
Chemistry findings in serum samples from clinical dogs indicated these animals had pan-elevation of Igs: IgA, >500 mg/dl; IgG, >5,000 mg/dl; IgM, 400 mg/dl (normal ranges are 20 to 150 mg/dl, 1,000 to 2,000 mg/dl, and 70 to 270 mg/dl, respectively). Hypergammaglobulinemia has been associated with CVL pathophysiology in disease progression (12) and suppression of the immune response to L. infantum (26). However, a relationship between IgG isotype profile and disease resistance versus susceptibility remains to be established. Conflicting reports have failed to provide a clear role for IgG1 or IgG2 production in disease development (25, 28). Based on our findings of detectable circulating parasites as disease progressed, we wanted to determine if this observation correlated with detection of specific antibody levels. Using whole parasite antigen we found that sera from control and noninfected groups contained minimal IgG1 and IgG2 antibodies when measured by ELISA, as optical density (OD) values observed were similar to background readings (OD, ∼0.01). The highest levels of anti-L. infantum IgG1 (Fig. 1A) and IgG2 (Fig. 1B) were produced by the infected susceptible and clinical groups. Overall, IgG levels increased as disease progressed; however, we did not observe a direct correlation between either IgG isotype and clinical status. Other nonantibody effector functions may therefore be more predictive of disease progression.
FIG. 1.
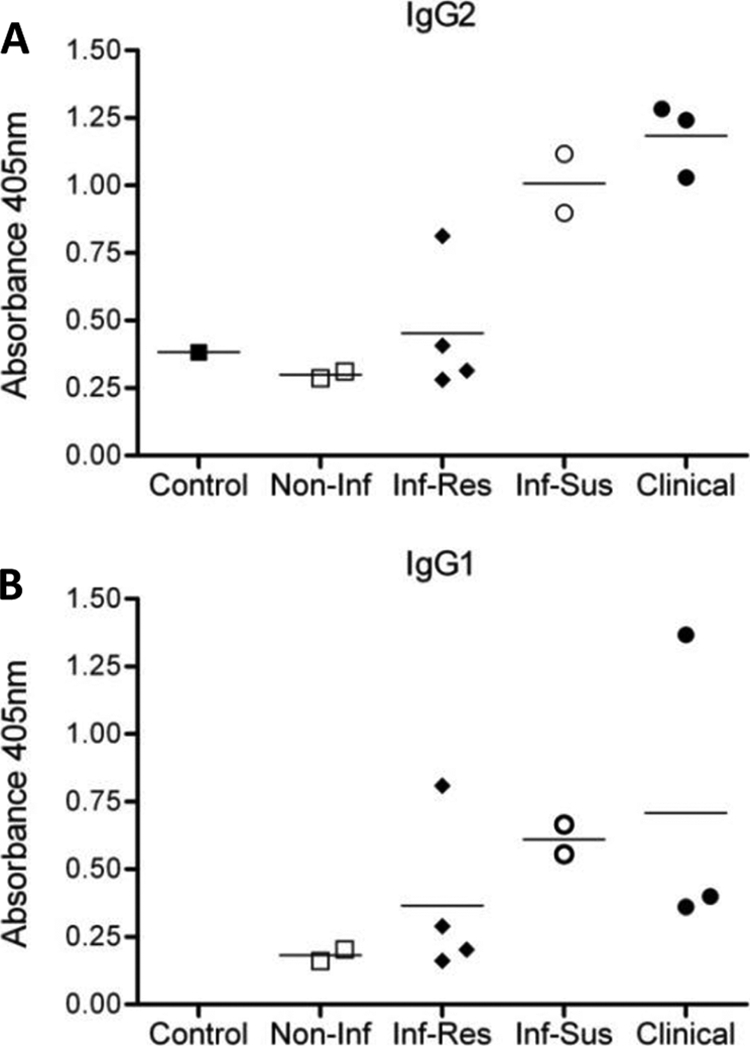
Anti-L. infantum IgG1 and IgG2 responses increase with CVL disease progression. L. infantum-specific IgG responses were measured via ELISA using sera from a control (one dog), noninfected (two dogs), infected resistant (four dogs), infected susceptible (two dogs), and clinical (three dogs) animals. Blood samples were collected and centrifuged to clarify serum. Results shown are OD values from the antigen-specific IgG1 (A) and IgG2 (B) ELISAs. Lines indicate mean values for each group.
L. infantum-specific PBMC proliferative response.
A key immunologic feature of late clinical VL is the inability of PBMC to generate a protective, L. infantum-specific immune response (31). This is characterized by the loss of the antigen-specific lymphoproliferative response and the loss of IFN-γ production. To identify whether this lack of antigen responsiveness as disease progresses occurred in our canine cohort, we analyzed the antigen-specific proliferative response of PBMC CD4+ T cells from all four groups. Blood samples were collected every 4 weeks during a period of at least 3 months for each dog. PBMC were isolated from whole blood samples, stained with CFSE, and stimulated with ConA, L. infantum antigen, or distemper vaccine or left untreated. PMBC were then analyzed for CD4+ T-cell proliferation via flow cytometry. CD4+ T cells from all dogs proliferated in response to stimulation with ConA, indicating that the CD4+ T-cell compartment was not mitogenically deficient (Fig. 2B). In response to distemper vaccine stimulation all groups except for the clinical dogs had a proliferative response, indicating that although mitogenically competent, clinical dogs were not capable of initiating antigen-specific proliferative responses (Fig. 2A and B). In response to L. infantum antigen stimulation, control (uninfected, nonfoxhound) and noninfected dogs showed a minimal level of proliferation in response to antigen restimulation (Fig. 2). While a significantly greater percentage of CD4+ T cells from infected resistant dogs proliferated in response to antigen restimulation than in noninfected dogs, infected susceptible dogs demonstrated the greatest percentage of proliferative CD4+ T cells, with a level significantly higher than in infected resistant animals (Fig. 2A). In contrast, the antigen-specific CD4+ T-cell proliferative response from clinical animals was significantly decreased compared to that of infected susceptible dogs. These data suggest that as disease progresses, there is an initial increase in antigen-specific lymphoproliferative responsiveness of CD4+ T cells that eventually dwindles. Appearance of clinical disease correlates with the loss of the antigen-specific lymphoproliferative response. Based on this observed loss of proliferative response in late disease, we wished to determine if cytokine production, specifically IFN-γ and IL-10, could be correlated with this lymphosuppressive change.
FIG. 2.
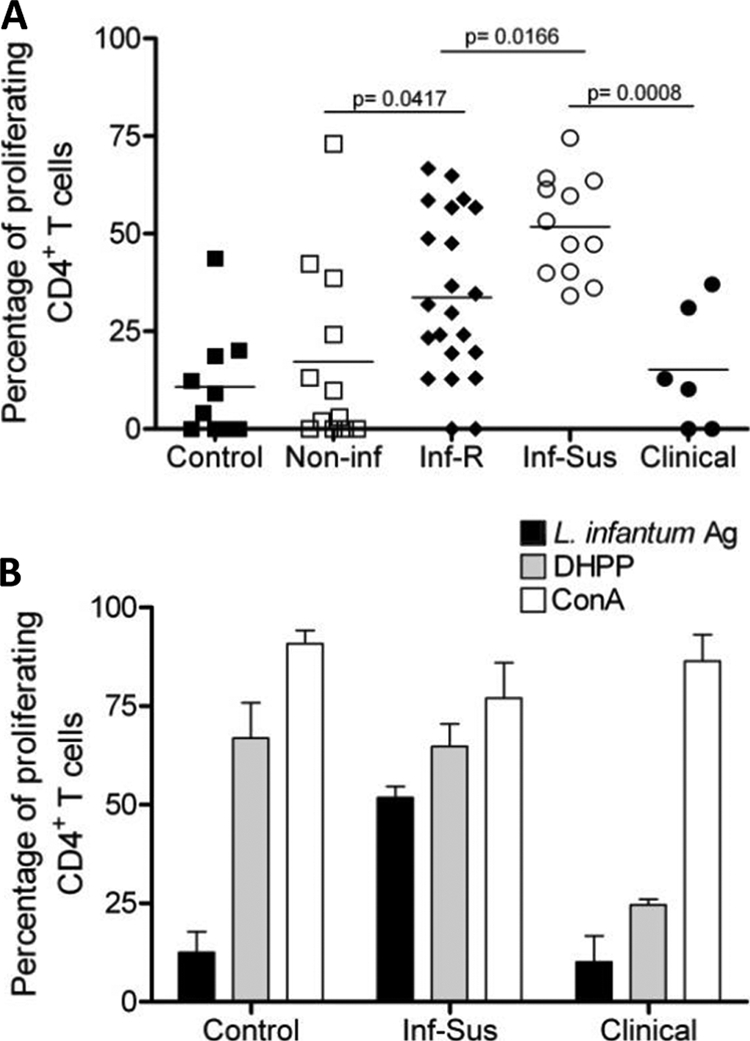
Decreased lymphoproliferative response in PBMC from L. infantum-infected, clinical dogs. (A) PBMC proliferative response evaluations from a control (one dog), noninfected (two dogs), infected resistant (four dogs), infected susceptible (two dogs), and clinical (two dogs) animals were repeated monthly over 3 to 6 months. PBMC were isolated, stained with CFSE, restimulated with freeze-thawed L. infantum antigen, and incubated for 7 days at 37°C. Cells were then harvested and stained with a PE-conjugated anti-CD4 antibody. PMBC CD4+ T-cell proliferation was assessed via CFSE dilution using flow cytometry. Each point is indicative of a blood draw from each animal over a 3- to 6-month period and subsequent proliferation assay. At least four separate proliferation assays were carried out over time on each dog in every group. Horizontal lines indicate the mean responses for each group. *, significant difference (P < 0.05). (B) PBMC proliferative responses to L. infantum antigen (Ag), distemper vaccine (DHPP), or ConA stimulation for the control (one dog), infected susceptible (three dogs), and clinical (two dogs) animals. PBMC were isolated and processed as described for panel A and stimulated with L. infantum antigen for 7 days, with DHPP for 10 days, or with ConA for 4 days. CD4+ T-cell proliferation was assessed via CFSE dilution using flow cytometry. At least three separate experiments were carried out for each dog in every group. Bars indicate the average proliferation for each group (± standard errors of the means).
Disease progression and antigen-specific PBMC IFN-γ and IL-10 production.
Treated individuals develop a cell-mediated immune response capable of offering protection from reinfection, as characterized by antigen-specific IFN-γ responses (7, 34). In contrast, individuals with advanced VL show a decrease in antigen-specific IFN-γ production and elevated levels of the immunoregulatory cytokine IL-10 in serum and increased IL-10 mRNA expression in lesional tissue (6, 10). The correlation between VL disease progression and IL-10 production in humans is now well established (22). In CVL, IFN-γ-mediated responses seem to predominate in L. infantum-infected but asymptomatic dogs (23). Similar to human disease, IL-10 mRNA expression has been positively correlated with parasitic load and progression of clinical disease in naturally infected dogs (15). In order to determine the correlation between disease and cytokine production in our cohort, culture supernatants from PBMC restimulated with L. infantum antigen were assayed for IFN-γ (Fig. 3A) and IL-10 (Fig. 3B) production. Production of IFN-γ and IL-10 from PBMC in the control group (Fig. 3A and B) was below the detection limit of the assay (16 pg/ml and 10 pg/ml, respectively). PMBC from infected resistant and infected susceptible animals produced comparable levels of IFN-γ (Fig. 3A). PBMC from infected resistant dogs produced significantly higher levels of IFN-γ than noninfected animal PMBC. PBMC from clinical dogs, however, produced significantly lower amounts of IFN-γ than infected susceptible and infected resistant dogs.
FIG. 3.
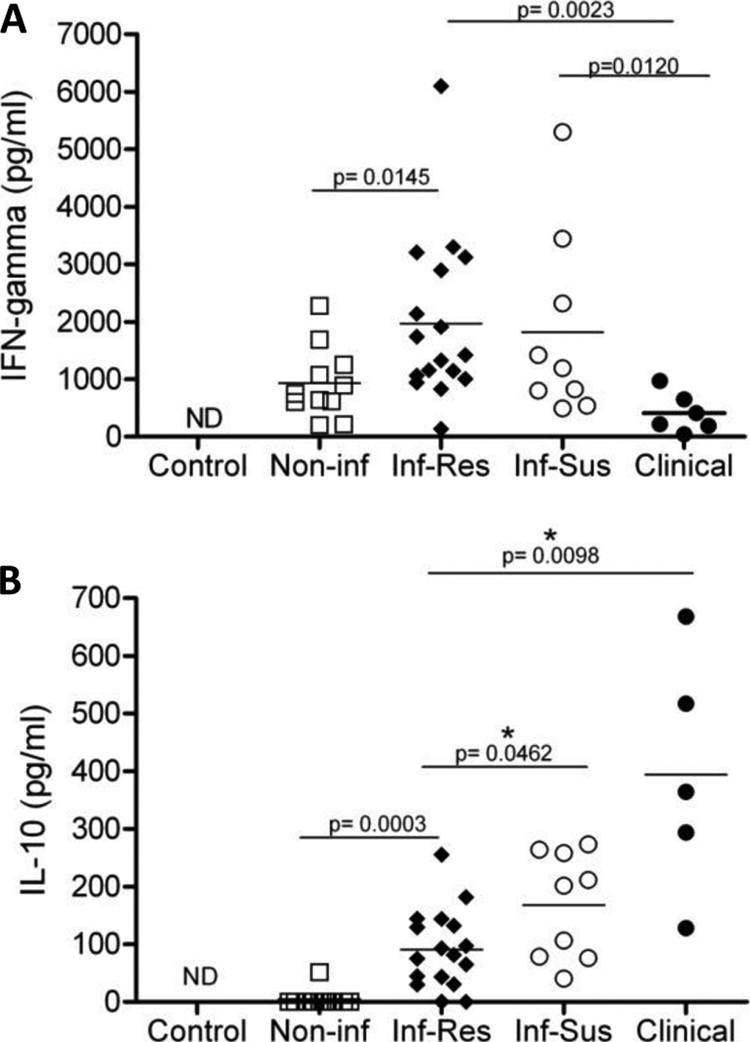
Disease progression correlates with decreased IFN-γ and increased IL-10 production. Shown are PBMC effector cytokine responses from the control (one dog), noninfected (two dogs), infected resistant (four dogs), infected susceptible (two dogs), and clinical (two dogs) animals. Culture supernatants were collected from PBMC cultures stimulated with L. infantum antigen for 7 days and analyzed via ELISA for IFN-γ (A) and IL-10 (B). Each point is indicative of one experiment. At least three separate experiments were carried out for each dog in every group. Lines indicate the mean responses for each group. *, significant difference (P < 0.05). ND, not detectable.
Analysis of IL-10 production from culture supernatants indicated a significant increase in the production of this cytokine with disease progression. PBMC from dogs in the clinical group produced the greatest amount of IL-10 compared to all other groups (Fig. 3B), with decreasing amounts detected from infected susceptible and then infected resistant dogs. All three groups were significantly different from one another. PBMC from noninfected dogs produced levels of IL-10 that were below the detection limit of the assay (10 pg/ml). These data demonstrate that clinical progression and loss of antigen-specific T-cell proliferation in our cohort were associated with decreased levels of antigen-specific IFN-γ production and increased production of IL-10 in response to L. infantum antigen restimulation.
DISCUSSION
During CVL, susceptibility to symptomatic infection has been associated with increased antibody production and loss of L. infantum-specific CD4+ T-cell function with a concomitant increase in immunosuppressive mechanisms. However, little is known regarding the mechanisms that control the balance between resistance to infection and susceptibility. Characterization of measurable immunopathological end points may provide a means to better predict disease development in infected dogs. Our studies using a cohort of naturally infected dogs show how changes in IgG production, lymphoproliferative responses, and effector cytokine production correlate with the appearance of clinical signs and disease progression.
In our study increases in serologic titer were associated with disease progression (Table 1). The highest titers (1:256 and 1:512) were observed in dogs displaying mild to severe clinical disease within the infected susceptible and clinical groups. Moreover, high antibody titers also correlated with the detection of L. infantum parasites in peripheral blood samples via qPCR (Table 1), indicating an increase in circulating parasites later in disease. Analysis of antigen-specific IgG1 and IgG2 in sera of the four groups of dogs showed an increase in both isotypes with disease progression (Fig. 1A and B). Infected susceptible and clinical dogs exhibited the highest OD values, indicating increased IgG1 and IgG2 levels compared to noninfected and infected resistant dogs, but there was no clear difference between isotypes regarding clinical state or progression.
During human VL, increased levels of anti-Leishmania IgG have been shown to have a negative correlation with delayed-type hypersensitivity (DTH) responses (17). Here we show that along with increased IgG in sera, L. infantum antigen responsiveness of PBMC CD4+ T cells significantly decreased in the clinical group of animals (Fig. 2). This loss of lymphoproliferation has been described as “immune exhaustion” due to unchecked levels of pathogen antigen (2, 19). Infected susceptible animals showed the most robust proliferative response compared to all other groups. Proliferation in the noninfected foxhound group may be attributed to nonspecific proliferation or perhaps a dwindling recall response. Animals in this group were donated as part of a litter of puppies born to a seropositive, qPCR-positive female. It is therefore possible that they may have been exposed to L. infantum parasites in utero at a very low dose, leading to exposure and some T-cell activation but perhaps not patent infection. Altogether, our data show that PBMC CD4+ T cells from L. infantum-infected dogs respond to antigen stimulation during the earlier stages of infection but lose that ability as they progress to clinical disseminated disease, negatively correlating with the increased levels of IgG in sera.
Antibody production is an important contributor to VL pathology due to antigen-antibody complex deposition. B-cell activation and increased IgG production are observed in conjunction with IL-10 overproduction during VL (22). To determine what effector cytokines were produced by the dampened T cells with limited antigen responsiveness in our cohort, we assessed IFN-γ and IL-10 production in cultured PMBC. We found that decreased proliferative responses in the clinical group were accompanied by significantly decreased IFN-γ production (Fig. 3A) and significantly increased IL-10 production (Fig. 3B). This profile matches observed changes in cytokine production in human cohort studies (10, 20, 21, 31) and dogs (23) in areas of endemicity. Our infected resistant and infected susceptible groups produced similar levels of IFN-γ; however, the infected susceptible group showed significantly increased production of IL-10 compared to the infected susceptible group. The observed increase in IL-10 production along with increased blood parasite burden may be specific factors which promote clinical disease.
The factors that determine disease progression in CVL remain poorly understood. It is clear that no one clinical parameter can be used to predict which infected dogs will likely become clinically ill. Our studies using our canine cohort of progressive CVL indicate that several key changes in clinical parameters should be considered, including a rise in IgG production and a progressive increase of antigen-specific PBMC proliferation followed by a decreased IFN-γ-mediated response, a dramatic increase in IL-10 production, and consistent detection of parasite kDNA in whole blood. Further studies are needed to fully understand the relationship between increased IgG, IL-10 production, and parasite load. While it has been shown that all three of these events precede clinical disease (16, 17, 23), the causal relationship between them is yet to be determined. Understanding which event drives the others may provide insights into the mechanisms leading to VL and for future immunotherapies.
Acknowledgments
This work was supported by the American Kennel Club CHF ACORN grants 799-A and 1220-A.
We thank Marie Bockenstedt, Jenna Bjork, Kevin Esch, Alex Osanya, and Clara Haydée Quevedo Salazar for their technical assistance. We thank the ISU LAR staff and the collaborating Foxhound Hunts for their support.
Footnotes
Published ahead of print on 23 December 2009.
REFERENCES
- 1.Barbieri, C. L. 2006. Immunology of canine leishmaniasis. Parasite Immunol. 28:329-337. [DOI] [PubMed] [Google Scholar]
- 2.Blackburn, S. D., and E. J. Wherry. 2007. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 15:143-146. [DOI] [PubMed] [Google Scholar]
- 3.Carvalho, E. M., A. Barral, D. Pedral-Sampaio, M. Barral-Netto, R. Badaro, H. Rocha, and W. D. Johnson, Jr. 1992. Immunologic markers of clinical evolution in children recently infected with Leishmania donovani chagasi. J. Infect. Dis. 165:535-540. [DOI] [PubMed] [Google Scholar]
- 4.Chappuis, F., S. Sundar, A. Hailu, H. Ghalib, S. Rijal, R. W. Peeling, J. Alvar, and M. Boelaert. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 5:873-882. [DOI] [PubMed] [Google Scholar]
- 5.Duprey, Z. H., F. J. Steurer, J. A. Rooney, L. V. Kirchhoff, J. E. Jackson, E. D. Rowton, and P. M. Schantz. 2006. Canine visceral leishmaniasis, United States and Canada, 2000-2003. Emerg. Infect. Dis. 12:440-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gama, M. E., J. M. Costa, J. C. Pereira, C. M. Gomes, and C. E. Corbett. 2004. Serum cytokine profile in the subclinical form of visceral leishmaniasis. Braz. J. Med. Biol. Res. 37:129-136. [DOI] [PubMed] [Google Scholar]
- 7.Garg, R., S. K. Gupta, P. Tripathi, S. Naik, S. Sundar, and A. Dube. 2005. Immunostimulatory cellular responses of cured Leishmania-infected patients and hamsters against the integral membrane proteins and non-membranous soluble proteins of a recent clinical isolate of Leishmania donovani. Clin. Exp. Immunol. 140:149-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaskin, A. A., P. Schantz, J. Jackson, A. Birkenheuer, L. Tomlinson, M. Gramiccia, M. Levy, F. Steurer, E. Kollmar, B. C. Hegarty, A. Ahn, and E. B. Breitschwerdt. 2002. Visceral leishmaniasis in a New York foxhound kennel. J. Vet. Intern. Med. 16:34-44. [DOI] [PubMed] [Google Scholar]
- 9.Gavgani, A. S., M. H. Hodjati, H. Mohite, and C. R. Davies. 2002. Effect of insecticide-impregnated dog collars on incidence of zoonotic visceral leishmaniasis in Iranian children: a matched-cluster randomised trial. Lancet 360:374-379. [DOI] [PubMed] [Google Scholar]
- 10.Ghalib, H. W., M. R. Piuvezam, Y. A. Skeiky, M. Siddig, F. A. Hashim, A. M. el-Hassan, D. M. Russo, and S. G. Reed. 1993. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J. Clin. Invest. 92:324-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghalib, H. W., J. A. Whittle, M. Kubin, F. A. Hashim, A. M. el-Hassan, K. H. Grabstein, G. Trinchieri, and S. G. Reed. 1995. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J. Immunol. 154:4623-4629. [PubMed] [Google Scholar]
- 12.Gibson-Corley, K. N., J. M. Hostetter, S. J. Hostetter, K. Mullin, A. E. Ramer-Tait, P. M. Boggiatto, and C. A. Petersen. 2008. Disseminated Leishmania infantum infection in two sibling foxhounds due to possible vertical transmission. Can. Vet. J. 49:1005-1008. [PMC free article] [PubMed] [Google Scholar]
- 13.Jones, D. E., M. R. Ackermann, U. Wille, C. A. Hunter, and P. Scott. 2002. Early enhanced Th1 response after Leishmania amazonensis infection of C57BL/6 interleukin-10-deficient mice does not lead to resolution of infection. Infect. Immun. 70:2151-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones, D. E., L. U. Buxbaum, and P. Scott. 2000. IL-4-independent inhibition of IL-12 responsiveness during Leishmania amazonensis infection. J. Immunol. 165:364-372. [DOI] [PubMed] [Google Scholar]
- 15.Lage, R. S., G. C. Oliveira, S. U. Busek, L. L. Guerra, R. C. Giunchetti, R. Correa-Oliveira, and A. B. Reis. 2007. Analysis of the cytokine profile in spleen cells from dogs naturally infected by Leishmania chagasi. Vet. Immunol. Immunopathol. 115:135-145. [DOI] [PubMed] [Google Scholar]
- 16.Manna, L., S. Reale, F. Vitale, and A. E. Gravino. 2009. Evidence for a relationship between Leishmania load and clinical manifestations. Res. Vet. Sci. 87:76-78. [DOI] [PubMed] [Google Scholar]
- 17.Miles, S. A., S. M. Conrad, R. G. Alves, S. M. Jeronimo, and D. M. Mosser. 2005. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J. Exp. Med. 201:747-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miro, G., L. Cardoso, M. G. Pennisi, G. Oliva, and G. Baneth. 2008. Canine leishmaniosis. New concepts and insights on an expanding zoonosis: part two. Trends Parasitol. 24:371-377. [DOI] [PubMed] [Google Scholar]
- 19.Mueller, S. N., and R. Ahmed. 2009. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 106:8623-8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy, M. L., U. Wille, E. N. Villegas, C. A. Hunter, and J. P. Farrell. 2001. IL-10 mediates susceptibility to Leishmania donovani infection. Eur. J. Immunol. 31:2848-2856. [DOI] [PubMed] [Google Scholar]
- 21.Nylen, S., R. Maurya, L. Eidsmo, K. D. Manandhar, S. Sundar, and D. Sacks. 2007. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J. Exp. Med. 204:805-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nylen, S., and D. Sacks. 2007. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol. 28:378-384. [DOI] [PubMed] [Google Scholar]
- 23.Panaro, M. A., O. Brandonisio, A. Cianciulli, P. Cavallo, V. Lacasella, P. Paradies, G. Testini, D. De Caprariis, V. Mitolo, and D. Otranto. 2009. Cytokine expression in dogs with natural Leishmania infantum infection. Parasitology 136:823-831. [DOI] [PubMed] [Google Scholar]
- 24.Petersen, C. A., and S. C. Barr. 2009. Leishmaniasis in North America: emerging or newly recognized? Vet. Clin. North Am. Small Anim. Pract. 39:1065-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinnell, R. J., O. Courtenay, L. M. Garcez, P. M. Kaye, M. A. Shaw, C. Dye, and M. J. Day. 2003. IgG subclass responses in a longitudinal study of canine visceral leishmaniasis. Vet. Immunol. Immunopathol. 91:161-168. [DOI] [PubMed] [Google Scholar]
- 26.Reis, A. B., A. Teixeira-Carvalho, A. M. Vale, M. J. Marques, R. C. Giunchetti, W. Mayrink, L. L. Guerra, R. A. Andrade, R. Correa-Oliveira, and O. A. Martins-Filho. 2006. Isotype patterns of immunoglobulins: hallmarks for clinical status and tissue parasite density in Brazilian dogs naturally infected by Leishmania (Leishmania) chagasi. Vet. Immunol. Immunopathol. 112:102-116. [DOI] [PubMed] [Google Scholar]
- 27.Roberts, L. J., E. Handman, and S. J. Foote. 2000. Science, medicine, and the future: leishmaniasis. BMJ 321:801-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez-Cortes, A., H. Fernandez-Bellon, A. Ramis, L. Ferrer, J. Alberola, and L. Solano-Gallego. 2007. Leishmania-specific isotype levels and their relationship with specific cell-mediated immunity parameters in canine leishmaniasis. Vet. Immunol. Immunopathol. 116:190-198. [DOI] [PubMed] [Google Scholar]
- 29.Rosypal, A. C., and D. S. Lindsay. 2005. Non-sand fly transmission of a North American isolate of Leishmania infantum in experimentally infected BALB/c mice. J. Parasitol. 91:1113-1115. [DOI] [PubMed] [Google Scholar]
- 30.Rosypal, A. C., G. C. Troy, A. M. Zajac, R. B. Duncan, Jr., K. Waki, K. P. Chang, and D. S. Lindsay. 2003. Emergence of zoonotic canine leishmaniasis in the United States: isolation and immunohistochemical detection of Leishmania infantum from foxhounds from Virginia. J. Eukaryot. Microbiol. 50(Suppl.):691-693. [DOI] [PubMed] [Google Scholar]
- 31.Sacks, D. L., S. L. Lal, S. N. Shrivastava, J. Blackwell, and F. A. Neva. 1987. An analysis of T cell responsiveness in Indian kala-azar. J. Immunol. 138:908-913. [PubMed] [Google Scholar]
- 32.Schantz, P. M., F. J. Steurer, Z. H. Duprey, K. P. Kurpel, S. C. Barr, J. E. Jackson, E. B. Breitschwerdt, M. G. Levy, and J. C. Fox. 2005. Autochthonous visceral leishmaniasis in dogs in North America. J. Am. Vet. Med. Assoc. 226:1316-1322. [DOI] [PubMed] [Google Scholar]
- 33.Solano-Gallego, L., A. Koutinas, G. Miro, L. Cardoso, M. G. Pennisi, L. Ferrer, P. Bourdeau, G. Oliva, and G. Baneth. 2009. Directions for the diagnosis, clinical staging, treatment and prevention of canine leishmaniosis. Vet. Parasitol. 165:1-18. [DOI] [PubMed] [Google Scholar]
- 34.White, A. C., Jr., M. Castes, L. Garcia, D. Trujillo, and L. Zambrano. 1992. Leishmania chagasi antigens recognized in cured visceral leishmaniasis and asymptomatic infection. Am. J. Trop. Med. Hyg. 46:123-131. [DOI] [PubMed] [Google Scholar]